Clostridium difficile is a Gram-positive, spore-forming bacillus. Since Bartlett et al. [Reference Bartlett1] implicated it as an important enteropathogen responsible for antimicrobial-associated colitis in 1978, C. difficile has been confirmed as the primary cause of antibiotic-associated diarrhoea and virtually all cases of pseudomembranous colitis [Reference Spencer2]. It is the current leading cause of nosocomial diarrhoea and numerous outbreaks have been reported in hospitals globally [Reference McEllistrem3]. Recent data from 106 laboratories of 34 European countries indicate that C. difficile diarrhoeal infection in member hospitals ranged from 0 to 36·3 cases/10 000 patient-days [Reference Bauer4]. However, data from China on the incidence of C. difficile infection are quite limited [Reference Cheng5, Reference Huang6] and multi-centre surveys especially in the mainland are lacking.
In modern epidemic research, genotyping of strains plays an important role in tracing of sources and identification of epidemiological clusters, as well as informing evolutionary paths and comparisons of lineages in a global context. For example, PCR ribotyping is used in several countries to define epidemic C. difficile strains, and has identified the widespread ribotype O27 to be highly associated with outbreaks of infections in hospitals [Reference Bishara7]. With strong internet support, MLST facilitates the separation of isolates by indexing variation in intragenic sequences of several (usually 6–10) housekeeping genes. This method has been widely used for population genetics studies and global epidemiological analysis of many procaryotic species owing to its stability and portability of data between laboratories [Reference Maiden8]. For C. difficile, Griffiths et al. [Reference Griffiths9] developed a new MLST scheme and set up an internet-accessible database to allow simple depositing, retrieval and comparison of data. However, the information contained in the database consists mainly of strains from European countries, with few representatives from Asia, particularly from China. Moreover, no systematic phylogenetic study on C. difficile isolates from different regions and patient age groups has been conducted in China, and geographical differences of gene diversity and evolution of C. difficile remain unknown in this country. In the current study, we analysed the genetic lineages of isolates from China using MLST described by Griffiths et al. [Reference Griffiths9] to (i) define sequence polymorphism of the strains; (ii) explore the genetic polymorphisms and relationships between strain lineages from different regions and host populations; and (iii) provide insights into the origins and evolutionary relationships among the strains.
A total of 104 clinical isolates from three different hospitals in different regions of China were analysed; 88 isolates were from adult (⩾18 years) hospitalized patients patients (Beijing n = 69, Guangzhou n = 16, Shandong n = 3). The other 16 isolates were from outpatient children (<1 year) in Beijing. Stool specimens from patients with diarrhoea were collected using Transwabs (MW&E Ltd, UK) and cultured on cycloserine-cefoxitin-fructose-egg yolk agar (CCFA) and the plates were incubated anaerobically at 37 °C for at least 48 h. Colonies that demonstrated a typical morphology (flat, yellow, ground-glass appearance) and odour on CCFA, were Gram-positive bacilli with subterminal spores, and positive by the commercially available latex agglutination test (Oxoid Ltd, UK) were classified as C. difficile. Isolates that could not be confirmed by these methods were further investigated by 16S rDNA amplification and sequencing. For DNA extraction, the isolates were subcultured overnight from a single colony onto brain heart infusion agar (containing 5% defibrinated sheep blood) at 37 °C under anaerobic conditions. Genomic DNA was extracted using the Qiagen DNeasy blood and tissue kit (Qiagen, Germany) according to manufacturer's instructions for Gram-positive organisms, and stored at −20 °C for further study. MLST was performed using a previously described scheme [Reference Griffiths9]. PCR of the seven loci were performed and amplicons were sequenced with forward and reverse primers; DNA sequences were submitted to the MLST database (http://pubmlst.org/clostridium difficile) to obtain the sequence type (ST). Assays for the detection of toxins A and B encoded by tcdA and tcdB genes, respectively, were performed as described previously [Reference Kato10, Reference Lemée11].
MLST generated 22 different STs from the 104 isolates (Fig. 1). ST117, ST118, ST119 and ST129 were novel. Ten types were represented by single isolates. ST37 (ribotype O17) was the dominant type and accounted for 25 isolates, the other two most frequent types were ST54 (ribotype O12) and ST35 (ribotype O46). These three STs were also the most frequent types responsible for many C. difficile diarrhoeal infection cases in Europe [Reference Lemée and Pons12]. No correlation was found between the geographical origin and the genotype. For example, 85 isolates from Beijing were genotyped into 21 STs; three Shandong isolates into three STs; and 16 isolates from Guangzhou into four STs (Fig. 1). Furthermore, ST2 contained isolates from Beijing and Guangzhou, and both ST35 and ST37 contained isolates from Beijing, Guangzhou and Shandong. In addition, there was no correlation between the genotype and the host population origin. As 16 isolates from children were typed into nine STs and 88 isolates from adults were distributed among 20 STs; strains belonging to sequence type 2, 3, 35, 37, 54, 55 and 118 isolates were recovered from both children and adults. However, there was an interesting correlation between STs and toxin types and all strains of ST37 were toxin type A−B+.

Fig. 1. Neighbour-joining tree constructed based on the composite sequence of seven housekeeping gene fragments. Bootstraps were generated using 1000 replicates. BJ, Beijing; SD, Shandong; GZ, Guangzhou; ◂, new sequence types.
A neighbour-joining tree was generated (Fig. 1) from concatenated sequences of the seven housekeeping genes by using Molecular Evolutionary Genetics Analysis (MEGA) version 5 (http://www.megasoftware.net/). Strains belonging to ST37 (A−B+) and ST5 (associated with ribotype O23 [Reference Griffiths9]) constituted single (distinct) lineages while the majority of the STs (including the four novel types) were clustered into one lineage and constituted a homogeneous population because of the very high degree of conservation, indicating that neither geographically specific nor host-population lineages were generated.
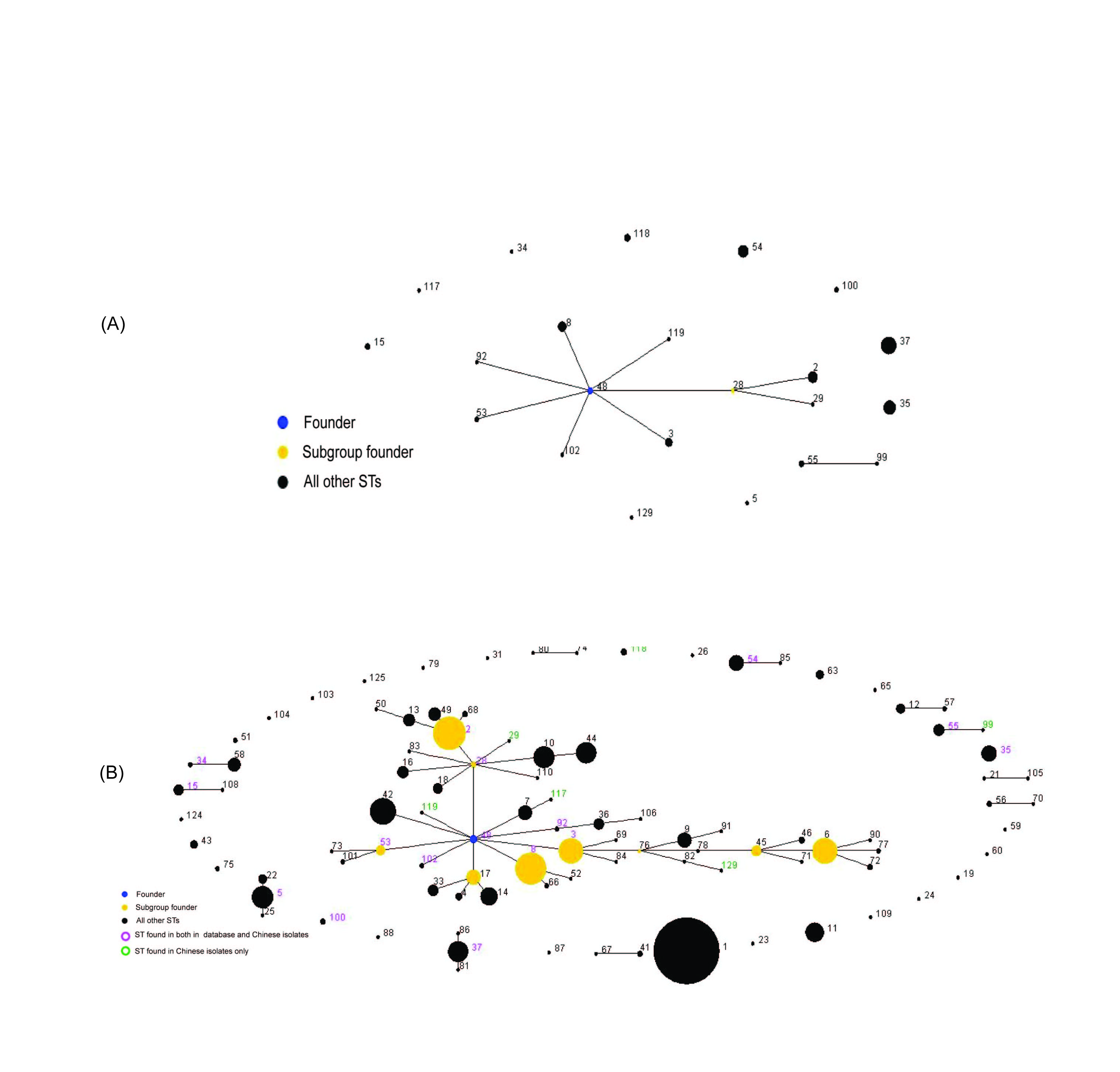
An analysis of the MLST data of 104 isolates (Supplementary Fig. S1) was performed using eBURST, version 3 (http://eburst.mlst.net/). The default eBURST setting was adopted, which defined a group as all members assigned to the same group and sharing identical alleles at >6 of the seven loci with at least one other member of the group; the primary founder of a group was defined as the ST that differed from the largest number of other STs at only a single locus. Two groups were generated and ST48 was identified as the group founder of the larger group. Ten STs (2, 3, 8, 28, 29, 48, 53, 92, 109, 119) were placed in the latter group, and ST55 and ST99 in the second group; the other STs being singletons. The first group comprised isolates from Beijing (27/85), Guangzhou (6/16), and Shandong (1/3). Moreover, the first group included isolates from children (4/16) and adults (30/88), suggesting that neither a geographically specific nor host population-specific group existed. A comparison of the 104 isolates from China with the 1423 isolates maintained in the MLST database created by Dingle (http://pubmlst.org/cdifficile/) (Supplementary Fig. S1), showed that our isolates were widely scattered in seven groups, and not specific to China, a finding which suggests the global spread of some lineages.
Lineage assignment showed that the isolates from children did not cluster in lineages distinct from that of adults, and no specific infant lineage was found. Based on the neighbour-joining tree, the strains from Guangzhou (all from the same Gastroenterology Department) are of a multi-evolutionary origin, although most strains (9/16) belonged to ST37 which might imply a common source of infection. The strains from Shandong of three different STs were most likely of sporadic origin. However, in Beijing, of the most frequent types (ST37, ST35, ST54, ST8), ST35 was widely distributed among general surgery, dermatology, intensive-care units, etc. in this hospital. Four of the eight ST8 strains from Beijing adults originated from the Department of Gastroenterology in 2010 and 2011, and 4/12 strains of ST37 were isolated from the Department of Hematology within 8 months in the same hospital. This finding indicates that C. difficile infections in these two departments were due mainly to distinct strains.
As previously reported by Lemée and co-workers [Reference Lemée and Pons12], most of the A−B+ variant isolates identified in the current study were ST37 but this toxin genotype was also found in sequence types 15, 48, 100, 117, 118 and 119. Furthermore, two toxin types (A−B+, A+B+) were identified in ST15. This may be the result of high genetic polymorphism in the Paloc region, due to instability within the same genetic background [Reference Dingle13]. We have previously observed from single nucleotide polymorphism analysis [Reference Cheng14], that isolates in the inter-group of toxin A+B+ and intragroup between A+B+ and A−B+, exhibited more diversity in the Paloc region of seven published C. difficile genomes.
In conclusion, this is to the best of our knowledge the first description of MLST analysis of C. difficile strains from China. We have confirmed the dominance of the ST37 (A−B+) strain in our collection of strains from different regions which constitute a distinct lineage. Four new STs were identified and high genetic polymorphism of toxin genes was found. There was no correlation between genotypes and geographical origin and between genotypes and host population.
SUPPLEMENTARY MATERIAL
For supplementary material accompanying this paper, visit http://dx.doi.org/10.1017/S0950268812000453.
ACKNOWLEDGEMENTS
This study was supported by the National Natural Science Foundation of China (no. 81101218) and by the Young Scholar Scientific Research Foundation of China CDC (no. 2011A101). This publication made use of the Clostridium difficile multilocus sequence typing website (http://pubmlst.org/cdifficile/) developed by Keith Jolley and sited at the University of Oxford.
DECLARATION OF INTEREST
None.