Iron is essential for life but can also catalyse the formation of reactive oxygen species (ROS) that can lead to cell damage. Therefore, iron must be safely bound by specialised proteins to keep ‘free’ iron levels as low as possible. To achieve this, the control of genes encoding proteins involved in iron uptake, utilisation, storage and export must be tightly coordinated. This challenging task is mainly performed by the iron-regulatory proteins (IRPs), which control intracellular iron metabolism, and hepcidin, which regulates body iron homeostasis (Ref. Reference Andrews and Schmidt1). Detailed reviews of IRP structure and regulation have recently been published elsewhere (Refs Reference Hentze and Kuhn2, Reference Hanson and Leibold3, Reference Cairo and Pietrangelo4, Reference Theil and Eisenstein5, Reference Hentze, Muckenthaler and Andrews6, Reference Wallander, Leibold and Eisenstein7, Reference Rouault8); here, we focus on the role of IRPs in maintaining intracellular iron homeostasis and the pathophysiological implications caused by alterations in this function.
The cloning of genes encoding the H and L subunits of the iron-storage protein ferritin led to the identification of iron-responsive elements (IREs) in the 5′ untranslated regions (UTRs). IREs were found to control gene expression in response to changes in the iron level (Refs Reference Aziz and Munro9, Reference Hentze10). Cytosolic proteins that specifically recognise and bind IREs (IRP1 and IRP2) were later identified (Refs Reference Leibold and Munro11, Reference Rouault12), and subsequently the identification of five IRE motifs in the 3′ UTR of the mRNA for transferrin receptor 1 (TfR1) (approved gene symbol TFRC) (Ref. Reference Mullner, Neupert and Kuhn13), which controls cellular iron uptake, indicated that IRPs might be the common regulators of genes involved in iron homeostasis. Indeed, several genes of iron metabolism are now known to be controlled post-transcriptionally through the IRE–IRP regulatory network (see below).
Soon after its identification, it was recognised that IRP1 (approved gene symbol ACO1) is a ‘moonlighting’ protein that can perform two entirely different functions: when present as an apoform, it is able to bind IRE and control gene expression; however, IRP1 can also assemble a [4Fe–4S] cluster and become the cytosolic counterpart of mitochondrial aconitase. The switch between the two forms is mainly regulated by the availability of iron in the so-called labile iron pool (LIP), a pool of metabolically available iron whose nature is difficult to characterise, but whose importance is widely recognised (Ref. Reference Breuer, Shvartsman and Cabantchik14). Therefore, this characteristic makes IRP1 both a sensor of iron levels within the cell and a regulator of cellular iron homeostasis (reviewed by Refs Reference Hentze and Kuhn2, Reference Hanson and Leibold3, Reference Cairo and Pietrangelo4, Reference Theil and Eisenstein5, Reference Hentze, Muckenthaler and Andrews6, Reference Wallander, Leibold and Eisenstein7, Reference Rouault8).
Two bands were evident in the first published example of an IRP–IRE bandshift assay (Ref. Reference Leibold and Munro11), and cloning of the human cDNA led to the discovery of the second IRP, which was very similar to IRP1 (Ref. Reference Rouault15). However, the role of IRP2 (approved gene symbol IREB2) was somewhat neglected until it was demonstrated that it regulates cellular iron homeostasis (Ref. Reference Schalinske16) and is itself regulated by specific pathophysiological stimuli (Ref. Reference Cairo and Pietrangelo4).
Cellular iron metabolism
The fine adjustment of intracellular iron levels is mainly achieved by means of a divergent but coordinated regulation of the iron-storage protein ferritin and the iron-uptake protein TfR1. Although transcriptional regulation of the ferritin heavy and light subunits (H-ferritin and L-ferritin, respectively) (see Refs Reference Cairo and Pietrangelo4, Reference Torti and Torti17 for reviews) and of the TFRC gene (Refs Reference Bianchi, Tacchini and Cairo18, Reference Lok and Ponka19) in response to iron has been described, these two proteins are mainly controlled at the post-transcriptional level through the IRE–IRP system. Over the past decade it has been shown that other genes involved in iron uptake, release and utilisation are also controlled by the IRE–IRP regulatory network (see below).
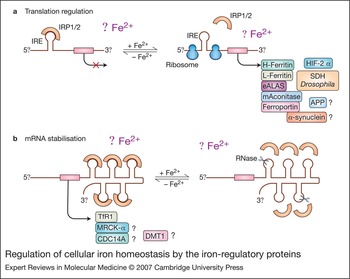
IRP1 and IRP2 are cytoplasmic proteins belonging to the aconitase superfamily and regulate intracellular iron metabolism by binding with high affinity and specificity to conserved IREs in the UTRs of mRNA (Fig. 1) (Refs Reference Hentze and Kuhn2, Reference Hanson and Leibold3, Reference Cairo and Pietrangelo4, Reference Theil and Eisenstein5, Reference Hentze, Muckenthaler and Andrews6, Reference Wallander, Leibold and Eisenstein7, Reference Rouault8). In spite of the predominantly cytosolic localisation of IRPs, microscopic and biochemical approaches identified a fraction (10%) of IRP1 (but not IRP2) that is associated in a phosphorylation-dependent manner with Golgi and endoplasmic reticulum membranes, although the role of membrane-bound IRP1 is still undefined (Ref. Reference Patton20). Under conditions of iron deficiency, IRPs actively bind to IREs and stabilise TfR1 mRNA while also decreasing translation of ferritin mRNA, eventually increasing the uptake and availability of iron within the cell. Conversely, high iron levels decrease IRE-binding activity, leading to efficient translation of ferritin mRNA and decreased stability of TfR1 mRNA, favouring iron sequestration over uptake (Fig. 1).
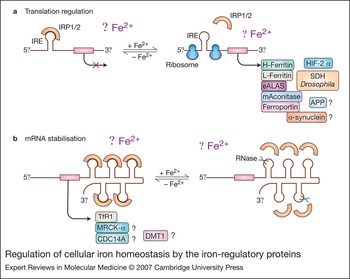
Figure 1. Regulation of cellular iron homeostasis by the iron-regulatory proteins. Under conditions of iron deficiency, iron-regulatory proteins IRP1 and IRP2 bind to the iron-responsive elements (IREs) located in either the 5′ or 3′ untranslated regions (UTRs) of the indicated mRNAs, thus repressing mRNA translation (a) or preventing mRNA degradation (b), respectively. Increased iron levels result in the loss of IRP affinity for IRE, causing increased translation of 5′ IRE-containing mRNAs (a, right) and degradation of 3′ IRE-containing mRNAs (b, right). The functional role of IRE in some mRNAs remains unclear. mRNAs containing non-canonical IREs are indicated with a question mark. Abbreviations: APP, amyloid precursor protein; CDC14A, cell division cycle 14 homologue A; DMT1, divalent metal transporter 1; eALAS, erythroid aminolevulinate synthase; HIF-2α, hypoxia-inducible factor 2α; mAconitase, mitochondrial aconitase; MRCKα, myotonic dystrophy-related CDC42-binding kinase α; SDH Drosophila, Drosophila succinate dehydrogenase.
IRP1 is the cytoplasmic counterpart of mitochondrial aconitase, the enzyme that converts citrate to isocitrate through a cis-aconitate intermediate in the tricarboxylic acid cycle by means of a catalytic [4Fe-4S] cluster (Ref. Reference Beinert and Kennedy21). In iron-replete cells the cluster is assembled and IRP1 displays aconitase activity; in iron-depleted cells, no cluster is formed and apo-IRP1 functions as an RNA-binding protein (Fig. 2). IRE binding is also controlled by the redox state of cysteine residues involved in cluster coordination (Refs Reference Hentze22, Reference Hirling, Henderson and Kuhn23). Therefore, it has been proposed that a reversible switch between a cluster-containing holoprotein and a cluster-deficient apoprotein allows aconitase/IRP1 to constantly sense iron levels and to adapt them to cell requirements without changes in protein levels. However, apo-IRP1 is also subject to iron-dependent degradation (Refs Reference Wang24, Reference Clarke25) (Fig. 2), and crystallographic studies did not predict the direct insertion of the cluster in the apo-IRP1 bound to mRNA (Ref. Reference Walden26). Therefore, the idea that a single molecule can reversibly assume both forms perhaps needs to be revised; IRP1 exists predominantly in an aconitase form (Refs Reference Kennedy27, Reference Chen, Schalinske and Eisenstein28, Reference Recalcati29) and de novo formation of either IRP1 or aconitase can be stimulated, depending on iron scarcity or availability, respectively.

Figure 2. Effect of iron and other signals on the IRE–IRP regulatory system. (a) Regulation of iron-regulatory protein IRP1. The conversion of IRP1 into different isoforms is controlled by several effectors. Hypoxia and high iron levels (Fe2+) favour the formation of the [4Fe–4S] cluster-bearing cytosolic aconitase (cAconitase). RNIs, DOXol and administration of extracellular H2O2 favour cluster disassembly and the acquisition of RNA-binding activity. ROS cause loss of both aconitase and IRE-binding activities by inducing the formation of a degradation-prone intermediate. Phosphorylation favours [4Fe–4S]-cluster-independent regulation of IRP1 in response to iron. (b) Regulation of IRP2. Increased iron levels and exposure to conditions favouring the formation of ROS and RNIs promote the degradation of IRP2 by the proteasome, whereas hypoxia leads to IRP2 stabilisation. Abbreviations: DOXol, doxorubicinol; IRE, iron-responsive element; IRP, iron-regulatory protein; RNI, reactive nitrogen intermediate; ROS, reactive oxygen species.
In response to increased iron levels, apo-IRP1 is more likely to be degraded than to assemble a [4Fe-4S] cluster, whereas the loss of all four iron atoms that convert cytoplasmic aconitase to the RNA-binding form can probably occur only in response to oxidative or nitrative stress (see below). In addition, it has been shown that protein-kinase-C-dependent phosphorylation controls both the RNA-binding and aconitase activities of IRP1 and influences the mechanisms of IRP1 regulation in response to iron (reviewed by Ref. Reference Wallander, Leibold and Eisenstein7) (Fig. 2).
IRP2 is highly homologous to IRP1 but lacks aconitase activity, probably because of its inability to assemble a [4Fe–4S] cluster. The protein accumulates in iron-deficient cells and is rapidly targeted for proteasomal degradation in iron-replete cells (Ref. Reference Iwai30) (Fig. 2). IRP2 binds consensus IRE sequences with an affinity and specificity similar to that of IRP1 (Refs Reference Hentze and Kuhn2, Reference Theil and Eisenstein5), but it has been shown to recognise an exclusive subset of IRE-like motifs (Refs Reference Henderson, Menotti and Kuhn31, Reference Butt32). Furthermore, growing evidence suggests that IRP2 may play a specific role independently of aconitase/IRP1. IRP2 is specifically modulated in response to stimuli and agents other than iron, such as hypoxia (Refs Reference Hanson, Rawlins and Leibold33, Reference Meyron-Holtz, Ghosh and Rouault34) and oxidative stress characterised by enhanced production of ROS (Refs Reference Cairo35, Reference Minotti36, Reference Corna37) or reactive nitrogen intermediates (RNIs) (Refs Reference Cairo38, Reference Cairo39) (Fig. 2). Moreover, IRP2 expression varies greatly between tissues (Refs Reference Hentze and Kuhn2, Reference Henderson, Seiser and Kuhn40). IRP2 is more sensitive than IRP1 to variations of iron in the diet (Ref. Reference Chen, Schalinske and Eisenstein28), is expressed at high levels in most cell lines (Ref. Reference Recalcati, Conte and Cairo41) and, when abundantly (Ref. Reference Recalcati42) or uniquely (Ref. Reference Schalinske16) expressed, can act as the major or only modulator of intracellular iron metabolism, as also indicated by studies in cells in which either IRP1, IRP2 or both were knocked down (Ref. Reference Wang43).
Gene-targeting experiments have provided additional information about the respective role of the two IRPs. Early embryonic lethality in mice doubly deficient for both IRPs (Ref. Reference Smith44) shows that the IRP–IRE regulatory system is essential. However, mice lacking either IRP1 or IRP2 are viable, thus indicating that the two IRPs can compensate for each other. Irp2 −/− mice display mild microcytosis and compromised haematopoiesis with abnormal body iron distribution (Refs Reference Galy45, Reference Galy, Ferring and Hentze46, Reference Galy47, Reference Cooperman48) and either late-onset neurodegeneration (Refs Reference LaVaute49, Reference Ghosh50) or a mild neurological phenotype (Ref. Reference Galy51) in different mouse lines; however, mice lacking IRP1 present with no detectable phenotypic abnormality (Ref. Reference Meyron-Holtz52). Therefore, IRP2 seems to dominate the regulation of iron homeostasis in animal models under normal conditions. Extensive investigation of the comparative expression of the two IRPs in human tissues and blood cells suggested that IRP2 is also the key regulator of intracellular iron homeostasis in humans (Ref. Reference Recalcati29). The above indications of the minor role of IRP1 have been challenged by recent results obtained in anaemic zebrafish, which showed that IRP1 regulates expression of IRE-containing mRNA for erythroid aminolevulinate synthase (eALAS) and hence has a role in haem synthesis (Ref. Reference Wingert53).
IRE-containing mRNAs
In addition to modulating ferritin and TfR1 levels, IRPs can regulate the mRNAs for other proteins. IRE structures have been detected in several mRNAs encoding proteins related to iron utilisation (eALAS; mitochondrial aconitase, approved gene symbol ACO2; Drosophila succinate dehydrogenase, gene symbol SDHB), uptake [divalent metal transporter 1 (DMT1), official symbol SLC11A2] and release (ferroportin, gene symbol SLC40A1) (Refs Reference Hentze and Kuhn2, Reference Hanson and Leibold3, Reference Cairo and Pietrangelo4, Reference Theil and Eisenstein5, Reference Hentze, Muckenthaler and Andrews6, Reference Wallander, Leibold and Eisenstein7, Reference Rouault8). All these mRNAs except that encoding one splicing form of DMT1 – which contains an IRE-like structure in its 3' UTR and is upregulated by iron deficiency (Ref. Reference Gunshin54) – have one IRE in their 5′ UTR and are therefore regulated at the translational level (Fig. 1), although the extent of regulation varies between different transcripts (Ref. Reference Schalinske, Chen and Eisenstein55). However, recent findings indicate that the influence of IRPs extends over a number of regulatory pathways not directly related to iron homeostasis. In particular, integrated strategies utilising a variety of approaches led to the identification of novel IRE-containing genes (Fig. 1). Myotonic-dystrophy-related CDC42-binding kinase α (MRCKα) is a kinase that acts downstream of small GTPases known to be involved in cytoskeletal regulation and has an IRE in its 3' UTR. This IRE may mediate a similar response to iron as TfR1, albeit of lower intensity (Ref. Reference Cmejla, Petrak and Cmejlova56).
A functional IRE has been found in a differentially spliced mRNA isoform encoding CDC14A (cell division cycle 14 homologue A), a phosphatase involved in the regulation of critical cell cycle proteins that has been suggested to be a tumour suppressor (Ref. Reference Sanchez57). Although it is not known how IRPs regulate CDC14A expression, this finding reveals interesting links between iron metabolism and cell cycle regulation, particularly in light of recent findings connecting iron-depletion-mediated growth suppression at the G1–S transition with a mechanism regulating cyclin D1 expression (Ref. Reference Nurtjahja-Tjendraputra58). A functional IRE has also been found in the mRNA for the hypoxia-inducible factor (HIF) 2α (gene symbol EPAS1), a transcription factor that is activated by lack of oxygen or iron (Ref. Reference Sanchez59). This result, which suggests a negative feedback control of the HIF-mediated response under conditions of limited iron availability, may shed further light on the link between iron and oxygen homeostasis (see below).
When considering that brain iron homeostasis is known to be disrupted in several neurodegenerative disorders (Ref. Reference Zecca60), the reported presence of an IRE-like motif in the 5′ UTR in mRNA of α-synuclein (Ref. Reference Friedlich, Tanzi and Rogers61), a presynaptic protein that accumulates in several brain disorders including Parkinson disease, and APP (amyloid precursor protein) (Ref. Reference Rogers62), is particularly intriguing. However, it should be kept in mind that the sequences are not canonical IREs, and IRE-like motifs found in other mRNAs have turned out to be nonfunctional (Refs Reference Kohler, Menotti and Kuhn63, Reference Recalcati64). Often, the iron regulation of these IRE-containing mRNAs is much lower than those of ferritin and TfR1 (Refs Reference Cairo and Pietrangelo4, Reference Hentze, Muckenthaler and Andrews6).
Oxidative stress, inflammation, xenobiotics and hypoxia affect IRPs
The regulation of IRPs under conditions of oxidative stress – that is, in the presence of an excess of ROS – has been the subject of intensive research and some debate. Early observations showed a rapid upregulation of IRP1 in cells exposed to exogenous H2O2 (reviewed by Refs Reference Hentze and Kuhn2, Reference Hanson and Leibold3, Reference Cairo and Pietrangelo4, Reference Cairo38), a transient effect that is permissive to later induction of ferritin (Ref. Reference Tsuji65) and that seems to be due to the pleiotropic effects of H2O2 on iron metabolism, rather than to a direct effect of oxidative stress on IRP1 activity (Ref. Reference Caltagirone, Weiss and Pantopoulos66). However, studies in several types of cell line, as well as in vivo, have shown that a number of conditions or agents known to increase cellular levels of H2O2 and O2·− were able to induce reversible inactivation of IRP1 (Refs Reference Cairo35, Reference Cairo67, Reference Tacchini68, Reference Smith69, Reference Gehring, Hentze and Pantopoulos70). This body of evidence has led to the conclusion that IRP1 downregulation, aimed at decreasing TfR1 levels while also increasing ferritin levels and hence diminishing the LIP, may be a common response to prevent enhanced formation of ROS (Fig. 2). In line with this, increased ferritin synthesis has been documented in a variety of cell types exposed to oxidative stimuli (Refs Reference Cairo and Pietrangelo4, Reference Torti and Torti17, Reference Cairo38), whereas reduced oxidative damage has been observed in cells overexpressing H-ferritin (Refs Reference Epsztejn71, Reference Cozzi72, Reference Orino73). IRP2 is an additional target of ROS produced under conditions of oxidative stress. Available data suggest that IRP2 is highly sensitive to ROS-induced downregulation; this may be triggered by oxidative modifications of sensitive residues and could lead to ubiquitin-dependent proteasomal digestion (Ref. Reference Iwai30) (Fig. 2). Thus, IRP2 promptly undergoes inactivation in rat liver exposed to glutathione depletion or ischaemia–reperfusion (Refs Reference Cairo35, Reference Tacchini68), as well as in cells exposed to menadione (Ref. Reference Gehring, Hentze and Pantopoulos70). IRP2 is not downregulated in cells exposed to exogenous H2O2 (Refs Reference Hentze and Kuhn2, Reference Hanson and Leibold3, Reference Cairo and Pietrangelo4, Reference Cairo38), confirming that extracellular H2O2 acts through mechanisms independently of oxidative stress.
On balance, there is solid evidence to conclude that both IRP1 and IRP2 serve as controllable targets of intracellularly produced ROS. This provides the cell with a regulatory loop to reduce the LIP and prevent further oxidative damage.
IRPs may also be direct or indirect targets of RNIs (Fig. 2); in fact, iron–sulphur clusters have long been recognised as molecular targets of nitric oxide (· NO). Therefore, several studies have addressed whether endogenously generated or exogenous · NO may influence iron metabolism by attacking the [4Fe–4S] cluster of cytoplasmic aconitase/IRP1. Several reports have shown that · NO does increase IRP1 activity, although the precise mechanism(s) of such activation remain unclear: some investigators have suggested that · NO attacks the [4Fe–4S] cluster of cytoplasmic aconitase, inducing its disassembly to form IRP1 (reviewed by Refs Reference Hentze and Kuhn2, Reference Hanson and Leibold3, Reference Cairo and Pietrangelo4, Reference Cairo38, Reference Drapier74), whereas others suggest that · NO acts by inducing cellular iron release and decrease of the LIP (Refs Reference Hentze and Kuhn2, Reference Hanson and Leibold3, Reference Cairo and Pietrangelo4, Reference Cairo38, Reference Drapier74), although these two events may not be mutually exclusive. Other RNIs, such as the nitrosonium ion (NO+), which nitrosylates thiol groups of proteins, may have other effects. In fact, it has been reported that treatment of human erythroleukaemia K562 cells with an NO+ donor decreased the RNA-binding activity of both IRP1 and IRP2 (reviewed by Ref. Reference Kim and Ponka75). Whereas IRP1 is susceptible to activation under defined conditions, IRP2 is almost invariably inactivated by RNIs, which causes redox modifications of the residues that are exposed by the [4Fe–4S] cluster-free IRP2 followed by proteasome-mediated protein degradation (Refs Reference Hentze and Kuhn2, Reference Hanson and Leibold3, Reference Cairo and Pietrangelo4, Reference Cairo38, Reference Drapier74, Reference Kim and Ponka76) (Fig. 2).
From a pathophysiological viewpoint, RNI-induced inactivation of IRP2 may be of greater relevance than a concomitant activation of IRP1-binding activity because IRP2 is highly expressed in macrophages, which are key cells of inflammatory processes. IRP2 downregulation may therefore account for the enhanced ferritin synthesis and reduced TfR1 mRNA content observed in cytokine-stimulated macrophages producing · NO or ONOO− (Refs Reference Recalcati42, Reference Kim and Ponka76). The described downregulation of IRP1 protein levels may contribute to the maintenance of such responses (Ref. Reference Oliveira and Drapier77). Total IRP activity (IRP1 plus IRP2) of human monocytes and macrophages tends to rise shortly after treatment with cytokines or · NO donors, but then decreases markedly accompanied by an enhanced ferritin content (Ref. Reference Recalcati78), similarly to the downregulation of IRP2 in mouse macrophage lines (Ref. Reference Recalcati42). The effects of RNIs on IRPs may therefore explain the iron-sequestration pattern that characterises macrophages under inflammatory conditions.
The cardiotoxicity induced by doxorubicin (DOX), an antitumour anthracycline that also causes a severe form of chronic cardiomyopathy, provides an excellent model to investigate whether IRPs may also be the target of xenobiotics (Refs Reference Minotti79, Reference Minotti, Cairo and Monti80). Three independent studies have shown that the combined action of an alcohol metabolite of DOX (DOXol), which delocalises iron from the [4Fe–4S] cluster of cytoplasmic aconitase, and O2·− and H2O2 derived from redox activation of DOX, eventually converts IRP1 into a ‘null’ protein, which lacks both aconitase and RNA-binding activities (Refs Reference Minotti81, Reference Minotti82, Reference Brazzolotto83) (Fig. 2). According to another study (Ref. Reference Kwok and Richardson84), the formation of the null protein is independent of the action of DOXol on the cluster and derives from the attack of anthracycline–iron complexes on aconitase/IRP1. IRP2 is not attacked by DOXol; only ROS seem to be active in this setting, triggering its degradation (Refs Reference Minotti36, Reference Corna37).
What are the pathological consequences of the actions of DOXol and ROS on IRP1 and IRP2? The decline of IRP2 that occurs in response to ROS formation might serve as a protective stratagem to facilitate the translation of ferritin mRNA, and to sequester free iron before it converts O2·− and H2O2 into more-potent oxidants. In fact, DOX treatment increases L-ferritin and H-ferritin synthesis in H9c2 cardiomyocytes (Ref. Reference Corna37) and in mouse hearts (Ref. Reference Corna85), with a prominent upregulation of H-ferritin, which has been proposed to have an antioxidant function (Refs Reference Torti and Torti17, Reference Harrison and Arosio86). IRP1 deficiency does not alter DOX-induced iron metabolism changes nor DOX-induced oxidative damage of the myocardium, showing that high cardiac IRP1 activity in DOX-treated animals does not counteract the potential cardio-protective effect of IRP2 downmodulation (Ref. Reference Corna85). However, the conversion of aconitase/IRP1 into a null protein, by making cells unable to sense iron levels, may have a major role in inducing chronic cardiomyopathy, which coincides with the gradual conversion of DOX to DOXol.
The ability of DOX to modulate the RNA-binding activity of both IRP1 and IRP2, and hence the expression of IRE target genes, could be involved in both the antitumour and cardiotoxic properties of the drug, and has produced some important clinical results. Indeed, it has been shown that combining DOX with other antineoplastic drugs (e.g. taxanes) enhances chronic cardiotoxicity because conversion of DOX to DOXol is accelerated (Ref. Reference Minotti82). Conversely, anthracyclines with a lower level of formation of their alcohol metabolites exhibit reduced cardiotoxicity in preclinical settings, and have entered clinical trials to assess their efficacy and safety compared with DOX (Refs Reference Minotti82, Reference Minotti87).
IRPs are also regulated by oxygen tension (reviewed and discussed in Refs Reference Hanson and Leibold3, Reference Rouault8), which differentially regulates the binding activity of the two IRPs, with a decrease of IRP1, accompanied by a rise in aconitase activity, and an increase of IRP2 at low oxygen concentrations. Why IRP1 and IRP2 respond in opposite ways to hypoxia remains to be established, but it should be kept in mind that at physiological oxygen tension (3–5%) IRP2 is the predominant RNA-binding protein, thus possibly explaining its major role in the regulation of iron metabolism (Fig. 2).
Clinical applications
Hyperferritinaemia with autosomal dominant congenital cataract – a disorder of iron metabolism characterised by early-onset, bilateral nuclear cataracts and elevated serum ferritin concentrations – is the best-characterised human disease involving the IRE–IRP regulatory system (reviewed by Refs Reference Sheth and Brittenham88, Reference Aguilar-Martinez, Schved and Brissot89). In patients affected by this disorder, serum iron and transferrin saturation are normal or low, and body iron (as evaluated by phlebotomy), is not increased, thus excluding iron overload as the underlying cause of hyperferritinaemia. Molecular studies of tissues from these patients have identified multiple point mutations in the IRE of L-ferritin mRNA, which affects the highly conserved CAGUGU motif in the IRE loop responsible for the high-affinity interaction with IRPs (reviewed by Refs Reference Sheth and Brittenham88, Reference Aguilar-Martinez, Schved and Brissot89). In cultured lymphoblastoid cells from affected patients, the mutation was found to abolish the binding of IRPs and results in constitutively high L-ferritin synthesis, which correlates with serum ferritin levels (Ref. Reference Cazzola90). The affected subjects show no haematological or biochemical abnormalities and the phenotype of this mutation is characterised only by an accumulation of L-ferritin in the lens, resulting in cataract, although a direct relationship between the mutation and the lens deposit of aggregated and crystallised ferritin has not been formally demonstrated.
A point mutation in the IRE of the human H-ferritin gene (approved gene symbol FTH1) has been found in members of a Japanese family affected by iron overload (Ref. Reference Kato91), but this finding has not been confirmed in other pedigrees.
Further support for the clinical implication of alterations in the IRE–IRP network is provided by the expression of ferroportin – the major or only iron exporter from cells – which can be altered upon loss of its 5′ IRE, as seen in polycythaemic mice (Ref. Reference Mok92). Mutations in the ferroportin mRNA 5′ UTR were detected in a patient with iron overload because of ferroportin disease (Ref. Reference Liu93), which may depend on altered IRE–IRP recognition. Similarly, the presence of alternative ferroportin transcripts without an IRE in erythroid cells (Ref. Reference Cianetti94) leaves open the possibility that alterations in ferroportin mRNA splicing may be relevant in pathological conditions of altered erythroid differentiation.
The relevance of dysregulated iron metabolism in the neurological disorders mentioned above makes the finding that polymorphisms in the promoter region of the IRP2 gene are statistically associated with Alzheimer disease potentially interesting (Ref. Reference Coon95). However, this preliminary report needs to be confirmed in a larger independent population, and the functional significance of the haplotype determined. In addition, the unexpectedly high IRP1 activity found in patients with Parkinson disease may account for the low ferritin levels present in neurons of the substantia nigra in spite of iron accumulation (Ref. Reference Faucheux96).
Research in progress and outstanding research questions
[4Fe–4S] cluster assembly
In recent years, significant advances have been made to define the processes and the molecular participants underlying the assembly of the IRP1 [4Fe–4S] cluster, indicating that iron availability is not the only requirement. In fact, detailed analysis of the pathways of [4Fe–4S] cluster assembly in mammalian cells has indicated a key role of mitochondria (Ref. Reference Lill97), although a cytosolic [4Fe–4S] cluster assembly machinery has also been identified (discussed in Ref. Reference Rouault8). Impairment of both pathways of [4Fe–4S] cluster assembly leads to induction of IRP1-binding activity and may also alter iron homeostasis. Indeed, it has been found that a mutation in mitochondrial glutaredoxin, a protein involved in [4Fe–4S] cluster biogenesis (Ref. Reference Wingert53), shifts the IRP1/aconitase balance in favour of the apoform and induces anaemia in zebrafish by repressing eALAS translation and hence haem formation. Future work will define the role of the flux of iron through the mitochondrial and cytosolic [4Fe–4S] assembly pathways in iron homeostasis, possibly leading to treatments for disease, because mutations in the human counterparts of the proteins involved in these processes are likely to be found in human pathological settings (Ref. Reference Camaschella98). Similarly, the impairment of [4Fe–4S] cluster formation that characterises Friedreich ataxia may result in uncontrolled IRP1 activation and pathological abnormalities of iron homeostasis (Refs Reference Seznec99, Reference Stehling100).
Structural studies
Insight into the structure–function relationship of IRPs has been recently provided by crystallography studies. The crystal structure of cytosolic aconitase has been solved (Ref. Reference Dupuy101). Walden et al. (Ref. Reference Walden26) also solved the crystal structure of IRP1 bound to ferritin IRE, detailing the molecular basis underlying the bifunctionality of this protein. Further investigation in this field, perhaps with a detailed crystal structure of IRP2 complexed with IRE, will hopefully determine the specificity of the IRE–IRP interaction, the preferential binding of individual IRPs to a subset of IREs and the effects of IRP modification on its binding activity and selectivity.
Role of cytosolic aconitase
Under normal conditions, most IRP1/aconitase is enriched with the [4Fe–4S] cluster and the ratio of aconitase to IRP1 may exceed 100:1 (Refs Reference Kennedy27, Reference Chen, Schalinske and Eisenstein28); this finding hence leaves open the question of the function of cytoplasmic aconitase. A role in modulating the levels of NADPH and citrate has been proposed for cytosolic aconitase (Ref. Reference Tong and Rouault102).
Mouse models
The role of IRPs in mammalian iron metabolism has been thoroughly studied in cell culture, but relatively little in vivo information is available. Further information regarding their tissue-specific regulation could be gained by the characterisation of knockout mice bearing conditionally targeted IRP1 and IRP2 loci generated by tissue-specific promoters used to control the cre recombinase (Ref. 46). This approach could give indications of the particular role of individual IRPs in specific organs or tissues, as suggested by their different abundances (Ref. 2), and would also be helpful in understanding their physiological role. Further information could be obtained by subjecting animals with tissue-specific or general deficiency of IRPs to stressful stimuli that have been demonstrated to affect IRP activity.
New targets
One main area for future research is expected to be the search for new IRP targets. Integration of bioinformatic, biochemical and genomic approaches has recently led to the identification of new IRP-regulated genes involved in a number of pathways; implementation of these strategies will help in the identification of other IRE-regulated genes and lead to a wider understanding of iron-regulatory networks.
Conclusion
In recent years, the regulation of iron homeostasis has revealed its increasing complexity and its multifaceted and unforeseen interactions with a number of molecular pathways and processes. The work highlighted here has shown that significant advances have been made in elucidating the role of the IRE–IRP network in the post-transcriptional control of key mRNA molecules involved in iron metabolism, but also indicates that further studies are warranted. In particular, we need to gain further insight into the IRP-mediated metabolic remodelling that occurs in response to changes in iron availability and other iron-independent effectors. This information might shed light on the role of iron in the molecular pathophysiology of a wide range of disorders.
Acknowledgements and funding
The authors thank the reviewers for insightful comments and suggestions. This work was supported by grants from Ministero dell'Istruzione, dell'Università e della Ricerca (PRIN) and from MIUR-FISR to G.C.