


On fait de la science avec des faits comme on fait une maison avec des pierres; mais une accumulation de faits n’est pas plus une science qu‘un tas de pierres est une maison. (Henri Poincaré ‘La Science et l’Hypothèse’Reference Poincaré1)
(Science is built upon facts, as a house is built of stones; but an accumulation of facts is no more a science than a heap of stones is a house)
1. Introduction: Some Basic Concepts
1.1. General Aspects and Language
We shall start by introducing some basic concepts, also questions of language, and follow in part an earlier publication (in German) from an earlier lecture.Reference Quack2 In Sections 2 and 3 we shall discuss the development of atomic and molecular models in chemistry. In Section 4 we shall discuss the development of models of the chemical bond. Section 5 deals with the origin of today's quantum mechanical theory of matter and Section 6 with models for processes in chemistry. In Section 7 we shall discuss the limitations of current models and some fundamental problems of current research in the context of symmetry, conservation laws and the violation of fundamental symmetries in relation to molecular chirality and the ‘standard model of particle physics’ (SMPP). Section 8 deals with some speculations on CPT symmetry violation in chiral molecules and a simple model for dark matter. We conclude in Section 9 with some brief remarks on understanding nature.
Table 1 summarizes some words related to the concept of natural law. The ambivalence of the use of such words is commented upon in the footnote referring to Ref. 3 in Table 1, and in relation to the word ‘model’ I remember the joke of a famous stereochemist from Zurich, Vladimir Prelog, who said in a lecture in about 1979 in Göttingen: ‘I like playing with models …, but that can be dangerous –, think of Profumo and his affair: he played too much with (photo-)models’.
Table 1 Natural law (‘Naturgesetz’) some wordsFootnote *

* Es erben sich Gesetz und Rechte
Wie eine ewge Krankheit fort …
…
Vom Rechte, das mit uns geboren ist
Von dem ist leider! nie die Frage
(Goethe, Faust)3
It is obviously useful to first introduce some of the basic concepts used in the natural sciences.
Assuming that there is an external reality, which is independent of us, whatever that may be, how do we understand it? We have several instruments of the human mind that allow us to picture and represent the observations or ‘observed facts’, related to that reality. As a rule, scientists assume the existence of such an external reality. There are exceptions to this rule, but I shall not discuss these further. Thus, the starting point is given by the ‘facts’ of reality. The scientific approach starts then frequently with the implicit assumption that these facts of reality follow some underlying structures, rules or ‘natural laws’, which ‘exist’ independently of our representations. Whether or not this is true, this is the actual practice of the scientists and engineers, their basic hypothesis. In order to uncover the structures we use some instruments of our mind to generate organized, well-ordered mental pictures or ‘representations’ of the facts: theories, hypotheses and models (Figure 1). We shall say a little more about all three below.
Figure 1 Organizing the ‘facts of reality’ by models, hypotheses and theories.
Before doing so, we shall address another difficult word, which we just used: ‘true’ or ‘truth’. This is obviously a very difficult concept as is well illustrated by the famous sentence of Pilatus, when he was confronted with God's truth: ‘![]() ’ ‘What is truth?’. It is interesting to analyse the Greek word for truth, which contains a negation in the Greek prefix α as in the word
’ ‘What is truth?’. It is interesting to analyse the Greek word for truth, which contains a negation in the Greek prefix α as in the word ![]() or
or ![]() leading to the meaning un-hidden, uncovered, unveiled, unforgotten. A judge in court or a scientist in an investigation will uncover a hidden truth. Of course, some may question the existence of truth, as implied by the words of Pilatus. However, a practising scientist (and also a judge) assumes that something of the kind exists, perhaps only approximately so. This is easily seen by contrasting it with an error or a plain lie. A ‘truthful’ witness in court may not be able to tell the ‘real’ truth, only some approximation to it, as seen and remembered by him. However, we can usually distinguish this from a plain lie, which falsifies the facts. Again this is actual practice, and we shall not address the very difficult question of whether it can happen that a witness who presents a plain lie might be closer to the true facts than the witness who to the best of his knowledge is ‘truthful’. Those scientists who think about some of these fundamental difficulties are aware of the problems, but generally proceed then with a more practical attitude, which is reflected in three citations which we reproduce here.
leading to the meaning un-hidden, uncovered, unveiled, unforgotten. A judge in court or a scientist in an investigation will uncover a hidden truth. Of course, some may question the existence of truth, as implied by the words of Pilatus. However, a practising scientist (and also a judge) assumes that something of the kind exists, perhaps only approximately so. This is easily seen by contrasting it with an error or a plain lie. A ‘truthful’ witness in court may not be able to tell the ‘real’ truth, only some approximation to it, as seen and remembered by him. However, we can usually distinguish this from a plain lie, which falsifies the facts. Again this is actual practice, and we shall not address the very difficult question of whether it can happen that a witness who presents a plain lie might be closer to the true facts than the witness who to the best of his knowledge is ‘truthful’. Those scientists who think about some of these fundamental difficulties are aware of the problems, but generally proceed then with a more practical attitude, which is reflected in three citations which we reproduce here.
1. Nissuna humana investigatione si puo dimandare vera scientia, se essa non passa per le matematiche dimostrationi e se tu dirai, che le scientie, che principiano e finischono nella mente habbiano verità, questo non si concede, ma si niega, per molte raggioni e prima, che in tali discorsi mentali non accade esperientia, senza la quale nulla da di se certezza.
(No human inquiry can claim the status of true knowledge without passing through mathematical demonstration: and if you say that sciences which begin and end in the mind possess truth, this cannot be allowed, but must be denied for many reasons: and first of all because experience does not enter into such mental exercises, and without it there is no certainty.) (Leonardo da Vinci, as cited by Cyril Hinshelwood)
2. ‘Every attempt to employ mathematical methods in the study of chemical questions must be considered profoundly irrational and contrary to the spirit of chemistry. If mathematical analysis should ever hold a prominent place in chemistry – an aberration, which is happily almost impossible – it would occasion a rapid and widespread degeneration of that science.’ (A. Comte, ‘Philosophie Positive’, 1830)
3. Ce qui fait le mérite d'une théorie nouvelle, ce n'est pas d’être vraie: il n'y a pas de théories vraies; c'est d’être féconde. (The merit of a new theory does not rely on the fact that it is true: there are no true theories; its merit depends on being fruitful.) (Louis Pasteur)
The first two citations deal with mathematical and scientific truth – very sceptically so in the case of Leonardo, more positively in the case of the third citation by Pasteur, who emphasizes the concept of fruitfulness of a theory in contrast to abstract ‘truth’, which cannot be achieved by any theory.
This will be the spirit of the present essay: while we do not deny some of these fundamental difficulties, we shall not pursue these in any detail, as this might possibly lead into ‘fruitless philosophical hair-splitting’. Rather, we shall describe the attitude and actual practice of the active scientist, including also some of the historical developments in the following sections.
1.2. Theory
A ‘theory’ (from Greek θεωρία = vision, view, knowledge) is generally understood to be an exact, true picture of reality. A ‘correct’ theory claims to get everything right in providing a precise image of reality within its range of applicability. It claims to be able to make exact predictions of the future (if at all possible) and to allow looking back into the past with similar accuracy. A theory is an attempt ‘to do it right’. With this claim, when confronted with reality, a theory is either true or false, it can be ‘falsified’ in the terms of Popper's language. In fact, viewed from some historical distance, theories are always false, but at least they try to be true. This is part of the content of Pasteur's motto. In this sense there is no such precise distinction against the other two concepts of hypothesis and model, the border is somewhat continuous. Nevertheless, there are some ranges where we can distinguish the concepts. For example, one might say that in chemistry quantum mechanics provides the claim to be a theory of the structure and dynamics of atoms and molecules (see Sections 2 and 3) as well as to provide a theory of the chemical bond (or at least ‘binding’) (Sections 4 and 6).
1.3. Hypothesis
A hypothesis is a ‘theory in the process of being developed’. It is a preliminary image of reality and thus the basis for a future theory. In contrast to theory, a hypothesis need not describe the part of nature under consideration completely or precisely. A hypothesis can be amended and made more complete. It is a preliminary theory. In this sense a hypothesis is otherwise rather similar to a theory. It also can be ‘falsified’ in the sense of Karl Popper.
1.4. Model
We shall say a bit more about the concept of ‘model’. From the history of the word the concept as an image has been formed from the Latin word modulus (measure, scale) via the middle-age Latin word modellus into the Italian renaissance word modello (copy, image, example, prototype). Here, one implies generally that this is not a 1:1 copy or picture but smaller or larger than the original, often also simplified. The relation between a model and reality is twofold. A model can be made as a copy of the originally pre-existing reality. But a model can also be a prototypical representation of a reality that has to be constructed later (such as the model of an architect who plans to build a house). Such a prototypical model can exist as a three-dimensional object, on paper, or just in our mind. This twofold use of the concept of model is also particularly common in Chemistry. A chemist can build a model after a molecule she found in nature or she can develop a model of a molecule, which is still to be constructed or synthesized by her as a ‘molecular architect’. Models are, of course, also by themselves part of the real world. Thus, one might say that one actually relates two different objects of reality in order to understand reality, to modify or to improve it.
Different from a theory, a model does not in general claim to represent reality with perfect accuracy. Rather, certain essential aspects of reality should be described well by the model, whereas ‘less important’ parts might be described less accurately, perhaps even wrongly or omitted completely. A model attempts to be a useful representation of reality not necessarily an ‘exact’, true picture. A good model frequently is a simplified description of reality. In relation to theory and hypothesis it can be used in different ways. First, in the initial phase of discovery, when we do not yet have a complete theory of the phenomena, a model helps to build appropriate hypotheses and theories. Thus, the path followed is:
 $${\rm{observed}}\,{\rm{facts}}\, \rightarrow \,{\rm{model}}\, \rightarrow \,{\rm{hypothesis}}\, \rightarrow \,{\rm{theory}}$$
$${\rm{observed}}\,{\rm{facts}}\, \rightarrow \,{\rm{model}}\, \rightarrow \,{\rm{hypothesis}}\, \rightarrow \,{\rm{theory}}$$
Secondly, in the phase, when we do have a complete theory of the phenomena, a model can be used to simplify the description, thus we proceed according to the scheme:
 $${\rm{theory}}\, \rightarrow \,{\rm{model}}\, \rightarrow \,{\rm{comparison}}\,{\rm{with}}\,{\rm{observed}}\,{\rm{facts}}$$
$${\rm{theory}}\, \rightarrow \,{\rm{model}}\, \rightarrow \,{\rm{comparison}}\,{\rm{with}}\,{\rm{observed}}\,{\rm{facts}}$$
Now the model provides a simplified representation of a theory, which itself can claim to be an exact representation of reality. The usefulness of the model in this case becomes obvious, when frequently the ‘exact’ theory cannot be carried through to describe the phenomena, for instance because of the mathematical difficulties in the ‘realization’ of the theory. For instance, we might think that quantum mechanics provides an exact theory of proteins, but it would be completely illusory today to carry out the necessary calculations on a computer. On the other hand, we can build simple classical models as graphical representations of proteins on a computer and we can also do classical mechanical molecular dynamics with appropriate force fields to describe the motion of proteins. Even if a mathematical-numerical treatment using exact theory is possible, a simplified model can help us to better understand the essential features of the exact numerical results, because our mind is more able to comprehend a simplified picture, which distinguishes the essential from the unessential features.
Such a model is neither ‘right’ nor ‘wrong’. It cannot be falsified in the sense of Popper, as we know anyway that it cannot be completely true. Rather, a model could prove ‘useful’ or ‘useless’, perhaps misleading. In fact, some opinions see theories in a similar situation as exemplified by the citation from Louis Pasteur, which we have given above. Thus theories become closer relatives of models and their claim for truth is taken less seriously. The distinction between theory and model is not sharp.
In any case, we now have a basic definition of these three concepts, even if there may not always be a sharp distinction between the notion of theory, hypothesis and model. In the following sections we shall describe some developments of models and theories in chemistry, including a historical perspective, with a number of examples. Chemistry can be divided into two branches, analytical and synthetic chemistry. Related to this, there can also be two approaches in the use of models. In the analytical approach the ‘analysis’ of chemical facts leads to a model or a theory. In the synthetic approach a molecular model (mental or practical, macroscopic) is used as a starting point to newly synthesize the molecule in the laboratory, the task of ‘molecular architecture’.
2. Atomic Models and Chemistry
That matter can be built from atoms is a model, which further developed into a firm hypothesis and finally a theory over the course of history. The basic atomic models of chemistry go back to the concepts of Demokritos and Leukippos about 400 BC. who also introduced the word ‘atom’ (![]() , indivisible). Originally, there was no sharp distinction between atom and molecule. With the advent of the Renaissance in Europe, the knowledge of the natural philosophy of ancient Greece became widespread. In Shakespeare, we can read (in ‘As you like it’, ca. 1601): ‘It is as easy to count atomies as to resolve the propositions of a lover’. The supposed small size and large numbers of atoms in macroscopic bodies gave rise to thought and investigations. In AD 1646, the monk Johann Chrysostomus Magnenus estimated the number of ‘atoms of incense’ (we would rather say ‘molecules’ today) in a small piece of incense by an experiment using smell in a way that was in principle correct (he gave a lower bound based on the assumption that at least one molecule was necessary to generate the sense of smell of incense in our nose4). He obtained a number that we would consider reasonable today, a fact that is not widely known. We can cite him here literally: ‘…fuissent in hoc thuris grano, pisi magnitudinem non superante, atomi elementales ad minimum 777 600 000 000 000 000, ex quibus patet quantae sit parvitatis atomus una, concjicique potest, quantus sit atomorum numerus in toto universo’ (English translation: ‘In this piece of incense, which itself was not larger than a pea, there were at least 7.776 × 1017 elementary atoms. From this one can see how small an atom is and one can guess how large the number of atoms might be in the whole Universe.’)
, indivisible). Originally, there was no sharp distinction between atom and molecule. With the advent of the Renaissance in Europe, the knowledge of the natural philosophy of ancient Greece became widespread. In Shakespeare, we can read (in ‘As you like it’, ca. 1601): ‘It is as easy to count atomies as to resolve the propositions of a lover’. The supposed small size and large numbers of atoms in macroscopic bodies gave rise to thought and investigations. In AD 1646, the monk Johann Chrysostomus Magnenus estimated the number of ‘atoms of incense’ (we would rather say ‘molecules’ today) in a small piece of incense by an experiment using smell in a way that was in principle correct (he gave a lower bound based on the assumption that at least one molecule was necessary to generate the sense of smell of incense in our nose4). He obtained a number that we would consider reasonable today, a fact that is not widely known. We can cite him here literally: ‘…fuissent in hoc thuris grano, pisi magnitudinem non superante, atomi elementales ad minimum 777 600 000 000 000 000, ex quibus patet quantae sit parvitatis atomus una, concjicique potest, quantus sit atomorum numerus in toto universo’ (English translation: ‘In this piece of incense, which itself was not larger than a pea, there were at least 7.776 × 1017 elementary atoms. From this one can see how small an atom is and one can guess how large the number of atoms might be in the whole Universe.’)
Another concept also developed starting from early Greek philosophy: the element. In modern chemistry, the concept of the element is related to a conserved quantity in a chemical reaction. The stoichiometric equation of the chemical reaction expresses this conservation law quantitatively. We would say today that a pure element consists only of atoms of the same kind (slightly modified today due to the existence of isotopes). Demokritos also has thought about the geometric shape of atoms and how they can be interconnected by ‘hooks and loops’. He even made simple experiments to find out about possible shapes of these elementary entities. Based on these early ideas a symbolic description of atoms and molecules using some geometrical figures such as triangles, circles (with different ‘content’, etc) was developed just before 1800 by Pierre Auguste Adet and Jean Henri Hassenfratz. At around the same time Lavoisier systematized the notion of the element, giving it essentially the modern definition, and showed that water in contrast to ancient thinking was not elemental, but composed of the elements hydrogen and oxygen. Dalton, around 1810, used simple geometrical symbols such as empty (oxygen) or filled (carbon) circles, including some inside symbols such as a point in the middle of the circle (hydrogen) or a vertical line in the circle (nitrogen) to represent the known elements. The modern notation was introduced by Berzelius shortly after 1810, including in some publications in 1813/1814. He used a letter abbreviation of the Latin name, such as H, C, N, O for hydrogen, carbon, nitrogen, oxygen. This abstract symbolism has been codified today in the nomenclature of the IUPAC (International Union of Pure and Applied Chemistry, see Table 2).
Table 2 Periodic table of the elements
This notation does not imply any particular geometry of the atoms. The modern atomic model with some kind of three-dimensional geometry arose 100 years later based on the work of Rutherford and the ‘old quantum theory’ of the atom of Bohr (1913).Reference Merkt and Quack5–Reference Bohr8 It is frequently shown in pictures today somewhat similar to a microscopic planetary system, with the atomic nucleus taking the place of the sun, and the electrons in the places of the planets. Before going into the spectroscopic origin of the modern quantum theoretical modelsReference Quack9 we shall briefly discuss the tedious route that led to the atomic and molecular theory of matter between about 1800 and 1900.
The basic empirical ‘laws’ of quantitative chemistry were formulated around 1800.
1. Law of the conservation of mass in a chemical reaction (Lavoisier 1785), for instance in the reaction (in modern notation)
 $${\rm{2HgO}}\, = \,{\rm{2Hg}}\, + \,{{{\rm{O}}}_{\rm{2}}}\eqno(1)$$
$${\rm{2HgO}}\, = \,{\rm{2Hg}}\, + \,{{{\rm{O}}}_{\rm{2}}}\eqno(1)$$
when mercury oxide (HgO) is decomposed to mercury Hg and oxygen.
2. Conservation of mass when heating a substance (for instance ice being melted to water and warmed further, Benjamin Thompson, count Rumford, around 1800).
3. Law of constant proportions (Joseph Louis Proust 1754–1826). In modern notation one has, for instance, in terms of mass ratios with some constant mass ratio of H and O
 $${\rm{2}}{{{\rm{H}}}_{\rm{2}}}\, + \,{{{\rm{O}}}_{\rm{2}}}\, = \,{\rm{2}}{{{\rm{H}}}_{\rm{2}}}{\rm{O}}\eqno(2)$$
$${\rm{2}}{{{\rm{H}}}_{\rm{2}}}\, + \,{{{\rm{O}}}_{\rm{2}}}\, = \,{\rm{2}}{{{\rm{H}}}_{\rm{2}}}{\rm{O}}\eqno(2)$$
or any multiples of this, the proportions stay constant.
4. Law of multiple proportions (John Dalton, 1808). In modern notation this states that for different compounds formed from some elements, the ratios of masses in the composition are related to simple integer multiples. For instance different nitric oxides satisfy ratios
 $$m({\rm{O}})/m({\rm{N}})\, = \,({\rm{n}}\times {\rm{0}}{\rm{.571}})\,:\,{\rm{1}}\eqno(3)$$
$$m({\rm{O}})/m({\rm{N}})\, = \,({\rm{n}}\times {\rm{0}}{\rm{.571}})\,:\,{\rm{1}}\eqno(3)$$
with integer  $$$n = {\rm{1, 2, 3, 4, 5}} $$$
corresponding to the compounds N2O, NO, N2O3, NO2 and N2O5 in modern notation.
$$$n = {\rm{1, 2, 3, 4, 5}} $$$
corresponding to the compounds N2O, NO, N2O3, NO2 and N2O5 in modern notation.
It may be noted that Dalton also formulated an incorrect law, today forgotten: the rule of greatest simplicity: ‘If two elements A and B form only one compound, then this is of the form AB’. This rule resulted from the dangerous use of a philosophical method, which is known as Occam's razor and led Dalton to the wrong formulation of water as OH.
5. Law of equivalent proportions: elements combine in ratios corresponding to certain ‘equivalent’ masses or some integer multiples of this, say,
 $$\frac{{m({\rm{O}})}}{{m({\rm{N}})}}\, = \,\frac{{m({\rm{O}})}}{{m({\rm{H}})}}\,:\,\frac{{m({\rm{N}})}}{{m({\rm{H}})}}\eqno(4)$$
$$\frac{{m({\rm{O}})}}{{m({\rm{N}})}}\, = \,\frac{{m({\rm{O}})}}{{m({\rm{H}})}}\,:\,\frac{{m({\rm{N}})}}{{m({\rm{H}})}}\eqno(4)$$
when looking at water H2O and ammonia NH3 for instance in modern notation.
6. The law for combining volumes of gases in reactions found by Joseph-Louis Gay-Lussac and Friedrich Wilhelm Alexander von Humboldt in a joint research on H2O in 1804/05. In modern notation this states
 $${\rm{2}}\,{\rm{volumes}}\,{\rm{(}}{{{\rm{H}}}_{\rm{2}}})\, + \,{\rm{1}}\,{\rm{volume}}\,{\rm{(}}{{{\rm{O}}}_{\rm{2}}})\, = \,{\rm{2}}\,{\rm{volumes}}\,{\rm{(}}{{{\rm{H}}}_{\rm{2}}}{\rm{O}})\,{\rm{(vapor)}}\eqno(5)$$
$${\rm{2}}\,{\rm{volumes}}\,{\rm{(}}{{{\rm{H}}}_{\rm{2}}})\, + \,{\rm{1}}\,{\rm{volume}}\,{\rm{(}}{{{\rm{O}}}_{\rm{2}}})\, = \,{\rm{2}}\,{\rm{volumes}}\,{\rm{(}}{{{\rm{H}}}_{\rm{2}}}{\rm{O}})\,{\rm{(vapor)}}\eqno(5)$$
 $$ {\rm{3}}\,{\rm{volumes}}\,{\rm{(}}{{{\rm{H}}}_{\rm{2}}})\, + \,{\rm{1}}\,{\rm{volume}}\,{\rm{(}}{{{\rm{N}}}_{\rm{2}}})\, = \,{\rm{2}}\,{\rm{volumes}}\,{\rm{(N}}{{{\rm{H}}}_{\rm{3}}}) \eqno(6)$$
$$ {\rm{3}}\,{\rm{volumes}}\,{\rm{(}}{{{\rm{H}}}_{\rm{2}}})\, + \,{\rm{1}}\,{\rm{volume}}\,{\rm{(}}{{{\rm{N}}}_{\rm{2}}})\, = \,{\rm{2}}\,{\rm{volumes}}\,{\rm{(N}}{{{\rm{H}}}_{\rm{3}}}) \eqno(6)$$
These laws are still valid today (with some restrictions, for example Laws 1 and 2 are only approximate, because of a small, so far not measured ‘mass defect’ 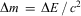 $$$\rDelta m\, = \,\rDelta E\,/\,{{c}^2} $$$
, with the energy release
$$$\rDelta m\, = \,\rDelta E\,/\,{{c}^2} $$$
, with the energy release 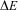 $$$\rDelta E $$$
and the speed of light c). They are most easily understood when assuming the formation of simple molecules from atoms as implied by the modern notation (not used at the time). They were used by Avogadro to derive a statement known as:
$$$\rDelta E $$$
and the speed of light c). They are most easily understood when assuming the formation of simple molecules from atoms as implied by the modern notation (not used at the time). They were used by Avogadro to derive a statement known as:
7. Avogadro's molecular hypothesis (Amedeo Avogadro 1811)Reference Avogadro10 ‘Equal volumes of different ideal gases at the same temperature and pressure contain equal numbers of molecules’.
This very powerful statement was only slowly appreciated towards the middle of the nineteenth century, in part due to the work of Cannizzaro. Avogadro's hypothesis can be considered to be the basic hypothesis of the kinetic theory of gases, and can be used to derive Avogadro's number (or Loschmidt's number), in modern notation the number of atoms in one mole of an element
 $${{N}_{\rm{A}}} = {\rm{6}}{\rm{.02214}}\times {\rm{1}}{{{\rm{0}}}^{{\rm{23}}}} \,{\rm{mo}}{{{\rm{l}}}^{{\rm{ - 1}}}} \eqno(7)$$
$${{N}_{\rm{A}}} = {\rm{6}}{\rm{.02214}}\times {\rm{1}}{{{\rm{0}}}^{{\rm{23}}}} \,{\rm{mo}}{{{\rm{l}}}^{{\rm{ - 1}}}} \eqno(7)$$
These laws were complemented by Faraday's laws of electrochemistry (1834).
8. Faraday's first law: the masses m obtained in electrolysis are proportional to electric current I and time t (i.e. charge
 $$${{Q}_{el}} = I \cdot t $$$
)
$$${{Q}_{el}} = I \cdot t $$$
)

 $$ m = A \cdot I \cdot t\, = \,A \cdot {{Q}_{{\rm{el}}}} \eqno(8)$$
$$ m = A \cdot I \cdot t\, = \,A \cdot {{Q}_{{\rm{el}}}} \eqno(8)$$
9. Faraday's second law: the ratio of masses obtained by the same electrical charge Q el in electrolysis is given by the ratios of the equivalent masses (point 5) of the corresponding substances.
10. This was finally complemented by the law of conservation of energy.Reference Helmholtz11
Combined, these lead to an atomic model of matter. Chemical elements are composed of atoms of the same kind. Chemical compounds are formed by combining these atoms to molecules containing some integer number of atoms of the different elements. For many aspects of chemistry this remains essentially valid today, with some necessary but rather straightforward extensions.
Nevertheless, while the majority of chemists (and physicists) accepted this atomic molecular model of matter at least by the middle of the nineteenth century, there remained some serious debates concerning alternative ‘continuum’ models of matter until about 1900 and even beyond. However, after 1900 much direct evidence for atoms and molecules finally settled these debates. By then, the picture of gases was given by atoms or molecules of perhaps spherical or somewhat more complex shapes flying around and colliding according to classical statistical mechanics as derived by Clausius, Maxwell and Boltzmann in the second half of the nineteenth century (based on much earlier work by Bernoulli and others). In the condensed phase, these spherical or non-spherical bodies would be densely packed together, which easily explained the difference by a factor of about 1000 in the density of the same compound as a solid or liquid compared with the gas (at 1 atmosphere pressure and room temperature). With this model, one could also easily derive microscopic properties from macroscopic measurements. For instance the root mean square velocity 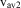 $$$ {{\rm v}_{\rm av\:2}} $$$
is obtained from measuring the pressure P (for instance 1 bar = 105 Pa) and the density ρ (for instance about 1 kg m−3 for air) by means of the equation:
$$$ {{\rm v}_{\rm av\:2}} $$$
is obtained from measuring the pressure P (for instance 1 bar = 105 Pa) and the density ρ (for instance about 1 kg m−3 for air) by means of the equation:
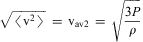 $$\root\of {\left\langle {{{\rm v}^2} } \right\rangle } \, = \,{{\rm v}_{\rm av2}}\, = \,\root\of {\frac{{{\rm{3}}P}}{\rho }} \eqno(9)$$
$$\root\of {\left\langle {{{\rm v}^2} } \right\rangle } \, = \,{{\rm v}_{\rm av2}}\, = \,\root\of {\frac{{{\rm{3}}P}}{\rho }} \eqno(9)$$
This gives about  $$$ {{\rm v}_{{\rm{av2}}}}\, \cong \,{\rm{500}}\,{\rm{m}}\,{{{\rm{s}}}^{{\rm{ - 1}}}} $$$
for the molecules in air. Similarly, one obtains other relations between macroscopic and microscopic properties such as the mean free path. The possibility of deriving such microscopic quantities for molecules from simple macroscopic properties is striking. Accurate results for N A became available only after 1900 following the work of Planck, Einstein, Perrin and Millikan. The order of magnitude of about 1023 to 1024 (mol–1) was obtained by Loschmidt and a little later the two Duprés after 1865.Reference Dupré and Dupré12 The numbers are huge. The number of molecules of water in 1 cm3 is about the same order of magnitude as the total number of stars in the Universe.
$$$ {{\rm v}_{{\rm{av2}}}}\, \cong \,{\rm{500}}\,{\rm{m}}\,{{{\rm{s}}}^{{\rm{ - 1}}}} $$$
for the molecules in air. Similarly, one obtains other relations between macroscopic and microscopic properties such as the mean free path. The possibility of deriving such microscopic quantities for molecules from simple macroscopic properties is striking. Accurate results for N A became available only after 1900 following the work of Planck, Einstein, Perrin and Millikan. The order of magnitude of about 1023 to 1024 (mol–1) was obtained by Loschmidt and a little later the two Duprés after 1865.Reference Dupré and Dupré12 The numbers are huge. The number of molecules of water in 1 cm3 is about the same order of magnitude as the total number of stars in the Universe.
It is instructive to summarize the historical situation of the determination of N A just after 1900.
1. Using the determination of the elementary charge e− and Faraday's constant
$$${{F}_A} $$$
with
$$${{N}_A} = {{F}_A}/e $$$
, there were several determinations between 1897 (by Townsend) and 1916 (by Millikan) with finally in this last year
$$${{N}_{\rm{A}}} = {\rm{6}}{\rm{.06}}\,\times \,{\rm{1}}{{{\rm{0}}}^{{\rm{23}}}} \,{\rm{mo}}{{{\rm{l}}}^{{\rm{ - 1}}}} $$$
.2. PlanckReference Planck13 determines around 1900 the Boltzmann constant k from his law for black body radiation, obtaining finally with the gas constant R,
$$$ {{N}_{\rm{A}}} = R/k = {\rm{6}}{\rm{.175}}\,\times \,{\rm{1}}{{{\rm{0}}}^{{\rm{23}}}} \,{\rm{mo}}{{{\rm{l}}}^{{\rm{ - 1}}}} $$$
.3. Perrin determines k from microscopic observation of the distribution of particles as a function of height, finding (1909)
$$${{N}_{\rm{A}}} = R/k = {\rm{6}}{\rm{.5}}\,\times \,{\rm{1}}{{{\rm{0}}}^{{\rm{23}}}} \,{\rm{mo}}{{{\rm{l}}}^{{\rm{ - 1}}}} $$$
.Reference Perrin144. Einstein determines k by means of his analysis of Brownian motion, finding in 1905
$$${{N}_{\rm{A}}}= {\rm{6}}{\rm{.17}}\,\times \,{\rm{1}}{{{\rm{0}}}^{{\rm{23}}}} \,{\rm{mo}}{{{\rm{l}}}^{{\rm{ - 1}}}} $$$
(also a less good value 4.15), then in 1908 the value 6.0 and in 1911 the value 6.56 as prefactor.5. Further, reasonably accurate values of N A were derived by X-ray crystallography after 1912 by von Laue, Bragg, Debye, Scherrer and Compton (in 1922). This is also one of the most accurate methods used today. The results given above indicate the accuracy achieved about 100 years ago.
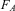

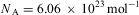
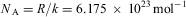
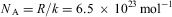
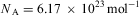
The accurate determination of N A or more generally the exact number of atoms in some macroscopic sample remains an important issue today. If we were able to reproducibly count this number for some specific sample (element or otherwise) we would be able to generate in the laboratory prototypes of exactly given mass defined by an appropriate definition of N A, for instanceReference Quack9, Reference Quack15
 $${{N}_{\rm{A}}} = {\rm{602,214,100,000,000,000,000,000}}\,{\rm{(mo}}{{{\rm{l}}}^{{\rm{ - 1}}}} {\rm{)}}\,{\rm{exactly}}\eqno(10)$$
$${{N}_{\rm{A}}} = {\rm{602,214,100,000,000,000,000,000}}\,{\rm{(mo}}{{{\rm{l}}}^{{\rm{ - 1}}}} {\rm{)}}\,{\rm{exactly}}\eqno(10)$$
assuming that the elementary units (atoms or molecules) have a unique, well-defined mass. Here, one mole might be defined by this number and would be consistent with the current definition (with 1 mole carbon corresponding to exactly 12 g 12C). However, it is still not possible to produce such mass prototypes with sufficient accuracy to provide a redefinition of the macroscopic mass unit kg. This is still defined by the ‘prototype kg’ in Paris, an arbitrary macroscopic body. Thus, starting out with old history and Avogadro's molecular hypothesis 200 years ago, we have reached here an unsolved and quite relevant problem of modern research combining the microscopic and the macroscopic world (see Refs Reference Quack9 and Reference Quack15 and references cited therein).
Other historical problems are related to just how the atoms combine to molecules, the question of molecular structure and the chemical bond. We have already mentioned Demokritos’ simple mechanical ideas on this matter and shall now turn to the development of ideas and models in more recent history after 1800.
3. Models of Molecules
The question of how to build molecules from atoms leads to the most fundamental models of chemistry. The basic concept was strongly influenced by the collaborative work of J.L. Gay-Lussac and A. von Humboldt in December 1804 (see above, Ref. Reference Gay-Lussac and Humboldt16).
In modern notation and including results derived from Avogadro's hypothesis we can write down their result on the synthesis of water from the elements quite naturally as an equation for molecules
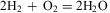 $${\rm{2}}{{{\rm{H}}}_{\rm{2}}}\, + \,{{{\rm{O}}}_{\rm{2}}} = {\rm{2}}{{{\rm{H}}}_{\rm{2}}}{\rm{O}}\eqno(2)$$
$${\rm{2}}{{{\rm{H}}}_{\rm{2}}}\, + \,{{{\rm{O}}}_{\rm{2}}} = {\rm{2}}{{{\rm{H}}}_{\rm{2}}}{\rm{O}}\eqno(2)$$
We know today following Avogadro's hypothesis that hydrogen and oxygen as gases consist of molecules H2 and O2 with two atoms each and water vapour is composed of molecules H2O. However, for a long time during the nineteenth century water was still formulated as OH. Otherwise modern notation for molecules is largely derived from the abstract notation with the symbols of the elements from Berzelius.
Berzelius still noted the number of atoms as an exponent. This was sometimes used until the end of the nineteenth century.17 Even in 1910 one could find the old Berzelius notation for the reaction corresponding to the explosion of picric acid17
 $${\rm{2}}\,{{{\rm{C}}}^{\rm{6}}} {{{\rm{H}}}^{\rm{2}}} {{({\rm{N}}{{{\rm{O}}}^{\rm{2}}} )}^{\rm{3}}} {\rm{OH}} = {\rm{C}}{{{\rm{O}}}^{\rm{2}}} \, + \,{{{\rm{H}}}^{\rm{2}}} {\rm{O}}\, + \,{\rm{11}}\,{\rm{CO}}\, + \,{\rm{2}}{{{\rm{H}}}^{\rm{2}}} \, + \,{\rm{3}}{{{\rm{N}}}^{\rm{2}}} \eqno(11)$$
$${\rm{2}}\,{{{\rm{C}}}^{\rm{6}}} {{{\rm{H}}}^{\rm{2}}} {{({\rm{N}}{{{\rm{O}}}^{\rm{2}}} )}^{\rm{3}}} {\rm{OH}} = {\rm{C}}{{{\rm{O}}}^{\rm{2}}} \, + \,{{{\rm{H}}}^{\rm{2}}} {\rm{O}}\, + \,{\rm{11}}\,{\rm{CO}}\, + \,{\rm{2}}{{{\rm{H}}}^{\rm{2}}} \, + \,{\rm{3}}{{{\rm{N}}}^{\rm{2}}} \eqno(11)$$
We follow today the notation with a right lower index introduced by J. v. Liebig in 1834. Formulae such as H2O for water, CH4 for methane or C2H4 for ethylene are not supposed to provide any structural model of the molecule, they just provide the composition of the molecule in terms of the numbers of atoms.
After about 1850, Loschmidt, Couper, Lothar Meyer and Kekulé used planar structural models, still sometimes used today, for instance for methane, CH4 (Figure 2).
Figure 2 Planar structural model for methane CH4.
One started to use a line drawn between atoms to symbolize a ‘bond’ and valence (Figure 3). This led to the concept of the double bond in order to have the valence 4 for the carbon atom in ethylene.
Figure 3 Structure of ethylene with a ‘double bond’.
Benzene was represented as a hexagon (Figure 4) with fluctuating double bonds (Kekulé) or ‘resonance’ structures (Pauling after 1930).
Figure 4 Resonance structure of benzene.
A further important step was the transition from a description in the plane to a three-dimensional model in space as proposed independently by le Bel and van't Hoff in 1874. The three-dimensional tetrahedral models built by van't Hoff for methane and its derivatives are particularly well known. They correspond to the approach of the molecular architect and similar spatial models are still used in everyday work by the organic chemist, for instance. The famous model built for DNA by Crick and Watson in the 1950s followed the same spirit. The three-dimensional models are much more realistic than the planar models and they can immediately explain some prominent observations in organic stereochemistry. For instance, a planar model for methylenechloride CH2Cl2 would predict incorrectly two different isomers ‘cis’ and ‘trans’ (Figure 5).
Figure 5 The incorrect planar model for CH2Cl2.
Only one isomer is actually found, as is obviously true for a tetrahedral model of CH2Cl2 and easily seen by inspection of a model analogous to the one shown in Figure 7. On the other hand, ethylene and its derivatives such as dichloroethylene C2H2Cl2 are actually planar, and thus cis- and trans-isomers do, indeed, occur (Figure 6).
Figure 6 Correct planar model for dichloroethylene with two isomers.
Figure 7 Correct, approximately tetrahedral structure of CHFClBr with two ‘enantiomers’, isomers that are the mirror image of each other.
Generally we call molecules with the same composition (here C2H2Cl2), but different structures (here cis and trans), ‘isomers’.
The tetrahedral model in space for methane derivatives with the four substituents sitting at the corners of a possibly distorted tetrahedron and the carbon atom in the middle of this tetrahedron explains also the very special kind of isomerism as observed for CHFClBr, for example, which in an (approximately) tetrahedral arrangement of the four substituent atoms H, F, Cl, Br around the central carbon atom has two ‘enantiomers’. These are isomers, which are the mirror image of each other.
Figure 7 shows a modern computer graphic of this model. Because of the special mirror symmetry, the two isomers can be distinguished by their geometry in the same way as we can distinguish left and right hands or a left- or right-hand glove, but they would be energetically exactly equivalent (provided the symmetry holds exactly, which is actually not the case, see below). Modern stereochemistry still uses this model to characterize this structural property of what we call today ‘chiral’ molecules (‘handed’ molecules, from the Greek χε $$${\rm \riota^{\hskip-2pt\tilde}} $$$
ρ, hand). This term was introduced by Lord Kelvin. An earlier word for this property was ‘dissymmetry’ (introduced by L. Pasteur). The modern convention to uniquely define the so-called ‘R’ and ‘S’ enantiomers has been introduced by Cahn, Ingold and Prelog 1956/57 (from ‘rectus’ and ‘sinister’). An older convention, still used today to some extent is the ‘D’ and ‘L’ nomenclature (from ‘dextro’ and ‘laevo’). The energetic consequences of the symmetry between enantiomers were recognized and pointed out by van't Hoff.Reference Bourgois18–Reference van't Hoff20
$$${\rm \riota^{\hskip-2pt\tilde}} $$$
ρ, hand). This term was introduced by Lord Kelvin. An earlier word for this property was ‘dissymmetry’ (introduced by L. Pasteur). The modern convention to uniquely define the so-called ‘R’ and ‘S’ enantiomers has been introduced by Cahn, Ingold and Prelog 1956/57 (from ‘rectus’ and ‘sinister’). An older convention, still used today to some extent is the ‘D’ and ‘L’ nomenclature (from ‘dextro’ and ‘laevo’). The energetic consequences of the symmetry between enantiomers were recognized and pointed out by van't Hoff.Reference Bourgois18–Reference van't Hoff20
For the R and S enantiomers of chiral molecules one would have exactly equal energies at an absolute temperature T = 0 Kelvin and thus a reaction enthalpy  $$${{\rDelta }_{\rm{R}}}H_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o} }}}} $$$
and Gibbs energy
$$${{\rDelta }_{\rm{R}}}H_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o} }}}} $$$
and Gibbs energy 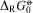 $$${{\rDelta }_{\rm{R}}}G_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o}}}} $$$
exactly zero by symmetry (and also at all other T)
$$${{\rDelta }_{\rm{R}}}G_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o}}}} $$$
exactly zero by symmetry (and also at all other T)
 $$R = S{\rm{;}}\; \; {{\rDelta }_{\rm{R}}}H_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o} }}}} = {{\rDelta }_{\rm{R}}}G_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o} }}}} = {\rm{0}}\,{\rm{(exactly}}\,{\rm{by}}\,{\rm{symmetry)}}\eqno(12)$$
$$R = S{\rm{;}}\; \; {{\rDelta }_{\rm{R}}}H_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o} }}}} = {{\rDelta }_{\rm{R}}}G_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o} }}}} = {\rm{0}}\,{\rm{(exactly}}\,{\rm{by}}\,{\rm{symmetry)}}\eqno(12)$$
Van't Hoff writes in conclusion of the first chapter of his paper (originally in French, translated here by us):
Such an equilibrium depends on the work [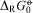 $$${{\rDelta }_{\rm{R}}}G_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o} }}}} $$$
, van't Hoff writes E in old notation], which such a transformation can produce. This work must be zero in this case in view of the exact mechanical symmetry of the two isomers, following the concepts developed. It follows that the equilibrium constant K, which determines the relative proportion of the two compounds [enantiomers] is equal to unity because of the following equation
$$${{\rDelta }_{\rm{R}}}G_{{\rm{0}}}^{{{\rm{ - \hskip -4.6pt o} }}}} $$$
, van't Hoff writes E in old notation], which such a transformation can produce. This work must be zero in this case in view of the exact mechanical symmetry of the two isomers, following the concepts developed. It follows that the equilibrium constant K, which determines the relative proportion of the two compounds [enantiomers] is equal to unity because of the following equation
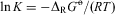 $$ \ln K = {\rm{ - }}{{\rDelta }_{\rm{R}}}{{G}^{{\rm{ - \hskip -4.6pt o} }}}} /(RT) \eqno(13)$$
$$ \ln K = {\rm{ - }}{{\rDelta }_{\rm{R}}}{{G}^{{\rm{ - \hskip -4.6pt o} }}}} /(RT) \eqno(13)$$
where T indicates the absolute temperature. It is thus clear that at equilibrium the relative amounts of the two isomers [enantiomers] must be equal. [We have rewritten equation (13) in modern notation here with the natural logarithm  $$$ \ln \,K = \ln \,{\rm{1}} = {\rm{0}} $$$
in this case.]
$$$ \ln \,K = \ln \,{\rm{1}} = {\rm{0}} $$$
in this case.]
Van't Hoff's simple models were perfectly adequate to recognize these properties of enantiomers. In this sense the modern computer graphics in Figure 7 adds nothing new, although the geometrical size relations and distances between the atoms are represented more realistically (see, however, below). Using the model, one can easily see that we have exactly these two isomers, which are mirror images of each other, and no more.
However, from a planar geometry one would expect incorrectly three isomers, depending on whether H is opposite to F, Cl or Br (Figure 8).
Figure 8 Incorrect planar model of CHFClBr showing three isomers.
These three isomers are not found, whereas the two energetically equivalent isomers are found precisely as predicted by van't Hoff's model. This could be taken as evidence in favour of the model.
It is quite remarkable in this context that until around 1950 it was not known whether our macroscopic models of molecules such as CHFClBr or other chiral molecules in nature, such as the chiral amino acid L-alanine, which is a building block of the proteins in our body, correspond to the microscopic molecules, which we find in nature, or to their mirror image, because methods of molecular structure determination available until that time could not answer this question. The answer was given around 1950 by J. Bijvoet using a special crystallographic method.Reference Bijvoet, Peerdeman and van Bommel21 Today we have also several other methods available to answer this fundamental question, such as measuring vibrational circular dichroism in infrared spectra, for instance, and comparing with quantum chemical ab initio calculations.
Models on the computer, such as the one shown in Figure 7, or physical models of plastic, steel and wood are ubiquitous in teaching and research in chemistry today. They prove enormously useful but in fact they do not, strictly speaking, correspond to our fundamental theoretical understanding of the structure and dynamics of chiral molecules, which we shall discuss further below.Reference Quack22–Reference Quack24 From the present point of view these classical mechanical macroscopic models are more a caricature than a true image of chiral molecules. Nevertheless, they remain useful, although with some serious limitations. Following the name of a well-known children's toy (‘Lego’) this kind of thinking about chemistry, by putting atoms together with sticks to provide molecules made of sticks and balls, is sometimes called ‘Legochemistry’ to express these limitations. On the other hand, it is widely used in synthesis planning in the pharmaceutical chemistry. Fitting chiral molecules together like key and lock, following a parable of Emil Fischer, or perhaps even better like hand and glove, fits some aspects of the very nature of chiral molecules. We might mention here an instructive book on molecular symmetry, structure and chirality with many nice pictures.Reference Heilbronner and Dunitz25
A historical remark might be useful to finish this section. At the time of van't Hoff's structural hypothesis, he was heavily criticized, indeed, severely attacked in print. H. Kolbe wrote a comment on the famous paper ‘La chimie dans l'Espace’ and its German translation ‘Die Lagerung der Atome im Raum’ by F. Hermann, which contains plenty of insults among some criticism.Reference Kolbe26 Nevertheless, this did not harm van't Hoff, who justly won the first Nobel prize in Chemistry in 1901.
From another point of view one must also remark that there was no a priori guarantee that methane and its derivatives have a tetrahedral structure such as in Figure 7. In principle, a planar structure such as in Figure 2 would have been similarly possible. The question had to be answered (and was answered) by experiment. Initially, this was shown by the stereochemical results discussed above, later much more precisely and definitively by physical chemical, for instance spectroscopic and crystallographic, techniques.
That such questions are not trivially answered by inspection can be seen from the theoretical prediction of a planar excited stateReference Pepper, Shavitt, Ragué Schleyer, Glukhovtsev, Janoschek and Quack27 for methane, not yet confirmed experimentally. The methyl radical CH3 could hypothetically be planar or pyramidal, we know today (from spectroscopic experiments after 1956) that it is planar in the ground state.Reference Herzberg28 Thus, van't Hoff's concepts were ingenious, but corresponded to a structural hypothesis, which had to be confirmed by experiment.
4. Models of Binding in Chemistry
As already discussed above, during the nineteenth century chemists essentially took over the mechanical picture of binding between atoms as formulated by Demokritos with his hooks and loops. One hook would correspond to a single bond, two hooks to a double bond, three hooks to a triple bond (Figure 9).
Figure 9 Representation of binding with a CC single bond in ethane (top), a CC double bond in ethylene (middle) and a CC triple bond in acetylene (bottom).
Each ‘bond’ is represented by one line for one ‘valence’. An atom has a fixed number of ‘valences’ (hooks), the hydrogen atom just one, the carbon atom four in the examples of Figure 9. With this kind of model one can nicely represent what we call today a covalent bond between atoms in molecules. Another description uses the electrostatic forces between ‘charged atoms’ (or ‘ions’) to bind these together, such as in Na+ Cl–, which forms ‘ionic crystals’. This seemed particularly natural after the theory of electrolytic dissociation due to Arrhenius (1884). G.N. Lewis around 1920 combined these ideas with the Rutherford-Bohr model of the atom. Each atom in a chemical bond tries to complete its electronic shell, for instance to establish a ‘stable octet’ of ‘8 valence electrons’. This can be done either by a transfer of electrons generating two ions such as in Na+ Cl– or H+ F– or by sharing the electrons, where each pair of shared electrons provides one covalent bond (Figure 10).
Figure 10 Electron pair and octet rule (ionic) models of the chemical binding. Each point represents an electron, each line represents two shared valence electrons.
This type of model is still much used today in elementary teaching, but also as a simple model of chemical binding quite generally.
A theory of chemical binding arose from quantum mechanics after 1925. In an abstract sense we would say today that molecules such as H2 or HF are bound, because the quantum mechanical ground state energy (and also the energy of some excited states) of the combined system with all particles ‘bound together’ in a small region of space is lower than the energies of the separated atoms H+H or H+F. The relevant energies are obtained from the solution of the general quantum mechanical equations of motion. In practice, one solves the time independent Schrödinger equation
 $$\hat{H}{{\rphi }_{\rm{k}}}\left( r \right) = {{E}_{\rm{k}}}{{\rphi }_{\rm{k}}}\left( r \right)\eqno(14)$$
$$\hat{H}{{\rphi }_{\rm{k}}}\left( r \right) = {{E}_{\rm{k}}}{{\rphi }_{\rm{k}}}\left( r \right)\eqno(14)$$
where 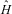 $$$\hat{H} $$$
is the Hamilton operator and
$$$\hat{H} $$$
is the Hamilton operator and 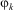 $$${{\rphi}_k} $$$
are the time independent wavefunctions for the ‘stationary states’ with energy eigenvalues
$$${{\rphi}_k} $$$
are the time independent wavefunctions for the ‘stationary states’ with energy eigenvalues 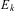 $$${{E}_k} $$$
. The ground state energy of the bound molecule would be the lowest eigenvalue
$$${{E}_k} $$$
. The ground state energy of the bound molecule would be the lowest eigenvalue 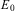 $$${{E}_0} $$$
with
$$${{E}_0} $$$
with 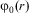 $$${{\rphi}_0}(r) $$$
being confined to values of the generalized coordinates r restricted to a region of space where the atoms are rather close together (thus ‘bound’). Of course, there may be, and in general are, many excited energy states with
$$${{\rphi}_0}(r) $$$
being confined to values of the generalized coordinates r restricted to a region of space where the atoms are rather close together (thus ‘bound’). Of course, there may be, and in general are, many excited energy states with  $$${{E}_{\rm{1}}},\,{{E}_{\rm{2}}},\,{{E}_{\rm{3}}}\:\, \ldots \,\,{{E}_{\rm{n}}} $$$
, which also have wavefunctions
$$${{E}_{\rm{1}}},\,{{E}_{\rm{2}}},\,{{E}_{\rm{3}}}\:\, \ldots \,\,{{E}_{\rm{n}}} $$$
, which also have wavefunctions  $$${{\rphi }_{\rm{1}}},{{\rphi }_{\rm{2}}} \ldots {{\rphi }_{\rm{n}}} $$$
corresponding to a bound molecule.
$$${{\rphi }_{\rm{1}}},{{\rphi }_{\rm{2}}} \ldots {{\rphi }_{\rm{n}}} $$$
corresponding to a bound molecule.
Simplified quantum mechanical model theories were constructed by L. Pauling with his ‘valence bond’ modelReference Pauling29 and by F. Hund and R. Mulliken with their molecular orbital model (MO-model) and also the Hückel-MO-model (HMO model) in the years between 1925 and 1950. All these models are still in use today and they can, for instance, describe the benzene structure as a symmetric, regular hexagon. These models are, in principle, mathematical models, simplifications of the Schrödinger equation for molecular systems consisting of many electrons and atomic nuclei. There are also many other simplifications or mathematical models derived from the quantum mechanical theory and since about 1960 graphical representations using molecular orbitals to describe certain types of binding have become commonplace. Figure 11 shows examples.
Figure 11 The nature of the chemical bond is not so simple: Heitler-London, Herzberg, Hund, Mulliken, Pauling …1926 ff … today. Various graphical representations of chemical binding in benzene, top, and nitric acid, bottom.
As in other mathematical model theories in physics and chemistry a very complicated differential equation from the complete mathematical theory is replaced by a much simplified equation (or even just a graphical picture). For instance, the complete molecular Hamiltonian  $$$\hat{H} $$$
is replaced by a simple model Hamiltonian. The concept of model is used here for a mathematical object. A brief history of the modern theory of the chemical binding can be found in the introductory chapters of Refs Reference Pauling29 and Reference Kutzelnigg30.
$$$\hat{H} $$$
is replaced by a simple model Hamiltonian. The concept of model is used here for a mathematical object. A brief history of the modern theory of the chemical binding can be found in the introductory chapters of Refs Reference Pauling29 and Reference Kutzelnigg30.
While, from an abstract point of view, the quantum mechanical understanding of chemical binding is straightforward, there has been some discussion of a deeper interpretation of the physical origin of the chemical bond (see, for example, Ref. Reference Ruedenberg31). The current state of affairs including relativistic theory is summarized in Ref. Reference Reiher and Wolf32.
5. Today's quantum mechanical theory of microscopic matter arises from the modelling of spectra
How did quantum mechanics as the current theory of molecular binding, structure and dynamics arise? Indeed, as is most beautifully exemplified by the development of experiment and theory in determining the binding energy in the molecule H2 over the last 85 years as summarized in Ref. Reference Sprecher, Jungen, Ubachs and Merkt33, quantum mechanics (in its extended forms including relativistic, quantum electrodynamical and other effects) can claim to be an accurate theory of chemistry at the atomic and molecular level.Reference Merkt and Quack5, Reference Reiher and Wolf32 Its predictions for the binding energy in molecular hydrogen H2 agree with experiment to within better than eight significant digits (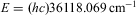 $$$E = (hc){\rm{36118}}{\rm{.069}}\,{\rm{c}}{{{\rm{m}}}^{{\rm{ - 1}}}} $$$
). Indeed, quantum mechanics is also the basic theory of all microscopic matter in the framework of the so-called ‘standard model of particle physics’.Reference Hoddeson, Brown, Riordan and Dresden34–Reference ‘t Hooft39
$$$E = (hc){\rm{36118}}{\rm{.069}}\,{\rm{c}}{{{\rm{m}}}^{{\rm{ - 1}}}} $$$
). Indeed, quantum mechanics is also the basic theory of all microscopic matter in the framework of the so-called ‘standard model of particle physics’.Reference Hoddeson, Brown, Riordan and Dresden34–Reference ‘t Hooft39
Historically, quantum mechanics had two major origins in attempts to find mathematical models for observed spectra. One of these spectra arose from the continuous heat radiation emitted by a ‘perfect black body’, which is in perfect thermal equilibrium at some temperature T, measured with ever increasing accuracy towards the end of the nineteenth century. At the turn of the centuryReference Planck40–Reference Planck41 Max Planck was able to model this spectrum with the mathematical form given by him in Ref. Reference Planck13, but slightly modified here to modern notation for the energy density 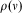 $$$\rho (\nu ) $$$
Reference Merkt and Quack5
$$$\rho (\nu ) $$$
Reference Merkt and Quack5
 $$\rho (\nu ) = \frac{{{\rm{8}}\pi h{{\nu }^{\rm{3}}} }}{{{{c}^{\rm{3}}} }}\;\frac{{\rm{1}}}{{{{e}^{h\nu /kT}} \,{\rm{ - }}\,{\rm{1}}}}\eqno(15)$$
$$\rho (\nu ) = \frac{{{\rm{8}}\pi h{{\nu }^{\rm{3}}} }}{{{{c}^{\rm{3}}} }}\;\frac{{\rm{1}}}{{{{e}^{h\nu /kT}} \,{\rm{ - }}\,{\rm{1}}}}\eqno(15)$$
where h is Planck's constant, ν the frequency of the radiation, c the speed of light and k the Boltzmann constant. When finding this mathematical model for the spectrum, Planck was able to explain it with the revolutionary hypothesis of energy quantization with energy quanta  $$$h \nu$$$
.Reference Planck40–Reference Planck41
$$$h \nu$$$
.Reference Planck40–Reference Planck41
The second spectroscopic model arose from the discovery by Bunsen und KirchhoffReference Kirchhoff and Bunsen42 in 1860 that the Fraunhofer lines in the spectrum of the sunReference Fraunhofer43 can be related to discrete line spectra arising from atoms of the elements absorbing or emitting radiation at specific frequencies ν. Balmer, in 1885, found a mathematical model for a series of lines arising from the hydrogen atom (now called Balmer series), with integer n > 2
 $$ {{\lambda }_{\rm{n}}} = {{\lambda }_{\rm{0}}}\;{{n}^{\rm{2}}} /({{n}^{\rm{2}}} \,{\rm{ - }}\,{{{\rm{2}}}^{\rm{2}}} ) \eqno(16)$$
$$ {{\lambda }_{\rm{n}}} = {{\lambda }_{\rm{0}}}\;{{n}^{\rm{2}}} /({{n}^{\rm{2}}} \,{\rm{ - }}\,{{{\rm{2}}}^{\rm{2}}} ) \eqno(16)$$
with the wavelengths 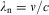 $$${{\lambda }_{\rm{n}}} = \nu /c $$$
.
$$${{\lambda }_{\rm{n}}} = \nu /c $$$
.
Bohr was able, in 1913, to show that this mathematical model (extended to a more general form including other series with 22 being replaced by m 2) could be explained by combining Rutherford's atomic model derived from scattering experiments, with a point-like nucleus and electrons with a classical mechanical ‘Kepler’-like model for the electron orbits and the energy (or action, angular momentum) quantization of Planck'sReference Merkt and Quack5–Reference Quack9 (Figure 12). Spectral lines  $$${{\nu}_{fi}} $$$
corresponded to transitions between stationary, quantized orbits in this model with quantized energies
$$${{\nu}_{fi}} $$$
corresponded to transitions between stationary, quantized orbits in this model with quantized energies 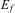 $$${{E}_f} $$$
and
$$${{E}_f} $$$
and 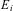 $$${{E}_i} $$$
$$${{E}_i} $$$
Figure 12 The Rutherford–Bohr atomic models with electrons orbiting around a nucleus analogous to a solar and planetary system (schematically, left Helium with two electrons, each shown on two positions on an orbit, right Hydrogen with one electron).
 $$\rDelta {{E}_{{\rm{f}}i}} = \left| {{{E}_{\rm{f}}}\,{\rm{ - }}\,{{E}_{\rm{i}}}} \right| = h{{\nu}_{{\rm{fi}}}}\eqno(17)$$
$$\rDelta {{E}_{{\rm{f}}i}} = \left| {{{E}_{\rm{f}}}\,{\rm{ - }}\,{{E}_{\rm{i}}}} \right| = h{{\nu}_{{\rm{fi}}}}\eqno(17)$$
This treatment was quickly extended by Sommerfeld including relativistic effects.Reference Sommerfeld44 It led to the models of chemical binding discussed in the previous section and to an understanding of the periodic system of elements with the ‘periods’ being characterized by the series of numbers for closed electron shells in the atom 2, 8, 8, 18, 18, 32 … (2n 2), when including the Pauli principle. Representing experimental spectra by mathematical models was certainly the crucial step in the development of modern quantum mechanics. It is also today one of the essential approaches towards understanding structure and dynamics at the quantum mechanical level.Reference Quack and Merkt45 In addition, it is the approach to extend the current frontiers of our understanding of molecules (see below, Section 7).
6. Models of Processes in Chemistry
The theory of microscopic processes in chemistry is based on the time-dependent Schrödinger equation.Reference Schrödinger46–Reference Schrödinger47
 $${\rm i}\frac{h}{{2\rpi }}\,\frac{{\partial\rpsi }}{{\partial t}} = \hat{H}\rpsi\eqno(18)$$
$${\rm i}\frac{h}{{2\rpi }}\,\frac{{\partial\rpsi }}{{\partial t}} = \hat{H}\rpsi\eqno(18)$$
with the time-dependent wave function  $$$\rpsi $$$
and the Hamilton operator
$$$\rpsi $$$
and the Hamilton operator  $$$\hat{H} $$$
(and
$$$\hat{H} $$$
(and  $$${\rm{i}}=\root\of {{\rm{ - 1}}} $$$
). It dates from the year 1926 and is known to be equivalent to other quantum mechanical equations such as the Heisenberg equations of motion.Reference Pleijel48–Reference Dirac51 This equation can be solved ‘exactly’ (on the computer) with reasonable accuracy only for very simple molecules with perhaps at most four to five not too heavy atoms. Many further simpler mathematical models have been derived from this and further developed for describing molecular processes and chemical reactions. Among these we can name classical molecular dynamics (molecular modelling), which describes the motion of atoms using classical (Newtonian) mechanics and forces derived empirically or semi-empirically in the framework of ‘force fields’, which can give rather accurate predictions for very complex molecules including proteins or other biomolecules.Reference van Gunsteren, Bakowies, Baron, Chandrasekhar, Christen, Daura, Gee, Geerke, Glattli, Hunenberger, Kastenholz, Ostenbrink, Schenk, Trzesniak, van der Vegt and Yu52
$$${\rm{i}}=\root\of {{\rm{ - 1}}} $$$
). It dates from the year 1926 and is known to be equivalent to other quantum mechanical equations such as the Heisenberg equations of motion.Reference Pleijel48–Reference Dirac51 This equation can be solved ‘exactly’ (on the computer) with reasonable accuracy only for very simple molecules with perhaps at most four to five not too heavy atoms. Many further simpler mathematical models have been derived from this and further developed for describing molecular processes and chemical reactions. Among these we can name classical molecular dynamics (molecular modelling), which describes the motion of atoms using classical (Newtonian) mechanics and forces derived empirically or semi-empirically in the framework of ‘force fields’, which can give rather accurate predictions for very complex molecules including proteins or other biomolecules.Reference van Gunsteren, Bakowies, Baron, Chandrasekhar, Christen, Daura, Gee, Geerke, Glattli, Hunenberger, Kastenholz, Ostenbrink, Schenk, Trzesniak, van der Vegt and Yu52
Further approaches use the so-called density functional theory (derived from the Schrödinger equation) to calculate forces between atoms ab initio and model the motion of the atoms under the influence of these forces in the framework of the so-called Car-Parrinello molecular dynamics, also using classical Newtonian mechanics for atomic motion.Reference Car and Parrinello53 The differential equations of chemical kinetics constitute another mathematical model for chemical reactions, including models for chain reactions, combustions, explosions, detonations and the complex chemical phenomena in the Earth's atmosphere.Reference Quack and Jans-Bürli54–Reference Marquardt and Quack58
In many cases the names ‘model’ and ‘theory’ are used here interchangeably. Thus one speaks of ‘transition state theory’, ‘RRKM theory’ (after Rice, Ramsperger, Kassel, Marcus), and ‘Quasi-equilibrium Theory’ of chemical reactions, even though these are really simplified models in reaction kinetics, or approximate theories.
On the other hand, a generalized version of these theories has used the more modest name ‘statistical adiabatic channel model’.Reference Quack and Troe55 Many of these approximate theories or models can be related to the time-dependent Schrödinger equation and we refer to the surveys given in Refs 5 and Reference Luckhaus and Quack56–Reference Quack61 for details and many further references. A problem of current interest concerns the flow of energy within molecules of the femtosecond to nanosecond time scales (10–15 to 10–9 s), which can be studied by modelling molecular motion using full quantum dynamics (Equation (18)),Reference Luckhaus and Quack56–Reference Quack61 describing ‘molecules in motion’. Very recent efforts study the motion of electrons on timescales of less than 10–15 s in atomic and molecular processes.Reference Wörner and Corkum62
7. Current Frontiers of Models in Chemistry and the Fundamental Laws of Physics
The underlying physical laws for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble.
It therefore becomes desirable that approximate practical methods of applying quantum mechanics should be developed, which can lead to an explanation of complex atomic systems without too much computation. (From P.A.M. Dirac (1929) as cited and discussed in Ref. Reference Quack63; emphasis added)
This famous citation from Dirac is widely quoted to demonstrate that the ‘theory of chemistry’ was then completed. However, this is not true, as we know today: for chiral molecules electroweak parity violating quantum chemistry introduces fundamentally new aspectsReference Quack23–Reference Quack24 and we shall address this now.
While the simple classical models of chemistry are very fruitful and widely used in chemistry still today, they reach their limits in many cases. Our modern understanding of chemical binding, molecular structure and dynamics is based on quantum mechanics, but often combined with a ‘quasi-classical’ picture of atoms in molecules. This has limits, for instance when ‘tunnelling’ becomes important, a process that is strictly forbidden, even meaningless, in a classical framework. We shall illustrate this with some recent research on chiral molecules, which is at the frontier of our current understanding of molecular structure and dynamics related to some of the most fundamental laws of physics. We have recently reviewed this topic from different points of view and refer to these more extensive reviews for details.Reference Quack9, Reference Quack24, Reference Quack61, Reference Quack63–Reference Quack, Stohner and Willeke65 Here we give only a very brief account of the basic ideas.
It turns out that the study of the structure and dynamics of chiral molecules by means of spectroscopy can provide a window towards some of the most fundamental laws of physics related to the symmetries of physics and the underlying conservation laws.Reference Quack9, Reference Quack22–Reference Quack24, Reference Quack61, Reference Quack63–Reference Mainzer67
We can formulate the following fundamental symmetries of physics, which leave the molecular Hamiltonian 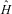 $$$\hat{H} $$$
invariant (see equation (18)):
$$$\hat{H} $$$
invariant (see equation (18)):
1. Any translation in space.
2. Any translation in time.
3. Any rotation in space.
4. Reflection of the particle coordinates at the origin (parity operation P or E*).
5. Time reversal or reversing momenta and spins of the particles (T for Tempus or time).
6. Every permutation of the indices of identical particles (the atomic nuclei, the nucleons, the electrons).
7. The replacement of all particles by their antiparticles (Charge conjugation C).
These symmetry operations form the symmetry group of the Hamiltonian operator.
A symmetry can be related to a ‘conservation law’ of physics and also to a fundamentally ‘non-observable property of nature’. When such a symmetry is broken or violated, this ‘non-observable’ property becomes observable and thus one can consider the discovery and study of such symmetry violations to fall among the most fundamental observations of physics and chemistry. We shall illustrate this with the example of parity symmetry (No. 4 in the list above) and chiral molecules. Figure 13 shows the corresponding symmetry operation.
Figure 13 Reflection  $$${{\hat{E}}^\ast} $$$
or Parity operation P (after Ref. Reference Quack23).
$$${{\hat{E}}^\ast} $$$
or Parity operation P (after Ref. Reference Quack23).
This symmetry operation transforms a right-handed Cartesian coordinate system into a left-handed system. It also transforms a chiral molecule into its enantiomer (Figure 7).
It was shown in the early days of quantum mechanics by Friedrich HundReference Hund68 that chiral molecules should show ground states (and other energy eigenstates) of well-defined parity, which are delocalized structures  $$${{\chi}_ + } $$$
and
$$${{\chi}_ + } $$$
and  $$${{\chi}_{\rm{ - }}} $$$
in a double well potential, as shown in Figure 14. By superposing them one can generate localized structures λ and ρ, corresponding to enantiomers
$$${{\chi}_{\rm{ - }}} $$$
in a double well potential, as shown in Figure 14. By superposing them one can generate localized structures λ and ρ, corresponding to enantiomers
Figure 14 Scheme illustrating de facto and de lege symmetry breaking with the example of potentials V(q) including parity violation in chiral molecules. The eigenstates of positive (+) and negative (–) parity in the symmetric potential (left-hand part) result in a delocalized probability density  $$${{\left|\rpsi \right|}^2} $$$
as a function of the inversion coordinate q but with a possibility of de facto localization near the left-hand
$$${{\left|\rpsi \right|}^2} $$$
as a function of the inversion coordinate q but with a possibility of de facto localization near the left-hand  $$$\left( {{{q}_{\rm L}}} \right)$$$
and right-hand
$$$\left( {{{q}_{\rm L}}} \right)$$$
and right-hand  $$$\left( {{{q}_{\rm R}}} \right)$$$
minima. With parity violation the potential is asymmetric (right-hand part) with localized eigenstates (at L or R). In the lower part we show the scheme illustrating localized wavefunctions
$$$\left( {{{q}_{\rm R}}} \right)$$$
minima. With parity violation the potential is asymmetric (right-hand part) with localized eigenstates (at L or R). In the lower part we show the scheme illustrating localized wavefunctions  $$$\rlambda$$$
and ρ and delocalized wavefunctions of well-defined parity
$$$\rlambda$$$
and ρ and delocalized wavefunctions of well-defined parity 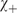 $$${{{\rm{\chi }}}_ + }$$$
and
$$${{{\rm{\chi }}}_ + }$$$
and  $$${\rm{ - }}{{{\rm{\chi }}}_{\rm{ - }}}$$$
. In the symmetric case (left-hand part) the wavefunctions of well-defined parity
$$${\rm{ - }}{{{\rm{\chi }}}_{\rm{ - }}}$$$
. In the symmetric case (left-hand part) the wavefunctions of well-defined parity  $$${{{\rm{\chi }}}_ + }$$$
and
$$${{{\rm{\chi }}}_ + }$$$
and  $$${\rm{ - }}{{{\rm{\chi }}}_{\rm{ - }}}$$$
are the eigenfunctions for the energies
$$${\rm{ - }}{{{\rm{\chi }}}_{\rm{ - }}}$$$
are the eigenfunctions for the energies 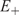 $$${{E}_ + }$$$
and
$$${{E}_ + }$$$
and  $$${{E}_{\rm{ - }}}$$$
(separated by
$$${{E}_{\rm{ - }}}$$$
(separated by 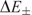 $$$\rDelta {{E}_ \pm }$$$
. Time-dependent wavefunctions
$$$\rDelta {{E}_ \pm }$$$
. Time-dependent wavefunctions 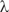 $$$\rlambda$$$
and ρ can be generated by superposition of
$$$\rlambda$$$
and ρ can be generated by superposition of  $$${{{\rm{\chi }}}_ + }$$$
and
$$${{{\rm{\chi }}}_ + }$$$
and  $$${\rm{ - }}{{{\rm{\chi }}}_{\rm{ - }}}$$$
. In the asymmetric case (right-hand part) the localized wavefunctions
$$${\rm{ - }}{{{\rm{\chi }}}_{\rm{ - }}}$$$
. In the asymmetric case (right-hand part) the localized wavefunctions 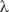 $$$\rlambda$$$
and ρ are eigenfunctions for the energies
$$$\rlambda$$$
and ρ are eigenfunctions for the energies 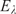 $$${{E}_\lambda}$$$
and
$$${{E}_\lambda}$$$
and  $$${{E}_\rho}$$$
separated by
$$${{E}_\rho}$$$
separated by 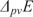 $$${{\Delta }_{pv}}E$$$
. Then the time-dependent wavefunctions of well-defined parity can be generated by superposition (after Ref. Reference Quack, Stohner and Willeke65).
$$${{\Delta }_{pv}}E$$$
. Then the time-dependent wavefunctions of well-defined parity can be generated by superposition (after Ref. Reference Quack, Stohner and Willeke65).
 $$ \lambda = \frac{{\rm{1}}}{{\root\of {\rm{2}} }}\left( {{{\chi }_ + }\, - \,{{\chi }_{\rm{ - }}}} \right) \eqno(19)$$
$$ \lambda = \frac{{\rm{1}}}{{\root\of {\rm{2}} }}\left( {{{\chi }_ + }\, - \,{{\chi }_{\rm{ - }}}} \right) \eqno(19)$$
 $$ \rho = \frac{{\rm{1}}}{{\root\of {\rm{2}} }}\left( {{{\chi }_ + }\,{\rm{ + }}\,{{\chi }_{\rm{ - }}}} \right) \eqno(20)$$
$$ \rho = \frac{{\rm{1}}}{{\root\of {\rm{2}} }}\left( {{{\chi }_ + }\,{\rm{ + }}\,{{\chi }_{\rm{ - }}}} \right) \eqno(20)$$
In a symmetric double well potential (with ‘parity conservation’) such superposition states can be transformed from one enantiomer into the other in a time given by the energy difference  $$$\rDelta {{E}_{\rm{ \pm }}} = {{E}_{\rm{ - }}}\,{\rm{ - }}\,{{E}_ + } $$$
.
$$$\rDelta {{E}_{\rm{ \pm }}} = {{E}_{\rm{ - }}}\,{\rm{ - }}\,{{E}_ + } $$$
.
 $$ {{t}_{\lambda {\rm{ - }}\rho }} = \frac{{\rm{1}}}{{{\rm{2}}\rDelta {{E}_ \pm }}} \eqno(21)$$
$$ {{t}_{\lambda {\rm{ - }}\rho }} = \frac{{\rm{1}}}{{{\rm{2}}\rDelta {{E}_ \pm }}} \eqno(21)$$
The transformation corresponds to a periodic motion with period 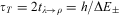 $$${{\tau }_T} = 2{{t}_{\lambda \rightarrow \rho }} = h/\rDelta {{E}_ \pm } $$$
. This quantum mechanical picture of chiral molecules introduced by Friedrich Hund in 1927 shows several features, which must appear most unusual in a classical mechanical framework.
$$${{\tau }_T} = 2{{t}_{\lambda \rightarrow \rho }} = h/\rDelta {{E}_ \pm } $$$
. This quantum mechanical picture of chiral molecules introduced by Friedrich Hund in 1927 shows several features, which must appear most unusual in a classical mechanical framework.
 $$)(--><$$>\hskip-19.3pc \rm Firstly, the\ states \ {{{\rm{\chi }}}_+ } \ and \ -{{{\rm{\chi }}}_{\rm{ - }}} $$
$$)(--><$$>\hskip-19.3pc \rm Firstly, the\ states \ {{{\rm{\chi }}}_+ } \ and \ -{{{\rm{\chi }}}_{\rm{ - }}} $$
 $$ {{\chi }_ + } = \frac{{\rm{1}}}{{\root\of {\rm{2}} }}\left( {\lambda \, + \,\rho } \right) \eqno(22)$$
$$ {{\chi }_ + } = \frac{{\rm{1}}}{{\root\of {\rm{2}} }}\left( {\lambda \, + \,\rho } \right) \eqno(22)$$
 $$ {\rm{ - }}{{\chi }_{\rm{ - }}} = \frac{{\rm{1}}}{{\root\of {\rm{2}} }}\left( {\lambda {\rm{ - }}\rho } \right) \eqno(23)$$
$$ {\rm{ - }}{{\chi }_{\rm{ - }}} = \frac{{\rm{1}}}{{\root\of {\rm{2}} }}\left( {\lambda {\rm{ - }}\rho } \right) \eqno(23)$$
are the ground and first excited quantum states of a chiral molecule and have the property of being at the same time, simultaneously one enantiomer and its opposite enantiomer.
Such states would be meaningless, unthinkable in a classical picture. This would in our macroscopic world correspond to one person being in one room and in another room at the same time (with a ‘probability’ = 50%, though). A similarly ‘unthinkable’ example, which is frequently quoted, is Schrödinger's cat, which in a quantum picture might be dead and alive at the same time, a somewhat cruel illustration. For chiral molecules such as hydrogen peroxide HOOH (Figure 15), substituted aniline C6H5NHD, and similar molecules,Reference Fehrensen, Luckhaus and Quack69–Reference Marquardt and Quack71 which have a relatively low potential barrier for interconversion such states have been well-studied by spectroscopy and are actually common. They have a well-defined ‘parity’, which is their symmetry under reflection, when they can be either symmetric (positive parity, index +) or antisymmetric (negative parity, index –). This property of parity can be observed by means of spectroscopic selection rules.Reference Fehrensen, Luckhaus and Quack69–Reference Marquardt and Quack71 Such states are actually ‘achiral’ (not chiral). On the other hand, the states λ and ρ have no well-defined parity, but are chiral. The second unusual effect is now that λ can be transformed to ρ in a short time, even if the energy is far below the potential barrier (the possible energies in the state λ are E + and E - in Figure 14, being far below the energy at qc in the left-hand part of Figure 14). For instance, the transformation in hydrogen peroxide happens on a time scale of picoseconds (10−12 s).Reference Fehrensen, Luckhaus and Quack69Figure 15 shows the enantiomers of hydrogen peroxide.
Figure 15 Image and mirror-image form of H2O2 (HOOH) in the chiral equilibrium geometry of the PCPSDE-potential hypersurface.Reference Kuhn, Rizzo, Luckhaus, Quack and Suhm72 Image and mirror image are enantiomers which cannot be converted into each other through a rotation in space but instead through an internal rotation about the OO-axis preferably via the trans geometry.Reference Kuhn, Rizzo, Luckhaus, Quack and Suhm72 White, H; blue, O (After Ref. Reference Quack73).
Classically, this process would be strictly forbidden by energy conservation (the lifetime of λ even at the maximum available energy E − for this enantiomeric state would be infinite, thus nothing would happen in a classical molecular dynamics simulation on time scales even of seconds or days). Thus, in a classical picture the extremely fast transformation would appear like magic. This effect was, indeed, discovered by F. Hund in this context and later named ‘quantum mechanical tunnel effect’ as if there were a tunnel through this barrier (but there is no real tunnel, of course).
These phenomena might appear already quite unusual; however, there are even more unusual properties to be discovered for chiral molecules resulting from more recent research. We know today that parity is not conserved, that is the corresponding symmetry is violated. For chiral molecules this has the consequence that the effective potential as on the right-hand side of Figure 14 is asymmetrical. This is in contradiction to van't Hoff's assumption of the energetic equivalence of the two enantiomers expressed by equation (12): there is actually a small energy difference. The theoretically predicted difference is very tiny,Reference Quack24, Reference Quack, Stohner and Willeke65, Reference Bakasov, Ha and Quack74–Reference Horný and Quack76 the value of 10 pJ mol–1 or 100 aeVReference Quack, Stohner and Willeke65 indicated in Figure 14 being typical. It would appear immeasurably small, an ‘impossible experiment’. However, we have proposed a spectroscopic experiment,Reference Quack77 which might prove such an energy difference, at a meeting in honour of Vladimir Prelog, one of the pioneers of classical stereochemistry on the occasion of his 80th birthday (Figure 16), who actually in his Nobel prize lecture still denied such an energy difference.78Figure 17 shows the scheme for such an experiment. In the initial steps a parity isomer of a stable chiral molecule (such as CHFClBr or ClOOClReference Quack and Willeke79) is prepared, which then evolves in time following the Schrödinger equation, but including parity violation with the parity violating energy difference  $$${{\rDelta }_{\rm pv}}E $$$
.
$$${{\rDelta }_{\rm pv}}E $$$
.
Figure 16 Prelog-Symposium 1986.
Figure 17 Sequence of steps in the experiment on molecular parity violation (after Ref. Reference Quack9).
While the theory of molecular parity violation seems now well establishedReference Quack9, Reference Quack24, Reference Quack, Stohner and Willeke65 and progress has been made towards experiment,Reference Quack24, Reference Quack64–Reference Quack, Stohner and Willeke65 many fundamental questions have to be answered still in this context. Sometimes even the mere possibility of preparing ‘parity isomers’ for stable chiral molecules is questioned, as to our classical intuition this seems impossible for complex molecules, say DNA or proteins, or in a simpler version molecular knots as shown in Figure 18, and in a less serious vain the question of the superposition of left and right skis has been raised.Reference Zack80
Figure 18 Left and right ‘molecular knot’. The extreme example of knots as topological enantiomers (after Ref. Reference Quack81).
More seriously, a very surprising experiment has been formulated in the general context of chirality, of which the outcome is believed by many to be known, but is not actually known in reality today.Reference Quack61
8. Open Problems: Speculations on CPT Symmetry Violation in Chiral Molecules, and Dark Matter from a Simple Model of Chiral Baryleptons
Up to this point, this paper is based on an account of the history of our understanding of ‘matter’ as seen by chemistry and its laws, based on solid experimental ‘facts’ and similarly solidly based theory related to our current understanding of the underlying laws of physics. We conclude here with some more speculative considerations related to currently open problems. We have recently reviewed the ‘42 open problems of current science’ as related in particular to the frontiers of spectroscopy.Reference Quack9 We shall briefly summarize here the first five of these and discuss them together.
1. Can we ‘see dark matter’ in astrophysical spectroscopy?
2. Can we see the left-right asymmetry in terms of the parity violating energy difference of the enantiomers of chiral molecules and is it important for the evolution of biomolecular homochirality?Reference Quack22–Reference Quack24
3. Can we synthesize ‘mirror-image’ D-amino acid life and will it function like L-amino acid life in a symmetrically equivalent fashion?Reference Quack82
4. Can we devise methods of kinetic spectroscopy to see the difference between time-forward and time-reversed kinetics?Reference Quack73
5. What are the relations between irregularity in spectra and irregularity in time-dependent molecular quantum dynamics and can these be related to a molecular theory of thought?Reference Quack83
These five open questions are all related to severe limitations of our current understanding of ‘matter’ as we observe it in our universe, certainly they are some basic questions of the ‘chemistry’ of the universe ranging from large-scale cosmological questions to small-scale biological questions. They can also be related to some basic asymmetries of fundamental laws as summarized in Table 3.
Table 3 Asymmetries in our world as observed today
The first open question concerning dark matter is perhaps the most striking one, as it is like a dark cloud hanging over all of current chemistry and physics. We have given in Table 2 the periodic system of the elements. We know from spectroscopic observations of stars, interstellar clouds, planets, and so on, that in the ‘visible’ universe, as seen to large distances in space and also back into time, this type of matter is everywhere the same and has been the same since the early years of the universe (after ‘recombination’ happened about 12 × 109 years ago, at a relatively short time after the putative big bang of current cosmology). Indeed, hydrogen, H, and helium, He, make up most of visible matter, the remaining elements being a quantitatively small but qualitatively important ‘impurity’. We can compare this to a good wine: This is made mostly of water and alcohol (ethanol) quantitatively, but the taste arises from small qualitatively important impurities.
We understand reasonably well, how the heavier elements have been and still are being ‘cooked’ by nuclear reactions in stars and supernovae, even if in detail many open questions may remain. However, we also know from gravitational effects very clearly seen in many astronomical observations that all this ‘visible’ matter is only a rather small fraction of the total amount of matter. The remaining part of matter is thus called ‘dark matter’. There is also a hypothetical ‘dark energy’, which is quite a different thing and which we shall not discuss further, as we think that its observational status is much less evident than ‘dark matter’.
Concerning dark matter, we know from the early observations of Fritz Zwicky many decades ago that it exists in and around our galaxies and in terms of its gravitational behaviour it is like ordinary matter and subject to Newton's laws. Disregarding nonstandard explanations such as modifications of these laws, we thus know that there is much extra matter but we have no idea what it is. There are many theoretical speculations what it might be, and we give our personal one below, without trying to refer here to all the others. But this is clearly something in the ‘chemistry of our universe’, which we do not understand at all. One can easily predict that the experimental proof as to the ‘nature’ of this kind of matter will be one of the most important discoveries of this century, if it happens in this century, which we do not know, of course. Attempts at such experiments exist and we give a new proposal for such an experiment below.
A second aspect of the summary in Table 3 is related to another enigma: Why is there such a prevalence of matter as opposed to antimatter in the universe? Antimatter in the form of positrons is well known in the radioactive β+ decay, and antiprotons can be ‘synthesized’ in accelerators, even antihydrogen atoms made of antiprotons and positrons have been synthesized at CERN. But in astronomy we see essentially only normal matter, no antimatter. In current cosmology one expects (almost) equal amounts of matter and antimatter being present very early in the big bang, most of this has been annihilated, but about a fraction of 10−9 has survived as ‘ordinary matter’ in the annihilation. That this can happen, because of a slight asymmetry between matter and antimatter is qualitatively understood in the framework of the Standard Model of Particle Physics (SMPP), but the quantitative understanding is still lacking.Reference Quack9, Reference Dine and Kusenko84
A quantitatively somewhat similar very slight asymmetry exists between enantiomers of chiral molecules, as discussed in Section 7. However, we have no understanding as to whether (and how) this might be related to the prevalence of L-amino acids and D-sugars in the biopolymers of life. The two phenomena might even be totally unrelated, we simply do not know (see open questions numbers 2 and 3 above and Refs Reference Quack9, Reference Quack23 and Reference Quack85).
The third slight asymmetry concerns the non-equivalence of time-forward and time-reversed processes. As we have discussed elsewhere,Reference Quack9, Reference Quack24, Reference Quack73 this phenomenon is similarly related to a fundamental open question (number 4 above). Ultimately it can be related to the question of the existence of an absolute molecular clock, which does not only define time intervals (as does the normal atomic clock), but also defines an absolute direction of time. We have shown that ultimately this is possible only when the combined symmetry operation CPT (i.e. the simultaneous operations C, P, and T of Section 7) is not an exact symmetry.Reference Quack24, Reference Quack73 The current SMPP assumes CPT to be an exact symmetry. We would predict that an observation of a violation of CPT symmetry might be another important discovery of this century, if it happens in this century. We do not know, of course, when, or if it is going to happen at all. Experimental attempts have been made (all unsuccessful so far, see Refs Reference Quack24 and Reference Quack86 for some reviews).
We summarize here an experimental scheme for chiral molecules and other chiral particles, which might shed light on several of the open questions raised here. Figure 19 presents this scheme, as proposed by us two decades ago, as a possible test for CPT violation in chiral molecules.Reference Quack61, Reference Quack64, Reference Quack86–Reference Quack90
Figure 19 Scheme for a CPT test with spectroscopy on chiral molecules L and R* (after Refs 87 and 88). The scheme also illustrates the speculative existence of a heavy right-handed neutrino (R), if L is considered to be the normal neutrino and R* the normal antineutrino.
In brief, the scheme shows a chiral molecule L (say L-alanine), and its enantiomer R (say D-alanine). If parity were an exact symmetry they would be energetically equivalent and thus the energy difference  $$$\rDelta {{E}_{{\rm{pv}}}}\, \equiv \,{{\rDelta }_{{\rm{pv}}}}E $$$
between the two exactly zero. With parity violation, we know that
$$$\rDelta {{E}_{{\rm{pv}}}}\, \equiv \,{{\rDelta }_{{\rm{pv}}}}E $$$
between the two exactly zero. With parity violation, we know that  $$$\rDelta {{E}_{{\rm{pv}}}} $$$
is not zero, in general and for alanine.Reference Quack64, Reference Berger and Quack91 One way to prove parity violation would be to measure
$$$\rDelta {{E}_{{\rm{pv}}}} $$$
is not zero, in general and for alanine.Reference Quack64, Reference Berger and Quack91 One way to prove parity violation would be to measure 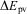 $$$\rDelta {{E}_{{\rm{pv}}}} $$$
to be not zero. This can now be extended to include molecules made of antimatter L* and R*. If CPT symmetry holds exactly, as in the current standard model SMPP, then L and R* would be energetically equivalent (
$$$\rDelta {{E}_{{\rm{pv}}}} $$$
to be not zero. This can now be extended to include molecules made of antimatter L* and R*. If CPT symmetry holds exactly, as in the current standard model SMPP, then L and R* would be energetically equivalent ( $$$\rDelta {{E}_{{\rm{cpv}}}} = {\rm{0}} $$$
) and similarly R and L*. One thus would have
$$$\rDelta {{E}_{{\rm{cpv}}}} = {\rm{0}} $$$
) and similarly R and L*. One thus would have
 $$ \left| {\rDelta {{E}_{{\rm{pv}}}}} \right| = \left| {\rDelta E_{{{\rm{pv}}}}^{{\rm{\ast}}} } \right| = \left| {\rDelta E_{{{\rm{cv}}}}^{{\rm{L}}} } \right| = \left| {\rDelta E_{{{\rm{cv}}}}^{{\rm{R}}} } \right| \eqno(24)$$
$$ \left| {\rDelta {{E}_{{\rm{pv}}}}} \right| = \left| {\rDelta E_{{{\rm{pv}}}}^{{\rm{\ast}}} } \right| = \left| {\rDelta E_{{{\rm{cv}}}}^{{\rm{L}}} } \right| = \left| {\rDelta E_{{{\rm{cv}}}}^{{\rm{R}}} } \right| \eqno(24)$$
Measuring a deviation from these exact equalities would prove CPT violation (for instance if one found 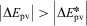 $$$\left| {\rDelta {{E}_{{\rm{pv}}}}} \right|\, \gt \,\left| {\rDelta E_{{{\rm{pv}}}}^{{\rm{\ast}}} } \right| $$$
.Reference Quack86–Reference Quack88 As it requires a measurement on chiral antimatter molecules, such an experiment is not to be expected in the near future, but it is possible.Reference Quack86 If successful it would contribute to answering one of the big open questions (related to irreversibility and CPT).
$$$\left| {\rDelta {{E}_{{\rm{pv}}}}} \right|\, \gt \,\left| {\rDelta E_{{{\rm{pv}}}}^{{\rm{\ast}}} } \right| $$$
.Reference Quack86–Reference Quack88 As it requires a measurement on chiral antimatter molecules, such an experiment is not to be expected in the near future, but it is possible.Reference Quack86 If successful it would contribute to answering one of the big open questions (related to irreversibility and CPT).
The scheme can, however, be read in a much more speculative way to answer the open questions on dark matter. Let us take L to symbolize normal neutrinos  $$${{\nu}_e} $$$
(which are left-handed, strictly left helical) and R* to symbolize the corresponding antineutrino
$$${{\nu}_e} $$$
(which are left-handed, strictly left helical) and R* to symbolize the corresponding antineutrino  $$${{\bar{\nu}}_e} $$$
, which is, indeed, right helical. These neutrinos have been detected experimentally. For instance, the famous ‘chlorine experiment’ of Davis is able to quantitatively detect on Earth the solar neutrinos
$$${{\bar{\nu}}_e} $$$
, which is, indeed, right helical. These neutrinos have been detected experimentally. For instance, the famous ‘chlorine experiment’ of Davis is able to quantitatively detect on Earth the solar neutrinos  $$${{\nu}_e} $$$
generated by nuclear reactions in the sun, through the ‘chlorine reaction’:
$$${{\nu}_e} $$$
generated by nuclear reactions in the sun, through the ‘chlorine reaction’:
 $${{v}_e}\, + \,{}_{{{\rm{17}}}}^{{{\rm{37}}}} {\rm{C}}{{{\rm{l}}}^{{\rm{17}} + }} \, \rightarrow \,{}_{{{\rm{18}}}}^{{{\rm{37}}}} {\rm{A}}{{{\rm{r}}}^{{\rm{18}} + }} \, + \,{{e}^{\rm{ - }}} \eqno(25)$$
$${{v}_e}\, + \,{}_{{{\rm{17}}}}^{{{\rm{37}}}} {\rm{C}}{{{\rm{l}}}^{{\rm{17}} + }} \, \rightarrow \,{}_{{{\rm{18}}}}^{{{\rm{37}}}} {\rm{A}}{{{\rm{r}}}^{{\rm{18}} + }} \, + \,{{e}^{\rm{ - }}} \eqno(25)$$
which acts as a detector. One, in practice, detects the radioactive decay of the newly produced Ar isotope, which decays by the reverse process. The experiment is quite difficult, as one finds only about one radioactive decay of  $$${}_{{{\rm{18}}}}^{{{\rm{37}}}} {\rm{Ar}} $$$
per day from about 600 tons of C2Cl4 exposed to the solar neutrino flux. Nevertheless, with time, unambiguous results have been obtained and reproduced.
$$${}_{{{\rm{18}}}}^{{{\rm{37}}}} {\rm{Ar}} $$$
per day from about 600 tons of C2Cl4 exposed to the solar neutrino flux. Nevertheless, with time, unambiguous results have been obtained and reproduced.
Interestingly, the normal enantiomers of the L-neutrino (R in the scheme of Figure 19) have not been found (nor has L* been found, similarly). There are several possible explanations for this, one being that they simply do not exist. Another speculative proposal is that they exist with very high mass  $$${{m}_{\rm{R}}} $$$
or energy
$$${{m}_{\rm{R}}} $$$
or energy
 $$ \rDelta {{E}_{{\rm{pv}}}} = {{m}_{\rm{R}}}{{c}^{\rm{2}}} \eqno(26)$$
$$ \rDelta {{E}_{{\rm{pv}}}} = {{m}_{\rm{R}}}{{c}^{\rm{2}}} \eqno(26)$$
in an energy range of GeV or TeV (or other) as nothing is known about the exact origin of this extreme ‘parity violating energy difference’ between the L and the hypothetical R neutrinos. The heavy R neutrinos would be stable and weakly interacting, as the light, (almost) massless L-neutrinos. Being generated in the big bang they would be contributing to dark matter today. This is one speculation for a so-called WIMP (weakly interacting massive particle). In extension of the word ‘Lepton’ for the light neutrino particles (L) and similar light particles, we might call the R-neutrino a barylepton, as it has some leptonic properties but may be heavy (‘barys’ in Greek). Of course, all of this is at present pure speculation.
The question thus arises, how to proceed in order to detect such particles experimentally. A rather conservative starting point would be first to try to detect the normal neutrinos left over from the big bang. These are expected to be very numerous but difficult to detect because, in contrast to the solar neutrinos, they have very low energy (low temperature). As we have proposed, one might accelerate  $$${}_{{17}}^{{37}} \rm Cl^{m+} $$$
ions to very high relativistic speed, such that the collision energy with the cold background neutrinos is similar to the solar neutrinos, with similar reaction probabilities. Obviously, a very high throughput ion accelerator would be needed to find some reaction events. However, one could also search for other reactions with perhaps higher cross-sections at lower energies and thus one could optimize the experiment. So far, the cold background neutrinos have not been detected and their detection would by itself be very interesting, because of information about the early phases of the big bang. It might be noted that the normal light neutrinos cannot contribute substantially to dark matter, unless they were much more numerous than expected from any model of the big bang.
$$${}_{{17}}^{{37}} \rm Cl^{m+} $$$
ions to very high relativistic speed, such that the collision energy with the cold background neutrinos is similar to the solar neutrinos, with similar reaction probabilities. Obviously, a very high throughput ion accelerator would be needed to find some reaction events. However, one could also search for other reactions with perhaps higher cross-sections at lower energies and thus one could optimize the experiment. So far, the cold background neutrinos have not been detected and their detection would by itself be very interesting, because of information about the early phases of the big bang. It might be noted that the normal light neutrinos cannot contribute substantially to dark matter, unless they were much more numerous than expected from any model of the big bang.
The next logical step in such a project would be to detect the R-neutrinos (or L*) at high energies in an accelerator, say at the Large Hadron Collider (LHC) at CERN. A more detailed theory of their properties would obviously be helpful. Once one has an idea about their reactions (from experiment or theory) one could then devise an experiment to measure the background R/L* neutrinos similar to the measurement of L/R* and by determining their mass and abundance, one would find out about their contribution to dark matter. We cannot expect, however, that such results will become available in the near future. Nevertheless, this highly speculative model might teach us something about the ‘chemistry of the universe’.
9. On Understanding Nature
We started this paper with an introductory discussion on how we understand reality (if at all) by means of theories, models and hypotheses. We conclude here by answering a question raised by Friedrich Hund at the very end of his inaugural lecture ‘Die Begreifbarkeit der Natur’ (‘The Comprehensibility of Nature’).Reference Hund92 We reproduce in Figure 20 his scheme including an English translation. He discusses that at the centre of our understanding of nature are physical chemical processes. From there one can try to go ‘upwards’ to understand mesoscopic and macroscopic processes, life, soul and mind (processes in the brain?Reference Quack83). One can also go downwards, towards the submicroscopic atom, elementary particles and more generally elementary matter towards the bottom. We cite Friedrich Hund's final question: can this be continued at the bottom? Our answer to this is yes: by Geist/Mind, λόγος, resulting in a circular scheme.Reference Quack93, Reference Quack94 Then we can take ‘Mind’ or ‘logos’ to be equivalent to our concept of (natural) law, which ‘governs’ the phenomena of Nature, and there are similar notions in Sanskrit (Buddhi). Dante in the last line of Canto 33 uses the anthropomorphic ‘l'amor che move il sole et l'altre stelle’.
Figure 20 On understanding nature.
Acknowledgement
This paper is based on a lecture presented at the Workshop ‘Basic Ideas in Science: Natural Law’. Academia Europaea and Klaus Tschira Stiftung, Heidelberg, 4–5 June 2012.
Thanks go to Ruth Schüpbach for preparing the manuscript from my handwritten notes. Part of this lecture was inspired by previous lectures at the Berlin Academies.Reference Quack2, Reference Quack93 The experimental and theoretical work quoted here owes its essence to my co-workers as mentioned in the literature cited here and more completely in Ref. Reference Quack24 and to financial support from ETH Zurich, SNF and ERC. I am grateful to Klaus Mainzer for his patience in waiting for this article. His book on symmetries has been a source of inspiration since I first read it a quarter century ago.Reference Quack95 The present paper is dedicated to Jack D. Dunitz on the occasion of his 90th birthday.
Martin Quack has been Professor of Physical Chemistry at ETH Zürich since 1983. He studied chemistry and chemical physics in Darmstadt, Grenoble and Göttingen. He obtained his doctoral degree (Dr ès sces techn) in 1975 from the École Polytechnique Fédérale de Lausanne after work in reaction kinetics (with Jürgen Troe). He was Max Kade postdoctoral fellow at the University of California Berkeley in 1976/77 (with W.H. Miller) and habilitated in Göttingen in 1978 with work on infrared laser chemistry. In 1982 he was appointed full professor (C4) at the University of Bonn and in 1983 Professor Ordinarius for Physical Chemistry at ETH Zürich. In the Hilary term 1988, he was also Hinshelwood lecturer and Christensen Fellow at Oxford University and St. Catherine's college and in 2005 Miller Visiting Professor at the University of California, Berkeley. His research concerns the foundations of molecular kinetics, high resolution spectroscopy, intramolecular primary processes on the sub-femtosecond to nanosecond time scales, infrared laser chemistry, theory of chemical reactions and fundamental symmetry principles in molecular processes. For his work in these areas he has received numerous honours including the Klung Prize (1984), Otto Bayer Prize (1991) and the Paracelsus Prize of the Swiss Chemical Society (2002). He has been elected fellow of the American Physical Society (1990), member of the German Academy of Sciences Leopoldina (1998) and the Berlin Brandenburg Academy of Sciences (1999). He has been a member of the Swiss National Research Council from 2002 to 2011 and president (1. Vorsitzender) of the Bunsen society for Physical Chemistry (DBG) in 2011 and 2012.


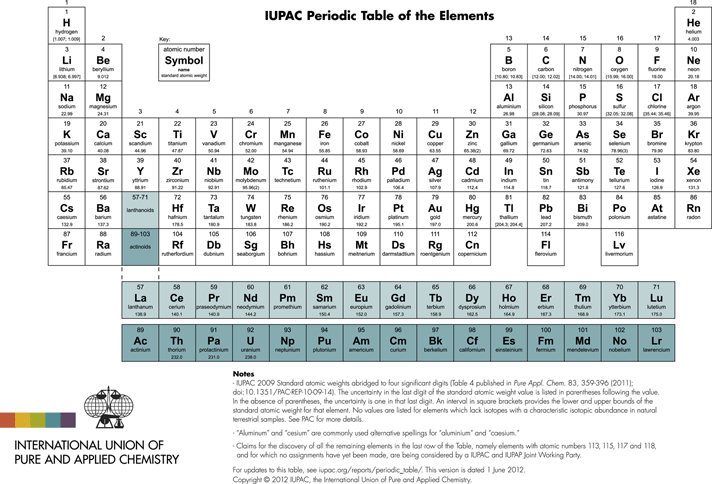


 Open access
Open access