


INTRODUCTION
Infection control programmes use a multi-pronged approach to the prevention and detection of healthcare-acquired infections. Reducing pathogen transmission is a key programme element [Reference Siegel1] but transmission pathways may be complex. Therefore, in addition to transmission estimates derived from infection prevalence [Reference Pelupessy, Bonten and Diekmann2], active surveillance for colonized patients and pathogen typing may be used to drive both empirical infection control practices and the mathematical models intended to inform and guide those practices [Reference Siegel1, Reference Austin3–Reference Forrester and Pettitt8].
Mathematical models of pathogen dynamics in hospitals have examined the relationship between rates of exogenous transmission and other factors underlying pathogen prevalence, such as the presence of asymptomatic carriers, antibiotic selection on endogenous flora and stochastic events [Reference Bonten, Austin and Lipsitch4, Reference Smith9, Reference Mathews and Woolhouse10]. These models show that small changes in transmissibility can have a major impact on pathogen prevalence [Reference Cooper, Medley and Scott11, Reference Lipsitch, Bergstrom and Levin12]. Since transmission plays a central role in prevalence, estimates of transmission parameters are critical to model-based formulation of infection control interventions, but the required combination of surveillance and genotyping to acquire such data has been considered economically and technically prohibitive [Reference Mathews and Woolhouse10]. Further, the lack of empirical transmission estimates represented a major limitation to model-based inference [Reference Pelupessy, Bonten and Diekmann2, Reference Cooper, Medley and Scott11]. More recently, detection of nosocomial transmissions has been strengthened by analyses based on Markov models and cluster detection methods while estimates and predictions from those models have gained accuracy with more suitable and diverse empirical data [Reference Grundmann5–Reference Forrester and Pettitt8, Reference McBryde13].
The ability to detect growing transmission clusters and potential impending outbreaks based on as few as two genetically identical isolates can be an invaluable prospective tool for infection control [Reference Mellmann14]. The goal of this study was to use genetic identity to quantify the magnitude of pathogen transmission throughout a medical centre and to evaluate potential risk factors associated with transmission. Our approach was based on identification of temporal clusters of genetically identical pathogen isolates. Because hospital populations are relatively small and undergo frequent turnover, random effects can have a major impact on pathogen prevalence [Reference Grundmann and Hellriegel15]. To minimize the influence of such stochastic processes, accurate estimation of transmission parameters should be based on long-term longitudinal data [Reference Grundmann and Hellriegel15, Reference DeRiemer and Daley16]. We illustrate the utility of our approach through analysis of a long-term, single species collection from a medical centre.
The analysis was conducted on Moraxella catarrhalis, a respiratory tract commensal and pathogen in adults. M. catarrhalis is considered an emerging pathogen because in the 1980s, the species experienced a remarkably rapid global acquisition of a chromosomally encoded β-lactamase [Reference Verduin17–Reference Wallace, Nash and Steingrube20]. Global and regional genetic diversity have been assessed in M. catarrhalis [Reference Walker and Levy21, Reference Verhaegh22] while genotype clusters have been used to infer outbreaks [Reference Patterson23, Reference Morgan24] and intrafamiliar transmission [Reference Watanabe25]. More recent studies have documented the dissemination of serum-resistant taxonomic clades [Reference Wirth26, Reference Attia27]. The availability of a long-term, genotypically characterized collection of a respiratory pathogen provided the raw material for a powerful retrospective analysis of pathogen transmission in a comprehensive medical centre and as such, represents an extension of studies based on single wards or acute care units.
METHODS
Study population and facility
Our approach was based on the tenet that pathogen transmissions between patients within a hospital will give rise to temporally delineated clusters of genetically identical isolates [Reference Mellmann14, Reference Grundmann28]. Temporally defined clusters were chosen as the standard because they are free of a priori spatial restrictions that often limit analyses to single units within a hospital. A highly discriminating genotyping system was used to distinguish identical from non-identical isolates [Reference Walker and Levy21, Reference Walker29]. Considering the extensive genetic diversity in the study population, genetic identity of isolates is particularly appropriate for transmission inference because it eliminates dependence on unknown mutation/evolutionary rate parameters that may arise from using measures of relatedness. Our definition of patient–patient transmission includes transmission pathways through unidentified intermediaries, e.g. patient–caregiver–patient, and patient–surface–patient.
We analysed a sample from a long-term (1984–1994) collection of M. catarrhalis comprised of all patient isolates from the James H. Quillen Veterans Affairs Medical Center (VAMC) at Mountain Home, Tennessee, USA. The VAMC encompasses an acute-care hospital, a nursing home, domiciliary, and several outpatient clinics. Samples from the collection have been well characterized phenotypically (antibiotic resistance phenotypes), genetically (three-locus PCR–RFLP genotypes) and epidemiologically (collection date and patient location) [Reference Walker and Levy21, 29–Reference Walker31]. The sample of 367 isolates was derived from 347 unique patients. More than one isolate from the same patient were included if those isolates differed in genotype (16 patients) or, when the isolates carried identical genotypes, if the isolation dates were separated by more than 100 days (one patient). Isolates were selected randomly within years with proportional representation across years to generate a sample representative of the population. All isolates were typable using three-locus PCR–RFLP. Details of the sample and identity of the genomic regions used in typing, PCR primers and cycling parameters, and RFLP procedures are given in Walker & Levy [Reference Walker and Levy21]. The typing system distinguished 148 three-locus genotypes in the 367 isolates. Only 10 genotypes were represented by 10 or more isolates; the most common genotype was found in 23 isolates, representing 6% of the total. The sample and typing system were particularly amenable to cluster analysis to infer transmissions because virtually all genotypes would be considered rare by the criterion of occurrence in <5% of the population. Moreover, the collection spans the transition to β-lactamase production, the primary antibiotic resistance factor in the species, and it encompasses a time when there was major renovation and new construction for the medical centre. Hence, the current analysis afforded an opportunity to assess the impacts of antibiotic resistance and facility configuration on pathogen transmission.
Cluster analyses
Temporal clusters
Temporal clusters of isolates were identified using the scan statistic based on a Poisson model as implemented by SatScan software version 7.02 [Reference Kulldorff32]. Significance was assessed with 999 Monte Carlo randomization replications. P values ⩽0·05 were considered significant. The scanning window was set at 3% of the total time (approximately equivalent to a 3-month season) because M. catarrhalis is a seasonal pathogen, but results were largely insensitive to a range of scanning windows. In addition, a 50% scanning window was used to search for long-running clusters. Analysis was first conducted on the entire sample to identify genotype-independent clusters followed by separate cluster analyses for each of the 52 genotypes represented by two or more isolates.
Spatial clusters
To evaluate the relative importance of spatial location in regard to bacterial transmission, the 36 facility locations from which isolates were derived were assigned x,y map coordinates. Vertical distance was specified through a z coordinate that corresponded to building floor. The scan statistic was used to identify spatial clusters regardless of isolate genotypes.
Epidemiological associations
Seasonal effects
Respiratory infections caused by M. catarrhalis and other bacterial pathogens tend to undergo seasonal fluctuations characterized by a winter peak [Reference Stone, Olinky and Huppert33]. Similarly, infectious transmissions may also occur with seasonal peaks. Logistic regression was used to test for seasonal differences in the proportion of isolates in clusters. To analyse the impact of seasonality on isolate recovery and clustering, fractional components of clusters that spanned seasons were assigned to seasons based on the number of clustered isolates within each season.
Antibiotic resistance
For each of the 40 significant temporal genotype clusters, we determined whether the clustered isolates were more or less likely to be β-lactamase producers relative to: (i) isolates of the same genotype whose occurrence was outside of that temporal cluster, and (ii) isolates of any genotype in temporal proximity to the cluster. For the latter, we chose as the reference isolates within ±50 days of the clustered isolates, a period of sufficient duration to ensure adequate samples sizes but not long enough to encompass significant genotype turnover or seasonal fluctuations. Differences in the frequency of β-lactamase producers between cluster and non-cluster isolates were assessed using Fisher's exact test.
Facility configuration
Patients in acute care were housed in multi-bed wards prior to completion of a major hospital renovation at the end of 1990, after which patients were assigned to rooms with 1–4 beds. The bed capacity for the hospital dropped from an average of 446 beds to 353 beds. A new nursing home was opened in 1992 at which time capacity more than doubled, from 58 to 120 beds. In 1993, domiciliary residents were transferred from several smaller buildings to a new single domiciliary but domiciliary beds remained stable at 540 beds. A gradual decline in clusters or cluster characteristics could be misinterpreted as a beneficial consequence of the hospital renovation if the time of the decline encompassed the renovation. Therefore, linear regression included variables to test for trends in clustering over the entire time, for a trend in patient days, and for a difference between the older and newer configuration eras.
Outbreak-prone genotypes
If a genotype was particularly prone to outbreaks, the number of isolates of that genotype should constitute a greater proportion of the clustered isolates relative to the entire sample. Similarity of genotypic prevalence in the sample and cluster subset of isolates was tested using a χ2 goodness-of-fit statistic. The expectation for each genotype was calculated as the product of the number of isolates clustered×the proportion a genotype represented in the sample of genotypes represented by four or more isolates.
Statistical methods
Logistic regression was used to test whether epidemiologically relevant potential risk factors were associated with clusters. The dichotomous response variable was whether or not an isolate was included in any cluster. Independent variables were season [winter (January–March), spring (April–June), summer (July–September), autumn (October-December)], β-lactamase status (producer or non-producer), and hospital configuration (pre- or post-alteration). Further analyses were conducted to dissect the relationship between each of the potential risk factors and the cluster attributes; number of clusters, cluster size, cluster duration, and the proportion of clustered isolates. ANOVAs were used to test for differences in cluster size and duration. The Mann–Whitney U statistic was used to compare durations between β-lactamase producers and non-producers. Frequency variables were analysed using Fisher's exact test for 2×2 contingency tables or heterogeneity χ2 for larger tables.
RESULTS
Genotype-independent clusters
Six genotype-independent, non-overlapping temporal clusters were identified (all P<0·003), four of which were comprised of relatively high numbers of isolates (27–39) (Figure 1a, d). Each of these four clusters was characterized by long persistence (107–152 days), winter occurrence and representation of diverse genotypes (17–24 different genotypes). These large winter season clusters accounted for 38% of the isolates but only 14% of the total time. Moreover, while 40% of the isolates were recovered in winter, winter accounted for 67% of the isolates in the four temporal clusters.

Fig. 1. (a) Number of isolates by year and season. The arrow on the x-axis points to the date of transition to the reconfigured facility. Timelines of: (b) genotype clusters of ⩾100 days, (c) genotype clusters of durations <100 days, (d) genotype-independent temporal clusters. Timescales as in (a). In panels (b) and (d), each arrow points to a cluster duration midpoint within a box whose width is scaled to duration. Number above the bar or arrow is the number of isolates in the cluster. For clusters <100 days, width of the bar corresponds to cluster durations of: ![]() =0–10 days;
=0–10 days; ![]() =11–30 days;
=11–30 days; ![]() =31–60 days;
=31–60 days; ![]() =61–99 days. A, Autumn; W, winter; Sp, spring, S, summer.
=61–99 days. A, Autumn; W, winter; Sp, spring, S, summer.
Only one significant spatial cluster was identified, a cluster of 30 isolates that encompassed all isolates from the only two wards in a building separate from the main hospital. This cluster spanned 5 years and included 20 different genotypes. These spatially separated wards were closed after the facility renovation.
Temporal genotype clusters
There were 40 significant temporal genotype clusters involving 33 different genotypes (Fig. 1b, c). Isolates in these clusters accounted for 142 (38·7%) of the 367 isolates. The mean prevalence was one cluster per 92 days or approximately four events per year and per 137 474 patient-days. The mean cluster size and duration was 3·55 days and 64·7 days, respectively, with size and duration medians of 3 days and 24·5 days, respectively. Thirty-eight percent of the clusters were comprised of two isolates and for eight of these, the two isolates were the sole occurrences of that genotype (Table 1). There was a positive relationship between cluster size and duration (r=0·49, P=0·001). For example, the four largest clusters were comprised of 7–10 isolates and these had a median duration of 129·5 days compared to a median duration of 15 days for two-isolate clusters.
Table 1. Characteristics of temporal genotype clusters

Outbreak-prone genotypes
Of the 52 genotypes with more than one isolate in the sample, 33 had isolates in clusters. Genotypic frequencies were similar in the entire sample and the subset of clustered isolates, i.e. there was no evidence of particularly transmissible or outbreak-prone genotypes (goodness-of-fit for 19 genotypes represented by four or more isolates: χ2=21·49, d.f.=18, P=0·26).
Epidemiological associations
Isolates were more likely to be in a cluster if they were isolated in winter or spring, isolated prior to the hospital reconfiguration, and were not β-lactamase producers (Table 2).
Table 2. Risk factors for variables associated with temporal genotype clusters
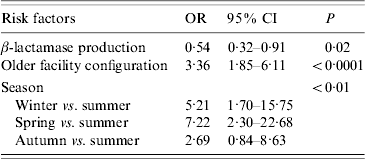
OR, Odds ratio; CI, confidence interval.
Seasonal effects
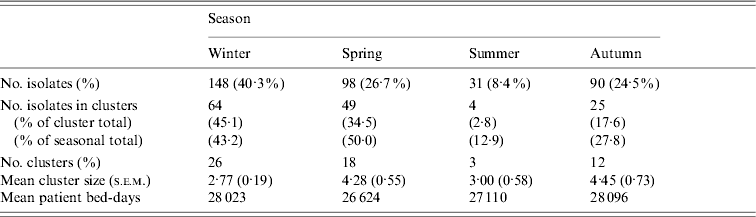
Whereas recovery of isolates from patients showed a marked seasonality (logistic regression, likelihood ratio=38·30, P<0·0001) there were no significant seasonal differences in cluster size (ANOVA: F=0·46, d.f.=3, 57, P=0·71) or cluster duration (ANOVA: F=0·60, d.f.=3, 57, P=0·62). Moreover, patient bed-days of care were similar across seasons (Table 3). However, significantly higher proportions of isolates were found within clusters in winter (43%) and spring (50%) compared to summer (13%) and autumn (28%) (Table 3). If the product of the total number of clusters and the mean seasonal proportion of isolates can be considered a seasonal expectation, the numbers of clusters per season were not significantly different from the expectations (χ2=4·08, d.f.=3, P=0·25).
Table 3. Seasonal characteristics of the sample and temporal genotype clusters
β-lactamase association
A higher proportion of β-lactamase non-producers (54·9%) were found in clusters compared to producers (34·4%) (P<0·001) but the β-lactamase status of isolates was not associated with either cluster size (means: producers 3·29, non-producers 3·50; P=0·75) or cluster duration (means: producers 107·5 days, non-producers 38·4 days; P=0·52). Of the 11 clusters for which it was possible to test for a difference in the frequency of β-lactamase producers in cluster isolates compared to non-cluster isolates of identical genotype, one cluster showed a significant difference. In that cluster, all four cluster isolates were non-β-lactamase producers while the remaining 10 isolates of identical genotype were producers. The isolates of four clusters were significantly different (all P<0·01) in the frequency of β-lactamase producers relative to isolates in the proximal temporal window. In all four cases, the clustered isolates were non-β-lactamase producers embedded in a temporal background of higher proportions of producers.
Facility configuration
For the cluster attributes of: clusters per year, proportion of isolates in clusters, and isolates per cluster, there were no significant temporal trends over the 10-year study period, no significant trends in patient bed-days of care, and no significant interactions, but for each cluster attribute there was a significant facility configuration effect (for each attribute, adjusted r 2=0·71–0·77, P<0·01). With the change in configuration, the number of isolates declined significantly from 44 to 23 per year (t=2·39, P=0·05). Under both configurations, cluster durations were similar (means 62–65 days for both older and newer) but there was a 65% reduction from 5·67 to 2·00 clusters per year, a 43% reduction from 3·71 to 2·11 isolates per cluster, and the proportion of clustered isolates showed a 70% reduction from 47·5% to 17·2% (Fig. 2).

Fig. 2. Trends in cluster attributes. The arrow on the x-axis corresponds to the time of the transition to the newer hospital configuration.
DISCUSSION
Genotype clusters and transmission
Identification of temporally delineated bacterial genotypic clusters represents a pathogen-centred approach to inferring pathogen transmission. Elucidation of pathogen transmission pathways and empirical estimates of transmission parameters garnered from long-term data are critical components of model-based prediction and resultant model-derived infection control recommendations [Reference Grundmann and Hellriegel15, Reference Levin34]. However, estimates of hospital transmission parameters often rely on cluster or outbreak criteria that are subjective [Reference Petersdorf, Oberdorfer and Wendt35, Reference Raboud36] with pre-defined spatial and/or temporal restrictions [Reference Grundmann28, Reference Jackson37, Reference Elias38]. Restricting analyses to particular wards or hospital units can minimize the rate of false positives, i.e. the incorrect determinations of transmission events, but spatial restrictions may incur the cost of missing transmission events mediated by transient contacts or patient transfers. These a priori constraints can be relaxed by application of a highly discriminating genotyping system to a population that harbours high genetic diversity. In such populations, identical genotypes shared between isolates (particularly when the genotype is rare) within a clinically relevant time-frame should, in itself, constitute strong epidemiological evidence of transmission.
Our ability to detect genotype clusters was influenced by several sample-related factors. While all patient isolates of M. catarrhalis were collected, only 367 or one-third were included in the analysis, a proportion expected to underestimate the proportion of clustered isolates and cluster sizes [Reference Glynn, Vynnycky and Fine39, Reference Murray40]. The analysis was conducted on a sample from a veterans' hospital population, a group that tends to be older than the general population, and predominantly male. Confining the analysis to a somewhat closed and homogenous host population of a pathogen species with no apparent geographic population substructure [Reference Enright and McKenzie19] minimized the potential confounding factor of immigration of pathogen genotypes that may be common elsewhere, but rare locally.
It should be noted that most of the statistical analyses presented in this study formally require the use of independent samples. In the case of infectious disease transmission, the isolates, of course, exhibit some level of dependence. In recent times, modern approaches, most commonly using Markov models, have explicitly modelled the dependencies in the data [Reference Grundmann5, Reference Cooper and Lipsitch6, Reference Forrester and Pettitt8, Reference McBryde13]. While we believe that some of the approaches would prove valuable in a further investigation of our data, it can also be shown that when the infectious transmission rate is small, then statistical methods requiring independence are approximately correct. However, in cases where there is an epidemic level of disease transmission, then modelling the dependencies in the data is critical. A preliminary analysis of the data showed the transmission rate to be low, so the statistical analyses presented here are considered reliable.
A pathogen genotype approach to inferring nosocomial transmission events can provide insight into the relative importance of the more traditional host-based transmission criterion of shared patient location. The following conclusions must be tempered by the knowledge that our spatial data were restricted to the location of the patient at the time the pathogen was isolated. Nevertheless, in only 20% (7/35) of the clusters were the clustered isolates all derived from patients in the same or adjacent locations (Table 4). Moreover, in 40% of the clusters there was a complete absence of spatial overlap in clustered isolates, and in no case involving clusters of a size above the minimum of two, were all clustered isolates from the same or adjacent locations. Similarly, clustered isolates were derived from patients in different wards in a shorter-term study of M. catarrhalis [Reference Ikram41], and in more than half of the MRSA clusters identified in a German hospital [Reference Mellmann14]. Further, for the VAMC, a genotype-independent spatial analysis was relatively uninformative in regard to transmission, in part because it appeared biased towards highlighting a cluster in a patient location somewhat distant from all others, but also because it failed to cluster genetically identical isolates.
Table 4. Spatial relationships in isolates within temporal genotype clusters. Table entries represent numbers of clusters

n.a., Not applicable.
Risk factors
Genotype-independent temporal clusters, during a period when patient bed-days of care remained relatively stable across seasons (Fig. 1d) support prior observations of seasonal peaks in prevalence [Reference Stone, Olinky and Huppert33, Reference Ikram41, Reference Keeling and Rohani42]. The finding of several large, seasonal, multi-genotype clusters showed our sample exhibited the population dynamics expected of this pathogen and thereby served to validate the cluster-based approach. During the winter and spring, genotype cluster sizes were larger and a higher proportion of isolates were found in clusters, but cluster durations were not longer and given the higher numbers of isolates, the frequency of clusters was no more than expected. Of particular relevance, the high proportion of isolates within genotype clusters during winter and spring suggests infectious transmission rather than independent acquisition as the major contributor to spread.
The emerging picture from the spatio-temporal analyses, i.e. the identification of many clusters for which chart data failed to show a shared patient location, suggests that evidence of direct patient contact in transmission is often lacking, perhaps particularly so in a retrospective study. Thus, a finding of identical genotypes in temporal proximity, especially if those genotypes are rare or absent overall (as in approximately half of the clusters at the VAMC), provides greater ability to infer transmission events than physical location of the patients. A similar approach was used to uncover tuberculosis transmission routes that would have been undetected in the absence of genotyping [Reference Ellis43]. Thus, it is not surprising that recent guidelines for infection control implicitly (or explicitly) acknowledge transmission pathways in addition to those of spatially defined outbreaks.
Under the older facility configuration, the association of more frequent recovery of M. catarrhalis with larger and more frequent cluster events suggests a hospital-specific source rather than a reflection of community prevalence. The VAMC hospital reconfiguration was accompanied by a 12·7% decline in patient bed-days of care. This decline was subsequently offset by an increase in nursing-home capacity and occupancy so that the combined number of hospital and nursing-home patient-days declined only 3·2% with pre- and post-reconfiguration yearly means of 140 242 and 135 838, respectively. Facility bed capacity remained relatively stable throughout the study period (yearly mean 1026, range 969–1138). After the facility transition, the mean number of isolates was approximately halved, the numbers of isolates in clusters was reduced by a factor of 6, and the proportion of isolates in clusters also was halved. Declines in these cluster parameters did not occur until after 1990, which coincided with the transfer to the new facilities. Thus, the timing and the precipitous nature of reductions in the number and severity of cluster events support a role for improved facility design in curtailing transmissions.
β-lactamase non-producers were 1·9 times more likely to be involved in genotype clusters (Table 2), but differences in β-lactamase status were not associated with other cluster parameters. This difference may arise from the fact that while only 22% of the sample were β-lactamase-negative isolates, they accounted for 31% of the clustered isolates. Non-producers comprised a much higher proportion of the sample prior to facility reconfiguration (26% vs. 13%), so the apparent higher risk for non-producers may instead be an effect of the facility renovation. This is also a probable explanation for the four clusters in which non-β-lactamase producers were more common in clusters relative to isolates in the proximal temporal window. Nevertheless, these clusters suggest that in this case, antibiotic resistance was not an essential determinant in pathogen transmission.
Although the number of pathogen transmission studies based on long-term longitudinal data is limited, certain characteristics related to transmission cluster size, duration and spatial location of hosts begin to emerge. Most transmission events are small, comprised of 2–3 isolates. This was observed for M. catarrhalis at the VAMC (65% small clusters), for MRSA in a German hospital (77%) [Reference Mellmann14], for vancomycin-resistant enterococci (VRE) in a Chicago hospital (60%) [Reference Stosor44], and for meningococcal disease across Germany (88%) [Reference Elias38]. Moreover, most clusters are not long-running but have median durations of <2 weeks [VAMC, 14, 38]. In those studies for which clusters were identified using genetic methods and no spatial constraints, in approximately half of the small clusters, patient hosts were located in different hospital units [VAMC, 14] or for meningococcal disease, in different states or counties in Germany [Reference Elias38].
Surveillance for increases in pathogen incidence will have the least success in early detection of small events, especially when the primary locations of patient hosts are spatially dispersed. For example, a numerical, genotype-independent algorithm had particular difficulty identifying small clonal transmission events in a Chicago hospital [Reference Hacek45]. Perhaps the preponderance of small transmission events is a testament to current infection control efforts, and the gains realized from infection control measures present the appearance that most transmission events are self-limiting. In the Chicago hospital, infection control, primarily hand washing and staff cohorting, reduced the prevalence of VRE by half and reduced the transmission parameter so that hospital transmission alone would not sustain outbreaks [Reference Pelupessy, Bonten and Diekmann2]. Stringent infection control measures will probably remain the primary defence against the short-lived, but relatively abundant small transmission events.
ACKNOWLEDGEMENTS
We thank S. Smiddy for help with data collation. Funding was provided by a grant from the Seattle Epidemiologic Research and Information Center of the Department of Veterans Affairs. This material is the result of work supported with resources and facilities at the James H. Quillen Veterans Affairs Medical Center, Mountain Home, Tennessee.
DECLARATION OF INTEREST
None.






