



INTRODUCTION
Bats (Order: Chiroptera) have been widely studied as reservoir hosts for pathogens of concern for human and animal health [Reference Hayman1,Reference Calisher2], with particular attention paid to RNA viruses in the Coronaviridae, Filoviridae, Rhabdoviridae, and Paramyxoviridae families [Reference Stoner-Duncan, Streicker and Tedeschi3–Reference Leroy5]. The association of bats with the human and animal diseases is in part explained by the high diversity of zoonotic viruses that circulate in bats. Per species, bats host more zoonotic viruses than all other mammalian orders and are more likely to share viruses among species, which may be due to aggregation of large colonies, migration, and the multi-species roosts of many bats [Reference Luis6–Reference Olival8]. However, whether bats are equally competent hosts of non-viral pathogens such as bacteria remains an open and understudied question [Reference Brook and Dobson9,Reference Mühldorfer10]. Bacteria such as Yersinia spp. and Leptospira spp. have been detected in bats [Reference Muhldorfer11,Reference Bunnell12], but the importance of these pathogens for human, wildlife, or domestic animal health remains unknown. For other bacteria such as Bartonella spp., phylogenetic analyses have suggested a potential role of bats in the transmission of zoonotic Bartonella sp. [Reference Wray13], such as Bartonella mayotimonensis, an etiologic agent of human endocarditis [Reference Veikkolainen14,Reference Lilley15].
Recent studies have also shown hemotropic Mycoplasma spp. (hemoplasma) infections in bats [Reference Mascarelli16–Reference Ikeda19]. Hemoplasmas are facultative intracellular erythrocytic bacteria without a cell wall that were formerly classified as Haemobartonella and Eperythrozoon spp., and were re-classified as Mycoplasma spp. based on their 16S rRNA gene sequences and cell morphologic properties [Reference Messick20–Reference Willi23]. These bacteria are thought to be transmitted through direct (blood and saliva) and possibly vector-borne contact [Reference Willi23–Reference Dean26] and can cause acute and chronic anemia in wildlife, humans, and domestic animals [Reference Groebel27–Reference Sykes30], particularly in immunocompromised hosts [Reference Pires dos Santos31,Reference Webster32]. Almost all Mycoplasma spp., including hemoplasmas, appear to show host specificity that seems to be a result of the host–pathogen interaction during evolution; however, potential zoonotic or inter-species transmission has also been reported [Reference Sykes30,Reference Pires dos Santos31,Reference Bosnic33–Reference Steer35]. Among bat species studied to date, 16S rRNA gene sequence analyses have shown that hemoplasmas identified in little brown bats (Myotis lucifugus) from the USA demonstrated the closest homology (~92%) with a hemoplasma detected in a human, Candidatus Mycoplasma haemohominis, and with Mycoplasma haemomuris detected in a small Japanese field mouse (Apodemus argenteus) [Reference Mascarelli16,Reference Sashida36]. Recent work on Neotropical bat species from Brazil found velvety free-tailed bats (Molossus molossus) were infected with hemoplasmas that shared close identity (93–96%) with a hemoplasma detected in mice, Mycoplasma coccoides [Reference Ikeda19]. Surveys of Schreibers’ bats (Miniopterus schreibersii) and one long-eared bat (Myotis capaccinii) also detected hemoplasma species with close identity (97%) to Candidatus Mycoplasma haemohominis [Reference Millán17]. These phylogenetic relationships between bat hemoplasmas and hemoplasmas from other species suggest possible cross-species transmission in history [Reference Pitcher and Nicholas37], which may be relevant for zoonotic transmission from bat species with frequent contact with humans.
No published data currently exist on evidence for hemoplasma infection in vampire bats or on the prevalence and diversity of these bacteria in hematophagous bats. Yet owing to their direct contact with mammals through blood feeding, vampire bats are an obvious candidate species for which to assess hemoplasma infection and phylogenetic relationships to genotypes previously described in other mammals, including humans and non-human primates. Three species comprise the subfamily Desmodontinae: the common vampire bat (Desmodus rotundus), the hairy-legged vampire bat (Diphylla ecaudata), and the white-winged vampire bat (Diaemus youngi). Vampire bats occur across diverse habitat types throughout Latin America, ranging from Mexico to northern Argentina [Reference Greenhall and Schmidt38]. While these species historically feed on wild mammals and birds, the most abundant species, D. rotundus, preferentially feed on livestock and poultry owing to the greater accessibility and reliability of these novel prey species [Reference Voigt and Kelm39–Reference Bobrowiec, Lemes and Gribel41]. D. rotundus also commonly feeds on humans, making it an important source of human rabies virus outbreaks [Reference Stoner-Duncan, Streicker and Tedeschi3,Reference Schneider42–Reference da Mendes44]. As biting is a possible transmission route for hemoplasmas in other mammals through exposure to infectious blood or saliva [Reference Willi24–Reference Dean26], vampire bat feeding behavior could possibly facilitate transmission to humans, domestic animals, and wildlife. Infection in vampire bats roosting in anthropogenic habitats could also enhance vector-borne transmission cycles [Reference Willi23]. The goals of our study were thus: (i) to identify hemoplasma species in vampire bats; (ii) to assess the position of detected sequences within the broader hemoplasma phylogeny; (iii) to identify risk factors for infection, including age and seasonality [Reference Schaer45]; and (iv) to determine hemoplasma presence in vampire bat saliva to assess the possibility of direct transmission of these bacteria.
MATERIALS AND METHODS
Vampire bat sampling
During 2015 and 2016, we sampled 224 vampire bats across 14 sites in Peru (Departments of Amazonas [AM], Apurimac [API], Ayacucho [AYA], Cajamarca [CA], and Loreto [LR]; n = 12) and in Orange Walk [OW] District (n = 2), Belize (Fig. 1). We sampled sites 1–2 times annually (Supplementary Table S1), in which bats were captured in mist nets or harp traps placed at exits of roosts, along flight paths, or outside livestock corrals from 19:00 to 05:00 [Reference Streicker46,Reference Becker47]. Upon capture, bats were held in individual cloth bags and issued a uniquely coded incoloy wing band (3·5 mm, Porzana Inc.). Bats were classified as subadult or adult based on the fusion of phalangeal epiphyses [Reference Delpietro and Russo48], and reproductive activity was indicated by the presence of scrotal testes in males and pregnancy or lactation in females. We obtained blood by lancing the propatagial vein with a sterile 23-gauge needle, followed by sample collection with heparinized capillary tubes. To screen for hemoplasmas by PCR, up to 30 µl blood was stored on Whatman FTA cards to preserve bacterial DNA [Reference Ahmed49]. The whole blood-impregnated FTA cards were stored in individual pouches at room temperature with desiccant until laboratory analysis. Thin blood smears were prepared on glass slides, stained with buffered Wright–Giemsa (Camco Quik Stain II, Fisher Scientific), and screened by a board-certified veterinary clinical pathologist (MSC) for hemoplasmas using a light microscopic examination of a representative area of the blood monolayer at 1000× magnification. For assessment of hemoplasmas presence in vampire bat saliva, we collected oral swabs from Peru; samples were preserved in 2 ml of RNAlater (Invitrogen) at −80 °C until laboratory analyses.
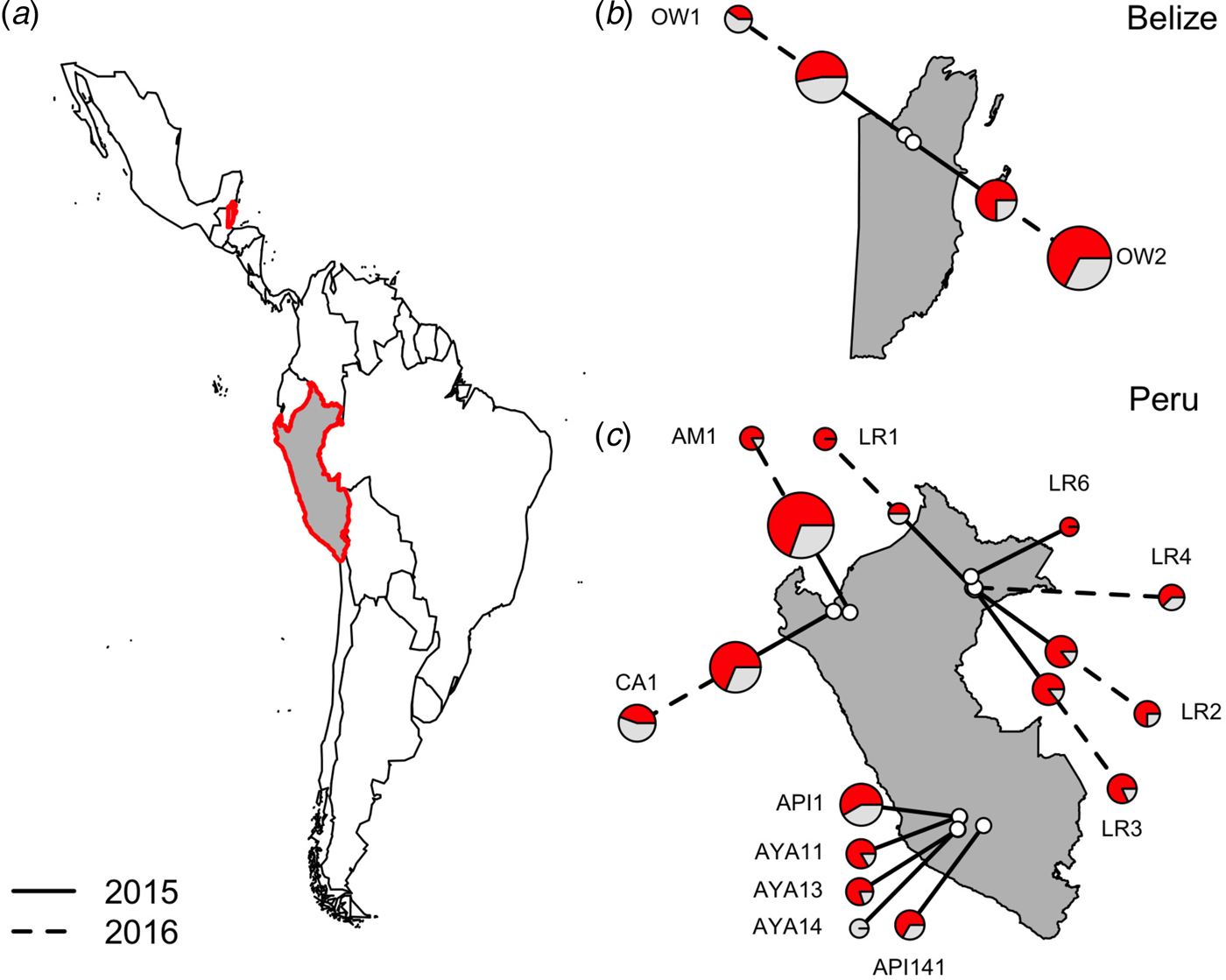
Fig. 1. Location of vampire bat sampling sites in Latin America, with Belize and Peru showed in gray with red outlines (a). Insets show the location of field sites (white) and the prevalence of hemoplasmas per site (b and c) across study years (solid line = 2015, dashed line = 2016), with red denoting the proportion of infected bats. Points are scaled by sample size.
All field procedures were approved by the University of Georgia Animal Care and Use Committee (A2014 04-016-Y3-A5) and the University of Glasgow School of Medical Veterinary and Life Sciences Research Ethics Committee (Ref08a/15). Bat capture and sampling were authorized by the Belize Forest Department under permits CD/60/3/15(21) and WL/1/1/16(17) and by the Peruvian Government under permits RD-009-2015-SERFOR-DGGSPFFS, RD-264-2015-SERFOR-DGGSPFFS, and RD-142-2015-SERFOR-DGGSPFFS. Access to genetic resources from Peru was granted under permit RD-054-2016-SERFOR-DGGSPFFS.
DNA extraction, PCR amplification, and sequencing of amplicons
Genomic DNA was extracted from 3 to 5 2 mm punches of blood preserved on Whatman FTA cards using QIAamp DNA Investigator Kits (Qiagen, Hilden, Germany) following the manufacturer's instructions. DNA samples were stored at −80 °C until use.
Primary screening for the presence of hemoplasmas was performed with PCR using previously published UNI_16S_mycF and UNI_16S_mycR universal primers for amplification of the partial 16S rRNA hemoplasma genes [Reference Volokhov50]. Based on our previously published data [Reference Volokhov50] and recent in silico analysis [Reference Christen51] using BLAST search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) of these universal primers against the different mycoplasma 16S rRNA gene sequences available in GenBank, these primers produce PCR fragments with size of approximately 1000–1035 bp (depending on the target Mycoplasma spp.). If samples were considered strongly positive for infection with hemoplasmas in the primary PCR screening, we also amplified the full-length 16S rRNA gene (approximately 1450–1560 nt) using PCR primers designed in this study (16SF-AGAGTTTGATCCTGGCTCAG and 16SR-CTCAAAACTGAAAGYCATCCGC) and then sequenced these amplicons (see GenBank accession numbers KY932674, KY932675, KY932677–KY932680, KY932687–KY932693, KY932695, KY932696, KY932701, KY932703–KY932710, KY932712, KY932716, KY932721, and KY932722). If samples were weakly positive (i.e., a weak band) in the primary PCR screening, we did not amplify the full-length 16S rRNA gene but instead sequenced amplicons of the partial 16S rRNA gene from the primary PCR (see GenBank accession numbers KY932676, KY932681–KY932686, KY932694, KY932697–KY932700, KY932702, KY932711, KY932713–KY932715, KY932717–KY932720, KY932723, KY932724). All PCRs in this study were qualitative and thus a load of hemoplasma DNA in individual blood samples was not quantified.
The 16S rRNA amplicons produced were directly sequenced (without cloning into a plasmid vector) by Macrogen (https://www.macrogenusa.com; Rockville, MD, USA). Prior to sequencing, PCR amplicons were purified by electrophoresis through 1·5% agarose gels and extracted with the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Amplicons were sequenced with the same primers used for PCR amplification and then with internal (walking) primers when needed. The amplification mixture for all PCR contained 5 µl of 10× HotStarTaq PCR buffer, 1·5 mM MgCl2, 200 mM dNTP mixture, 1 mM of each primer, and 2·5 U HotStarTaq Plus DNA Polymerase (Qiagen, Hilden, Germany) in a final volume of 50 µl, including 3 µl of DNA template. The Vent DNA Polymerase Kit (New England Biolabs), which contains high-fidelity thermophilic Vent DNA polymerase, was also used for the amplification of PCR products for subsequent sequencing. The absence of PCR inhibitors in isolated blood DNA was confirmed by PCR amplification of the D. rotundus mitochondrial 16S rRNA gene as an extraction positive control (with primers 16S_Desmodus_F-AACAGCAAAGCTTACCCCTTGTACC and 16S_Desmodus_R-GTCTGAACTCAGATCACGTAGGAC). Negative (no DNA added) controls were run for each PCR, and Candidatus Mycoplasma haemozalophi [Reference Volokhov50] was used as a positive control.
All PCR reactions were conducted under the following conditions: a polymerase activation step at 95 °C for 5 min (or 15 min for HotStarTaq only) followed by 45 cycles of 95 °C for 30 s, 60 °C for 60 s, and 72 °C for 60 s, with a final extension at 72 °C for 10 min. PCR products were detected by electrophoresis through 1% TAE-agarose gels containing ethidium bromide followed by UV visualization.
To avoid the potential presence of chimeric sequences or PCR-derived variants in the data, all hemoplasma 16S rRNA PCR products for phylogenetic analyses were directly amplified from blood DNA samples of vampire bats with two different DNA polymerases (HotStarTaq and Vent) and were directly sequenced without cloning [Reference Ashelford52,Reference Hugenholtz and Huber53]. All gene sequences prior to the downstream phylogenetic analysis were subjected to the chimeric sequence analysis using DECIPHER [Reference Wright, Yilmaz and Noguera54] and UCHIME [Reference Edgar55]. All sequences available from this study have been deposited in GenBank under the accession numbers KY932674–KY932724.
Phylogenetic analyses
The 16S rRNA sequences determined in this study were compared to those available in GenBank using procedures, algorithms, and methods for phylogenetic tree inference as described elsewhere [Reference Volokhov50,Reference Volokhov56,Reference Volokhov57]. Briefly, the sequences of the 16S rRNA genes were compared with the GenBank nucleotide database. Nucleotide sequences were aligned using the publicly available Clustal X software (http://www.clustal.org). Inter- and intra-species similarity were generated using BioEdit software (http://www.mbio.ncsu.edu/BioEdit/bioedit.html). Genetic distances were calculated by using the Kimura two-parameter and Tamura–Nei models, and phylogenetic trees were constructed in MEGA 6 software using the minimum evolution algorithm (http://www.megasoftware.net).
Statistical analyses
We first calculated hemoplasma prevalence and 95% confidence intervals using the Wald method in the prevalence package of R [58]. We then tested if hemoplasma genotypes detected via sequencing were associated with geography, bat demography, or time using Chi-squared tests with p values generated via a Monte Carlo procedure with 1000 simulations [Reference Hope59]. We used the Benjamini and Hochberg correction to adjust p values for multiple comparisons [Reference Benjamini and Hochberg60].
We used generalized mixed effects models (GLMMs) with binomial errors and a logit link to determine risk factors for hemoplasma infection status (positive or negative) [Reference Zuur61]. Bat ID was included a random effect to account for multiple sampling of a small number of individuals (n = 6); the site was not included as a random effect owing to repeatedly failed model convergence. Using a reduced dataset free of missing values (n = 220), we compared a set of GLMMs with country, bat age, sex, reproductive status, year, and season (spring, summer, fall) as fixed effects alongside possible two-way interactions; we limited the number of models to roughly 50% our sample. We compared models with Akaike information criterion corrected for small sample size (AICc) and calculated marginal and conditional R 2 values to assess fit [Reference Nakagawa and Schielzeth62,Reference Burnham and Anderson63]. We performed model averaging to compute mean odds ratios (OR) and 95% confidence intervals across the set of GLMMs whose cumulative Akaike weight (w i ) summed to 95%; mean ORs were standardized with partial standard deviation [Reference Cade64]. We used the MuMIn and lme4 packages for model averaging [Reference Barton65,Reference Venables and Ripley66].
Assessment of hemoplasmas in saliva
To examine the possibility for direct transmission of hemoplasmas through biting or grooming, we used metagenomic data from a parallel study to screen vampire bat saliva samples from the same regions of Peru where blood samples were collected. Although a PCR-based screening of saliva samples would have made for the most comparable set of results, untargeted metagenomic sequencing has been found equally or more sensitive for pathogen detection compared with conventional PCR [Reference Graf67–Reference Plaire69]. Five saliva pools were shotgun sequenced, each of which contained nucleic acid extractions from saliva swabs often vampire bats from 1 to 2 colonies within each department (Amazonas, Cajarmarca, Loreto, Ayacucho) or across two neighboring departments (Ayacucho and Apurimac). Pooled samples represent the same regions of Peru, though not necessarily the same colonies or individuals, tested for hemoplasmas in blood through PCR.
Total nucleic acid was extracted from swabs using a modified protocol with the BioSprint 96 One-For-All Vet Kit (Qiagen, Hilden, Germany) and a KingFisher Flex 96 machine (Thermo Fisher Scientific). Swabs were incubated twice consecutively in tubes containing lysis buffer (Buffer RLT) and Proteinase K for 15 min at 56 °C; volume from the two tubes was combined prior to the addition of other extraction reagents, and the manufacturer's protocol was subsequently followed. Extractions were quantified using a Qubit RNA HS Assay Kit (Thermo Fisher Scientific) and pooled at approximately 120 ng RNA per sample. Pools were treated for 5 min at 35 °C with 2U DNase I (Ambion) and cleaned with a 1·8× ratio of Agencourt RNAClean XP beads (Beckman Coulter). Pools were then depleted for host ribosomal RNA using the Ribo-Zero rRNA Removal Kit (Human/Mouse/Rat) (Illumina) per the manufacturer's instructions. Prior to library preparation, cDNA synthesis was performed using the Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) and the NEBNext mRNA Second Strand Synthesis Module (New England Biolabs). Samples were library prepared using the KAPA DNA Library Preparation Kit for Illumina (KAPA Biosystems), at which point they were individually barcoded with primers designed based on the NEBNext Multiplex Oligos for Illumina Index Primers (New England Biolabs). The libraries included in this study were combined in equimolar ratios with other metagenomic libraries for sequencing on an Illumina NextSeq500 at the University of Glasgow Centre for Virus Research.
Reads were demultiplexed according to barcode and quality filtered using TrimGalore [Reference Martin70,71] with a quality threshold of 25, minimum read length of 75 bp, and clipping the first 14 bp of the read. Low complexity reads were filtered out using the DUST method and PCR duplicates removed using PRINSEQ [Reference Schmieder and Edwards72]. We screened cleaned reads for hemoplasma-like sequences using nucleotide BLAST [Reference Altschul73] against a custom database composed of the PCR-generated hemoplasma sequences from this study, retaining only the best alignment for a single query-subject pair. The hemoplasma-like reads were then de-novo assembled using the assembly-only function of SPAdes [Reference Bankevich74], and contigs greater than 300 bp were screened for sequences closely matching Mycoplasma species using nucleotide BLAST in Genbank.
RESULTS
Hemoplasma genotype detection and phylogenetic analysis
Hemoplasma infection was detected by 16S rRNA PCR in 150/223 (67%; 95% CI = 0·61–0·73) of common vampire bats (D. rotundus) but was not found in our single sample from a hairy-legged vampire bat (Diphylla ecuadata). We did not detect hemoplasmas in any blood samples with light microscopy. Hemoplasma infection prevalence as assessed by PCR ranged from 0 to 100%, with a mean 67·53% bats per site infected with at least one genotype (Fig. 1).
Figure 2 shows the inferred phylogenetic position of the hemoplasma sequences identified in vampire bats among known hemotropic Mycoplasma species using partial sequences of the 16S rRNA genes (871–890 bp). Vampire bat hemoplasmas represented three main genotypes (Supplementary Table S2, Fig. 2). One other sample (D141; GenBank accession number KY932724) showed 97% similarity to the 16S ribosomal RNA gene of Mycoplasma moatsii strain MK405 (NR_025186), a non-hemotropic Mycoplasma spp. isolated from grivet monkeys (Cercopithecus aethiops); however, we were not able to amplify the full-length 16S rRNA gene from this sample. Inter-laboratory contamination with M. moatsii was excluded as we do not handle this species in our laboratory; the same sequence was also repeatedly amplified from the same blood sample. Vampire bat hemoplasma genotypes 1 and 2 were closely related (97–98% inter-genotype similarity, Supplementary Table S2) and similar to hemoplasmas detected in common bent-wing bats (M. schreibersii) in Spain (86–87% similarity to GenBank accession numbers KM538691–KM538698), little brown bats (Myotus lucifugus) in the USA (88–89% similarity to KF713538), wild Japanese monkeys (Macaca fuscata) (93–94% similarity to AB820288), tufted capuchins (Sapajus apella) in the Brazilian Amazon (88–90% similarity to KT314160–KT314164), and to a hemoplasma detected in a human patient with hemolytic anemia and pyrexia in the USA (94% similarity to GU562823). Genotype 3 was most similar to M. coccoides (93–95% similarity to AY171918), Candidatus Mycoplasma turicensis (93–94% similarity to DQ157153), a hemoplasma detected in a capybara (Hydrochoerus hydrochaeris) in Brazil (92–93% similarity to FJ667774), and hemoplasmas detected in velvety free-tailed bats (M. molossus) in Brazil (90–93% similarity to KY356747–KY356751). No chimeras were detected from these 16S rRNA gene sequences.
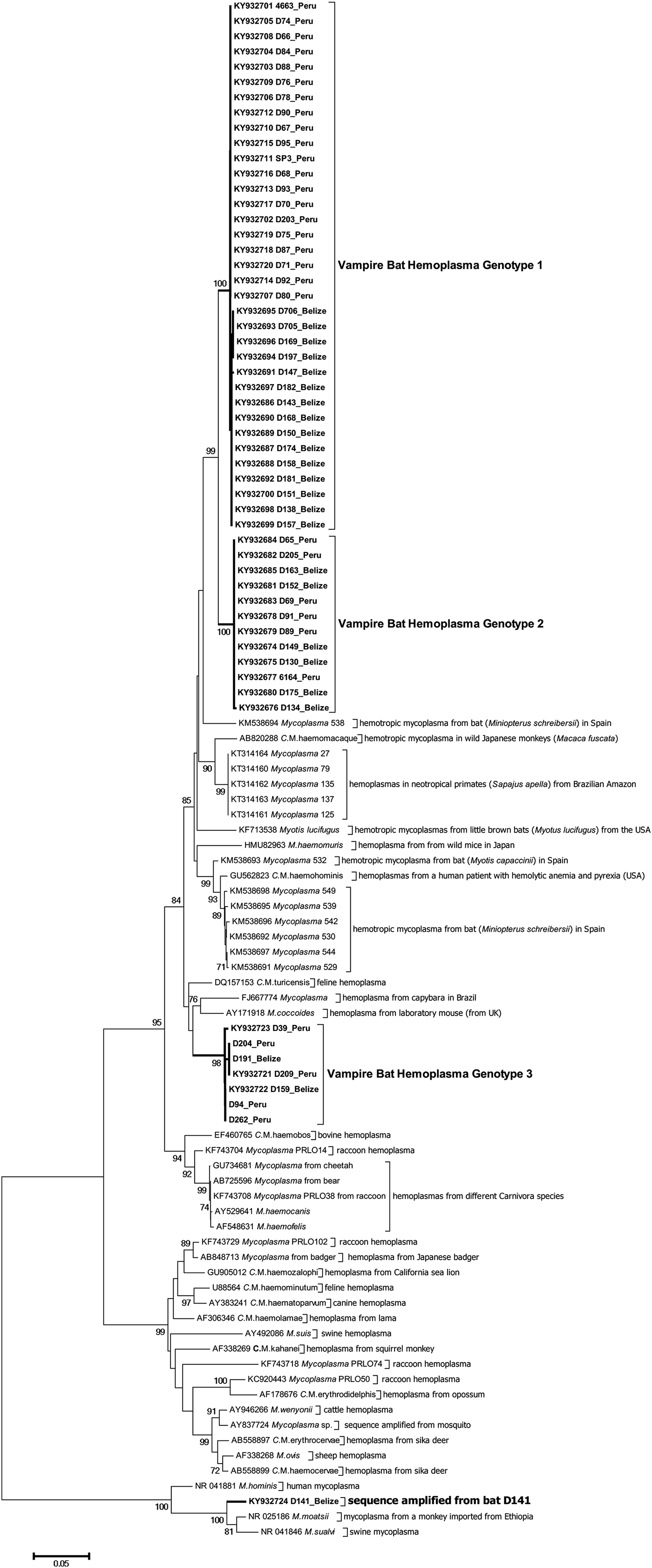
Fig. 2. Dendrogram showing phylogenetic relationships based on nucleotide sequence data for the 16S rRNA gene among the hemoplasma genotypes detected in common vampire bats (Desmodus rotundus) with other hemotropic Mycoplasma spp. The tree was constructed using the minimum evolution method in MEGA 6. Accession numbers for sequences downloaded from GenBank are shown alongside individual bat ID and country of sampling. The Desmodus rotundus samples sequenced in this study are displayed in bold.
Within vampire bats, hemoplasma genotype 1 was the most common sequence identified (Fig. 3), infecting 68% of positive bats (100/150). Genotypes 2 and 3 infected 22% (33/150) and 9% (14/150) of positive bats, respectively. The number of genotypes detected per site ranged from one to three, but no coinfection with multiple genotypes was observed. Genotype 1 was detected across all sites, genotype 2 was detected in both Belize sites and 40% of Peru sites (primarily northern and eastern Amazon), genotype 3 was detected in both Belize sites and 40% of Peru sites. The M. moatsii-like hemoplasma sequence was detected in only one bat in Belize. The three novel genotypes showed no association with country (χ 2 = 3·09, P = 0·31), site (χ 2 = 30·14, P = 0·31), department (χ 2 = 13·68, P = 0·31), age (χ 2 = 0·28, P = 0·88), season (χ 2 = 8·65, P = 0·21), or year (χ 2 = 1·48, P = 0·59) but were associated with reproduction (χ 2 = 11·32, P = 0·03) and were marginally associated with sex (χ 2 = 7·12, P = 0·10). Non-reproductive bats showed greater infection with genotype 1 but less infection with genotype 2 than reproductive bats, while males tended to harbor more infection with genotypes 1 and 2 than females (Supplementary Fig. S1).
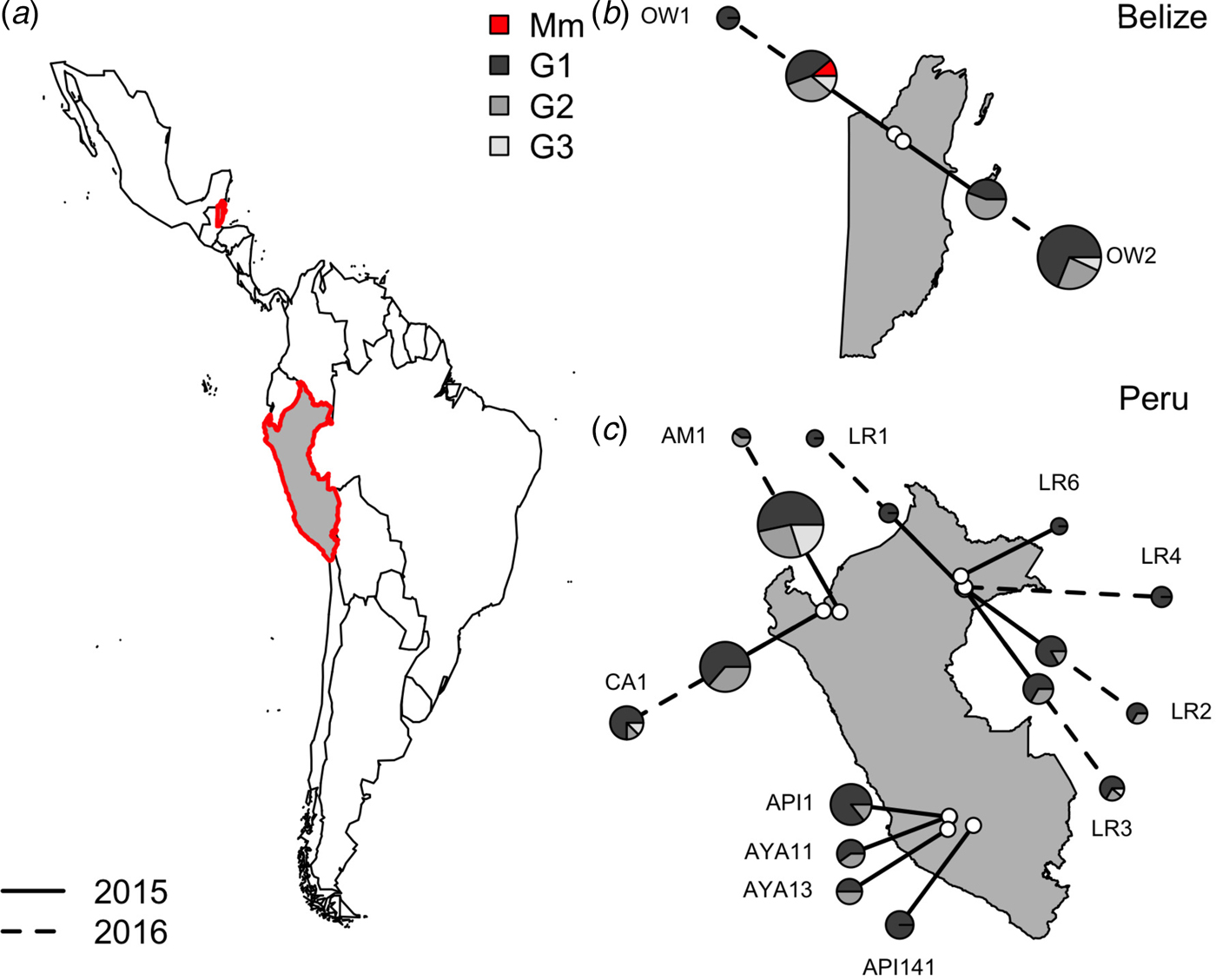
Fig. 3. Location of vampire bat sampling sites in Latin America, with Belize and Peru showed in gray with red outlines (a). Insets show the location of field sites (white) and the composition of hemoplasma genotypes per site (b and c) across study years (solid line = 2015, dashed line = 2016). Points are scaled by sample size. Mm denotes the M. moatsii-like hemoplasma.
Risk factors for hemoplasma infection in vampire bats
All hemoplasma genotypes and vampire bat species were pooled for analyses of infection prevalence. The 95% confidence set of GLMMs contained 58/124 of the original models (Supplementary Table S3), with variable importance as follows: reproductive status (95%), age (75%), season (65%), the interaction between season and reproductive status (48%), sex (44%), country (27%), year (18%), the interaction between season and sex (7%), the interaction between sex and reproductive status (7%), and the interaction between country and year (<1%). These models explained between 9% and 21% of the variation in hemoplasma infection (Supplementary Table S3). The average odds ratios for the effect of being non-reproductive (OR = 1·45, 95% CI = 1·05–2·02) and being non-reproductive in the spring (OR = 0·68, 95% CI = 0·50–092) on infection was different than one (Fig. 4a ), although the 95% confidence interval for subadults only just overlapped with one (OR = 1·50, 95% CI = 1·00–2·26; Fig. 4a ). A weaker effect was observed for males (OR = 1·21, 95% CI = 0·85–1·66). Hemoplasma infection prevalence was thus greatest for non-reproductive bats, especially those sampled in the fall, and for subadult bats (Fig. 4b , c ). The odds of hemoplasma infection did not vary across the two countries and two years (Fig. 4a ). In a small sample of recaptured bats (n = 6), we observed two bats move from infected to uninfected status within 367–371 days, while two individuals remained infected across 369–424 days (Supplementary Fig. S2).
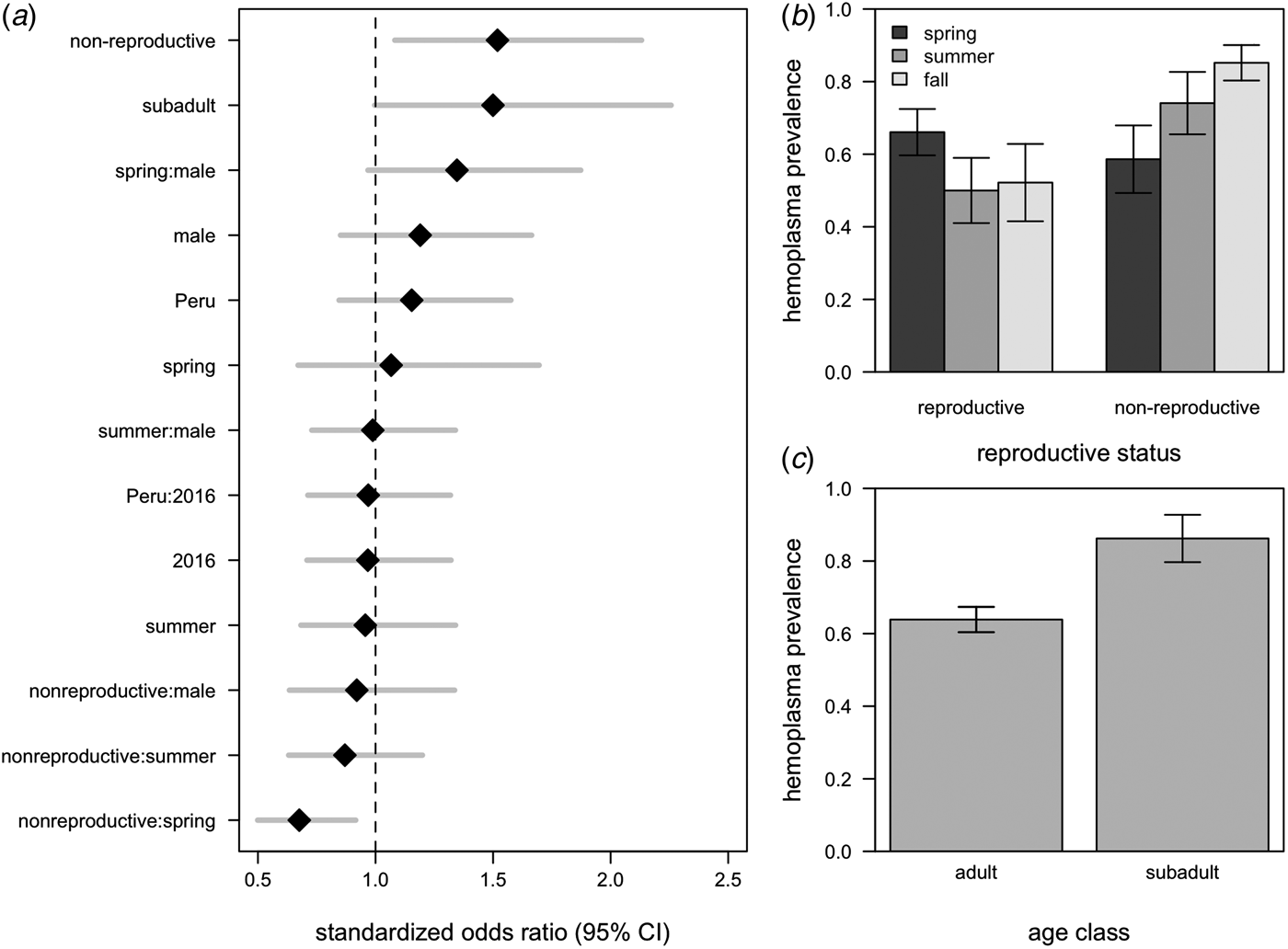
Fig. 4. Averaged odds ratios and 95% confidence intervals for all variables within the 95% GLMM set, standardized by partial standard deviation (a). The dashed line shows where the odds ratio equals 1. Raw hemoplasma infection prevalence and 95% confidence intervals for bat reproductive status by sampling season (b) and age class (c).
Comparison of Mycoplasma species detected in blood and saliva
Based on the saliva metagenomic data, we identified contigs closely matching to Mycoplasma species in all oral swab sample pools from Peru (Supplementary Table S4). We consistently found long contigs (>1300 bp) with high coverage likely belonging to non-hemoplasma Mycoplasma species present in vampire bat saliva. Across all study regions, we also identified shorter contigs (400–500 bp) with high sequence identity (98–100%) to the hemoplasma sequences of genotypes 1 and 3 that were detected in the blood of D. rotundus. This suggests the possible presence of similar hemoplasmas in both vampire bat saliva and blood, although the shorter length of these contigs prohibited conclusive identification.
DISCUSSION
Our study describes a novel and genetically diverse hemoplasmas in common vampire bats. Infection prevalence was relatively high for this species (67%) compared with that of from other bats, with all three novel genotypes being geographically widespread. The odds of hemoplasma infection were greatest for non-reproductive bats sampled in the fall and for subadult bats and did not vary between 2015 and 2016 both within and across countries, suggesting individuals important to transmission and endemic infection dynamics. Salivary metagenomics also showed the presence of non-hemotropic Mycoplasma species and hemoplasma genotypes phylogenetically similar to those identified in blood, providing indirect data for possible direct transmission of hemoplasmas in vampire bats through biting or grooming.
The genus Mycoplasma currently comprises 20 hemoplasma species (NIH NCBI Taxonomy). Except for M. haemocanis, M. haemofelis, and M. haemomuris, hemoplasmas have the provisional taxonomic status ‘Candidatus’ because they are uncultivated and as a result are incompletely characterized bacterial species [Reference Volokhov56,Reference Murray and Stackebrandt75,Reference Brown, Whitcomb and Bradbury76]. Here, PCR screening using the previously published UNI_16S_mycF and UNI_16S_mycR universal primers [Reference Volokhov50] and amplification of the full-length 16S rRNA gene using PCR primers designed in this study illustrate these primers can be used for detection of multiple hemotropic Mycoplasma spp. genotypes in vampire bats. This is also the first study in which non-hemotropic Mycoplasma species were detected in vampire bats using metagenomics. Mycoplasma species are a part of the normal oral, intestinal, and genital microflora in many animals. We know of no published references on non-hemotropic Mycoplasma species in bats, with the exception of a high presence of Mycoplasma spp. 16S rRNA gene sequences detected in intestinal biopsy samples of Cynopterus spp. bats [Reference Banskar, Mourya and Shouche18]. Based on our metagenomics data, we cannot infer the significance of finding non-hemotropic Mycoplasma in bat saliva on the health of these animals or potential inter-species transmission. More research is needed on the normal Mycoplasma microflora in bats.
Phylogenetic studies of the 16S rRNA gene of closely related Mycoplasma species (including hemoplasmas) propose to use the arbitrary interspecies sequence similarity value of ⩽97% as a minimum level indicating a separate, genetically distant species [Reference Brown, Whitcomb and Bradbury76,Reference Pettersson77]. Data based on the expanded analysis of the 16S rRNA gene sequences of the species within the family Mycoplasmataceae generally support this proposition [Reference Volokhov56]. All three vampire bat hemoplasma genotypes demonstrated low levels (i.e. <97%) of sequence identity to previously described genotypes (or hemoplasma species) detected in other animal species, which suggests that these vampire bat hemoplasmas are novel hemoplasma genotypes or putatively new hemoplasma species not yet described in other animals [Reference Volokhov56,Reference Volokhov57].
The vampire bat genotypes are paraphyletic to each other and appear to have common ancestry with hemoplasmas from other bats, rodents, humans, and non-human primates, suggesting that hemoplasmas have a history of host shifts between closely and distantly related species during evolution. Additionally, we observed no geographic clustering for genotypes 2 and 3, suggesting vampire bat hemoplasmas are broadly distributed across Latin America. However, for hemoplasma genotype 1, sequences from Belize and Peru had geography-specific single-nucleotide polymorphism (SNPs) and varied by 2·2% (20 SNPs of 871 nt analyzed sequence); these sequences fell into two country-specific groups (Fig. 2); this might imply more regionally constrained transmission cycles of this hemoplasma genotype.
Hemoplasma infection prevalence observed here in vampire bats (67%) was intermediate compared with that in other bat species. Hemoplasma prevalence in Schreibers’ bats (M. schreibersii) and one long-eared bat (M. capaccinii) in Spain was 97% [Reference Millán17], while only 47% of little brown bats (M. lucifugus) from the eastern and northeastern USA and only 14% of velvety free-tailed bats (M. molossus) were infected with hemoplasmas [Reference Mascarelli16,Reference Ikeda19]. However, the sensitivity of our PCR has not been quantified, so prevalence in vampire bats could conceivably be higher than we detected. Hemoplasmas have not been cultured in vitro, and their detection in many species has used PCR with or without analysis of Romanowsky–Giemsa and acridine orange-stained blood smears [Reference Messick20,Reference Volokhov57]. Prior work on bats has relied on PCR only but either with blood preserved in EDTA or with spleen, liver, or heart tissues [Reference Mascarelli16,Reference Millán17,Reference Ikeda19]. We instead used blood preserved on Whatman FTA cards to facilitate room-temperature sample storage in remote, tropical field conditions. During primary PCR screening, we found only 30% of positive samples produced a strong band through gel electrophoresis; other positive samples produced average or weak bands and we were mostly unable to amplify the full-length 16S rRNA gene from such samples. Similar problems with amplification of hemoplasma-specific PCR products were recently identified in other bat species in Brazil [Reference Ikeda19]. Two possibilities for the high number of weak band samples in primary PCR are that hemoplasma concentrations in vampire bat blood are low or that the use of current sample collection or storage methods is inefficient for hemoplasma characterization in bats. We did not detect hemoplasmas in blood using light microscopy, though the sensitivity of this method is relatively low compared to PCR [Reference Tasker and Lappin78]. Further, manual staining in field conditions often results in stain precipitate, which makes a definitive detection of hemoplasmas through microscopy difficult.
Within vampire bats, we found the odds of hemoplasma infection to be greatest for non-reproductive bats sampled in the fall and for subadult bats. Overall higher prevalence in non-reproductive bats is surprising, given that animals often down-regulate costly immune function during reproductive events and are more susceptible to infection [Reference Martin, Weil and Nelson79,Reference Plowright80]. This pattern could possibly reflect seasonal birth pulses and the influx of immunologically naïve bats [Reference George81,Reference Amman82], which is corroborated by the OR for non-reproductive bats being greatest in the fall, which could represent a time lag after the spring births in vampire bats [Reference Delpietro83]. Seasonal birth pulses could also explain the trend for prevalence to be greater in subadult bats, though the marginally significant averaged effect of age is likely due to controlling for other factors in the GLMMs. This trend is similar to findings on rabies virus exposure, in which younger vampire bats also showed higher seroprevalence [Reference Streicker84]. Subadults could also experience greater exposure to hemoplasmas if vectors are more attracted to younger bats [Reference Christe, Arlettaz and Vogel85] or if vampire bat hemoplasmas are transmitted vertically [Reference Fujihara86]. Unlike with feline and canine hemoplasmas [Reference Walker Vergara87,Reference Soto88], the odds of infection did not vary by sex, suggesting sex-biased parasitism may not occur with vampire bat hemoplasmas despite males playing a key role in the spatial dynamics of vampire bat rabies [Reference Streicker46]. More extensive sampling of vampire bats over time, alongside infection trials, is necessary to elucidate the transmission routes of these hemoplasmas. We note that even our top GLMMs only explained up to 21% of the variation in infection status (Supplementary Table S3), which highlights the roles that coinfection with other pathogens [Reference Willi89], differences in host physiology [Reference Walker Vergara87,Reference Hawley and Altizer90], or landscape factors such as food availability [Reference Volokhov57,Reference Becker, Streicker and Altizer91] could also play in determining vampire bat susceptibility and exposure to hemoplasmas.
Hemoplasma infection prevalence did not differ between years, across countries, or generally by season. While more years of data are necessary to corroborate this result, these findings suggest hemoplasmas are endemic within vampire bat populations. Along with relatively high prevalence, this stable temporal trend corroborates other work suggesting bats to be reservoirs of hemoplasmas and potentially other bacterial infections [Reference Brook and Dobson9,Reference Mühldorfer10,Reference Millán17]. The repeated sampling of a small number of recaptured bats in our study sheds further light on the infection dynamics of bat hemoplasmas. We observed two bats move from infected to uninfected within 117–123 days; this could again reflect hemoplasma DNA loads that were too low to be detected by our PCR but could also suggest vampire bats can clear hemoplasma infection. Future longitudinal sampling paired with mathematical models could help infer if vampire bats undergo cycles of latency and reactivation with hemoplasmas or obtain partial immunity from infection [Reference Blackwood92,Reference Plowright93].
To conclude, this study identified novel hemoplasma genotypes in vampire bats that were phylogenetically related to hemoplasmas reported in other mammals, including bats, rodents, humans, and non-human primates. These hemoplasma sequences clustered into three novel genotypes were most prevalent in young and non-reproductive bats, and were relatively stable in prevalence over time. Future studies should: (i) explore the host range and specificity of hemoplasmas among bat species and (ii) evaluate the pathogenicity of hemoplasmas in vampire bats with hematological and immunological assays. Given the close association between these vampire bat genotypes and those from humans, rodents, and non-human primates, future studies should aim to elucidate the potential for pathogen exchange between vampire bats and sympatric wildlife, humans, and domestic animals. Our metagenomic data identifying Mycoplasma species and similar hemoplasma genotypes in vampire bat saliva suggest the possibility for acquisition of hemoplasmas from reservoir hosts or for direct transmission of hemoplasmas through biting during aggressive encounters with conspecifics [Reference Greenhall, Schmidt and Lopez-Forment94], blood sharing [Reference Wilkinson95], and feeding on prey [Reference Greenhall and Schmidt38], but infection trials are needed to confirm this transmission route and its zoonotic potential.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S095026881700231X
ACKNOWLEDGEMENTS
For assistance with site identification and field logistics, we thank Patricia Mendoza, regional offices and hospitals of the Ministry of Health in Chiclayo and Iquitos in Peru, and staff of the Wildlife Conservation Society Peru, Programa de Conservación de Murciélagos del Peru, and Lamanai Field Research Center. We also thank residents of communities along the Chiriaco, Marañon, Tahuayo, Nanay, and Yanayacu rivers in Peru for accommodations and transportation during fieldwork. For assistance with bat sampling and research permits, we thank Jorge Carrera, Pierre Castro, Miluska Ramos, Marcela Oversluijs, Cindy Quino, Carlos Tello, Nestor Falcon, Carlos Shiva, John Claxton, Ornela Inagaki, Brock Fenton, Nancy Simmons, Mark Howells, Neil Duncan, John Hermanson, Alexandra Bentz, and staff of the Instituto Nacional de Salud Peru and Lamanai Field Research Center. We thank Annie Page-Karjian, Cecilia Nachtmann, and Katherine Smith for assistance with DNA extractions and thank Ana da Silva Filipe, Chris Davis, and Alice Broos for assistance with metagenomics labwork. We also thank anonymous reviewers for providing critical review comments on an earlier draft of this manuscript.
DJB was funded by an NSF Graduate Research Fellowship, ARCS Foundation Award, Sigma Xi, the Odum School of Ecology, the American Society of Mammalogists, the UGA Graduate School, the Explorer's Club, and a NSF Doctoral Dissertation Improvement Grant (DEB-1601052). SA acknowledges support from NSF DEB-1518611, RJO was supported by the UK Medical Research Council (MC_UU_12014/12), and DGS was supported by a Sir Henry Dale Fellowship, jointly funded by the Wellcome Trust and Royal Society (102507/Z/13/Z).
DECLARATION OF INTEREST
None.






