



Introduction
In 1993, a revolutionary discovery of the first microRNA (miRNA) while studying nematode Caenorhabditis elegans was made by Ambros et al. (Ref. Reference Rawat1). This finding revealed an essential part of the non-coding genome that plays a key role in post-transcriptional gene regulation (Ref. Reference Rawat1). Fast track 29 years, now over 2600 mature human miRNA sequences have been identified (Ref. Reference Kozomara, Birgaoanu and Griffiths-Jones2). MicroRNAs are small, endogenous, single-stranded, non-protein coding RNA molecules of 19 to 24 nucleotides. MiRNAs account for approximately 3% of the human genome and are evolutionary conserved across mammals (Refs Reference Mishra, Yadav and Rani3, Reference To4). The canonical miRNA biogenesis pathway (Fig. 1) starts with RNA polymerase II-mediated transcription of the primary miRNA gene (pri-miRNA). The pri-miRNA is characterised by a hairpin structure with a 5′ cap and polyadenylation site at the 3′ (Ref. Reference Bautista-Sánchez5). Drosha, an RNase III protein, then cleaves the pri-miRNA, releasing a precursor loop (pre-miRNA) of approximately 70–100 nucleotides (Ref. Reference Iorio and Croce6). Upon release, the pre-miRNA is exported to the cytoplasm via Exportin 5 (Ref. Reference Iqbal7). In the cytoplasm, pre-miRNA is cleaved by an RNase III called Dicer, to produce a double-stranded mature miRNA of approximately 22 nucleotides in length (Ref. Reference Acunzo8). The non-canonical pathways utilise different combination of proteins during the biogenesis steps. These pathways can be further classified as Drosha- and Dicer- independent pathways. For instance, miRNAs produced from introns of messenger RNAs or mirtrons can bypass Drosha-mediated processing (Ref. Reference Ruby, Jan and Bartel9). 7-methylguanosine (m7G)-capped pre-miRNA have been identified which are nascent RNAs directly exported to the cytoplasm through exportin 1 without the need for Drosha (e.g. pre-miR-320) (Ref. Reference Xie10). Another example is the biogenesis of miR-451 which is independent of Dicer processing but requires Drosha and Argonuate 2 (AGO2) protein (Ref. Reference Cheloufi11).
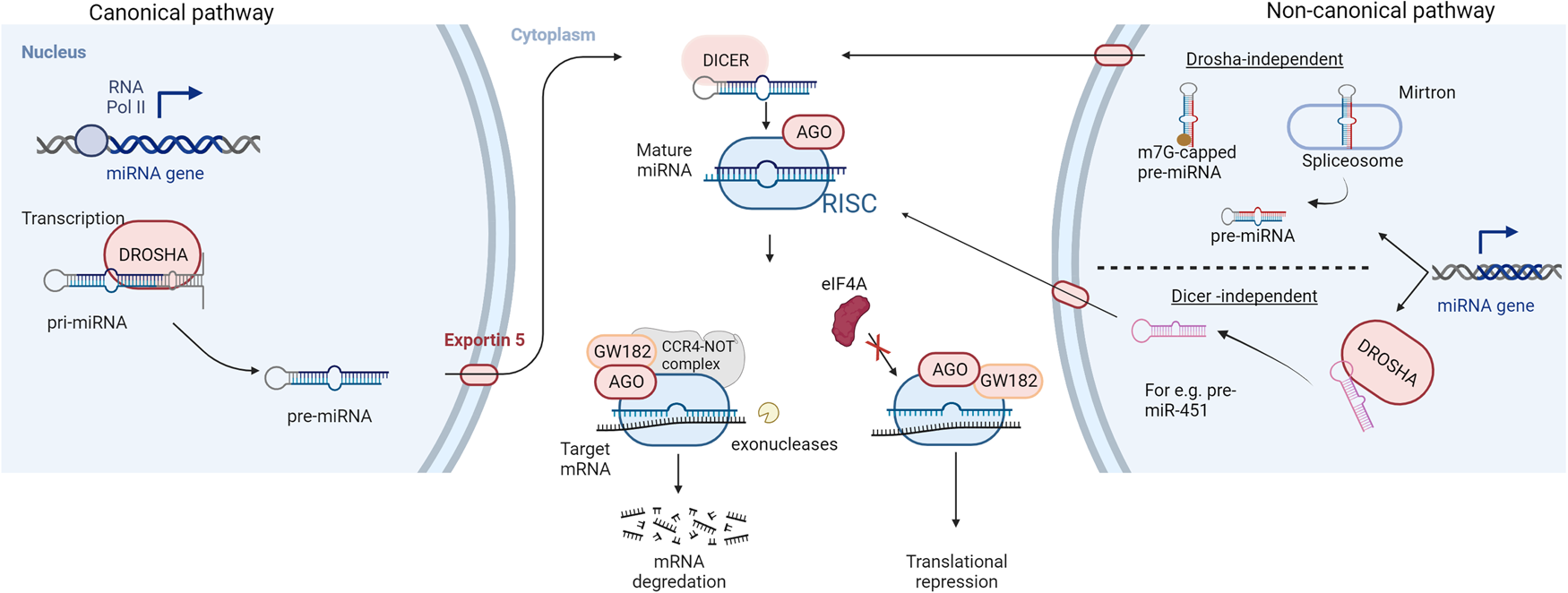
Fig. 1. Biogenesis and mechanism of action of miRNAs: RNA polymerase II-mediated transcription forms the primary miRNA (pri-miRNA) which is cleaved by an RNase III enzyme (DROSHA) to produce a precursor miRNA (pre-miRNA) in the canonical pathway of miRNA biogenesis. The pre-miRNA is exported to the cytoplasm via exportin 5, for further processing by RNase III DICER to form a mature miRNA duplex. Non-canonical pathways are independent of Drosha or Dicer processing. The miRNA duplex is then unwound whereby the guide strand along with Argonaute (AGO) proteins form a miRNA-induced silencing complex (RISC). The RISC complex binds to target sequences of mRNA leading to translation repression or degradation. AGO recruits GW182 which forms a complex with CCR4-NOT making the target mRNA susceptible to cleavage by exonucleases while hindrance to the binding of eukaryotic initiation factor-4A (eIF4A) to the target mRNA leads to translational inhibition.
The double-stranded mature miRNA is unwound, and the opposite strand is degraded. The remaining strand, the mature or the guide strand, is the final single-stranded miRNA molecule which forms a complex with the AGO proteins called the RNA-induced silencing complex (RISC) (Ref. Reference Mishra, Yadav and Rani3). There are four mammalian AGO proteins (AGO1–4) but only AGO2 is able to cleave the target mRNA complementary to the miRNA (Ref. Reference Liu12). The RISC complex binds to target messenger RNAs (mRNAs) which possess sequences complementary with the miRNA (Ref. Reference Gurbuz and Ozpolat13). The binding of the RISC complex to mRNAs is mediated by a 6–8 nucleotide long region within the miRNA called the seed sequence or miRNA binding site (Ref. Reference Hayes, Peruzzi and Lawler14). The resulting miRNA-mRNA duplex leads to an inhibition or in some cases enhancement, of translation (Ref. Reference Hayes, Peruzzi and Lawler14). The miRNA target sites are usually located at the 3′-untranslated region (3′-UTR) of mRNA and the binding of the RISC complex leads to gene silencing by translation repression and mRNA decay (Ref. Reference Jonas and Izaurralde15). Mechanistically, the AGO protein recruits the GW182 which interacts with the polyadenylate-binding protein PABPC to induce mRNA deadenylation (Ref. Reference Braun16). This promotes decapping of the mRNA and makes it susceptible to degradation by 5′-3′ exoribonucleases (Ref. Reference Braun17). For translational repression, GW182 recruits carbon catabolite repressor protein 4 complexes which in turn recruit RNA helicases like DDX6 (Refs Reference Fabian18, Reference Mathys19). MicroRNA-mediated inhibition of mRNA translation initiation results from the interference with the eukaryotic initiation factors eIF4A-I and eIF4A-II (Ref. Reference Jonas and Izaurralde15). Although exact molecular details remain to be uncovered, existing evidences suggest that the RISC complex dissociates initiation factors from target mRNAs inhibiting the assembly of the translation initiation complex (Refs Reference Fukao20, Reference Fukaya, Iwakawa and Tomari21).
Although a perfect complementarity with mRNAs is optimal for the function of miRNAs, some miRNAs can regulate mRNAs with partial complementarity (Ref. Reference Ganju22). Consequently, miRNA-mediated regulation of gene expression affects almost every fundamental cellular process, such as development, differentiation, proliferation, metabolism and apoptosis (Ref. Reference Barger and Nana-Sinkam23). Unsurprisingly, the dysregulation of miRNA profile also significantly correlates with the onset and progression of cancer (Ref. Reference Xue24). The impact of individual miRNAs on cancer often differs between cancer types. A miRNA can be tumour suppressive, oncogenic or a regulator of metastasis (Ref. Reference Kim25). This review will focus on the role of miRNA in cancer metastasis. We will investigate miRNA dysregulation in different stages of the metastatic process and examine molecular drivers affected. We will summarise pre-clinical studies that have successfully employed miRNA-based therapies for metastatic cancer and highlight their limitations. Finally, we will examine miRNA-based therapies for the treatment of advanced and metastatic cancer that are currently in clinical trials.
Metastasis
The development of secondary tumours in distant organs, i.e. metastasis, is a hallmark of cancer (Ref. Reference Rajasegaran26). The spread of cancer cells to secondary sites is the main cause of cancer-related morbidity and mortality (Ref. Reference Dillekås, Rogers and Straume27), yet we are only beginning to unravel the molecular mechanisms that drive metastasis (Ref. Reference Fares28). Several phases are involved in the development of secondary tumours (Ref. Reference Seyfried and Huysentruyt29). First, cancer cells loose adhesion factors and detach from the primary tumour allowing penetration and intravasation into the circulatory and lymphatic systems (Ref. Reference Seyfried and Huysentruyt29). The cells in the vasculature called circulating tumour cells (CTCs), exploit mechanisms like cell cycle arrest, to evade and survive immune surveillance (Ref. Reference Zeeshan and Mutahir30). There is significant research aimed at studying and characterising CTCs which is beyond the scope of this review. Secondly, this process is followed by the extravasation and infiltration of the cells into distant capillary beds (Ref. Reference Welch and Hurst31). Finally, invasion and proliferation of the tumour in distant organs occurs (Ref. Reference Welch and Hurst31). Metastasis and the establishment of secondary tumours is a very inefficient process as the majority of tumour cells in circulation are eliminated (Ref. Reference Labelle and Hynes32). The establishment of a microenvironment for cancer cells to seed, known as a premetastatic niche, is essential for the development of secondary cancer (Ref. Reference Liu and Cao33) (Fig. 2). The most common locations of metastasis in the body are liver, bone, lung, nervous system, pleura and peritoneum (Ref. Reference Riihimäki34). In this review, we will focus on the role of miRNAs in metastasis. We will discuss different stages in the metastatic process and summarise miRNAs that have been reported to be involved in each of these steps.
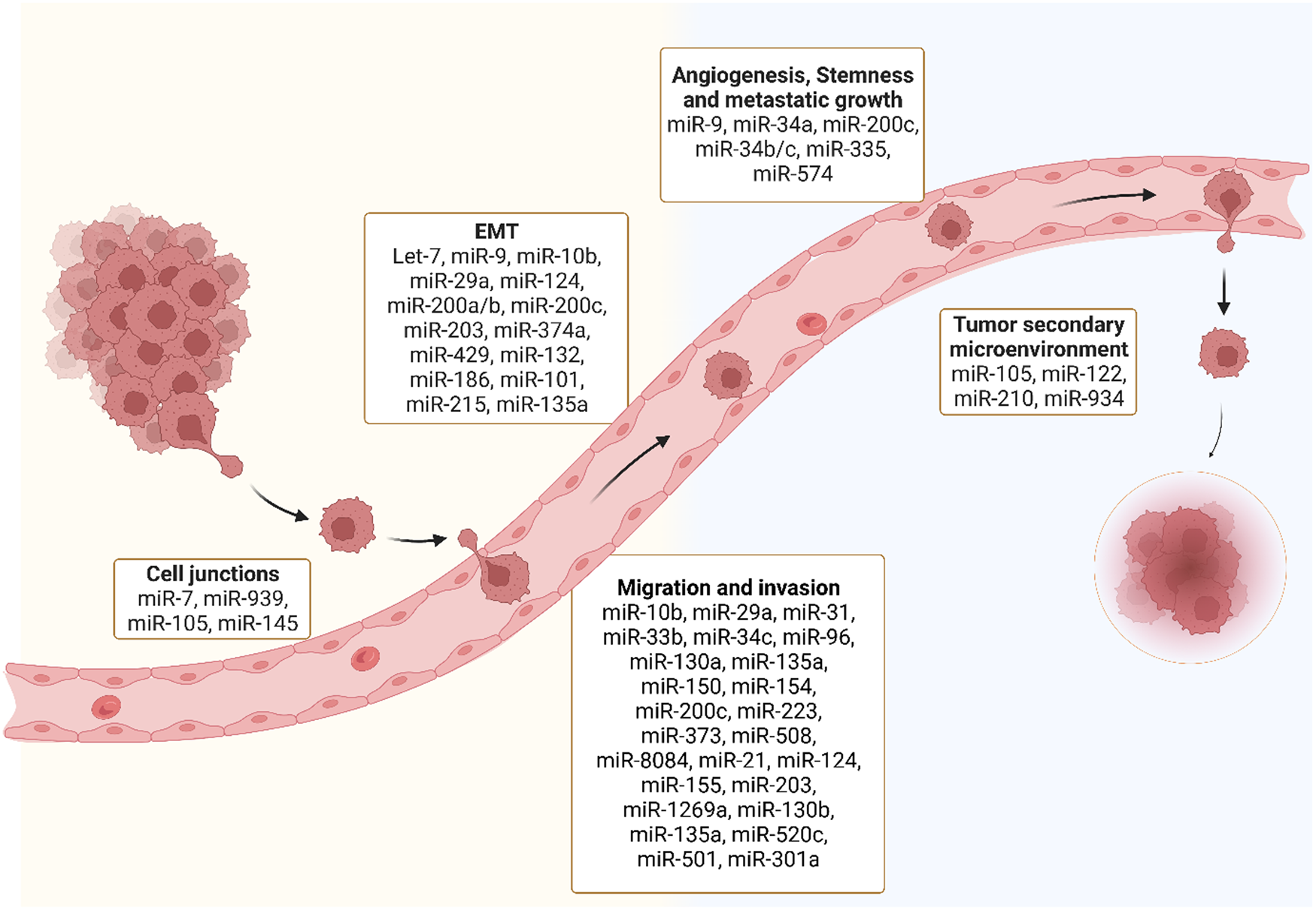
Fig. 2. MiRNAs and different stages of metastasis: Several miRNAs are dysregulated throughout different stages of the metastatic process including disruption of tight junctions, epithelial to mesenchymal transition (EMT), migration and invasion, angiogenesis, stemness and metastatic growth and tumour secondary microenvironment.
MicroRNA dysregulation in cancer metastasis
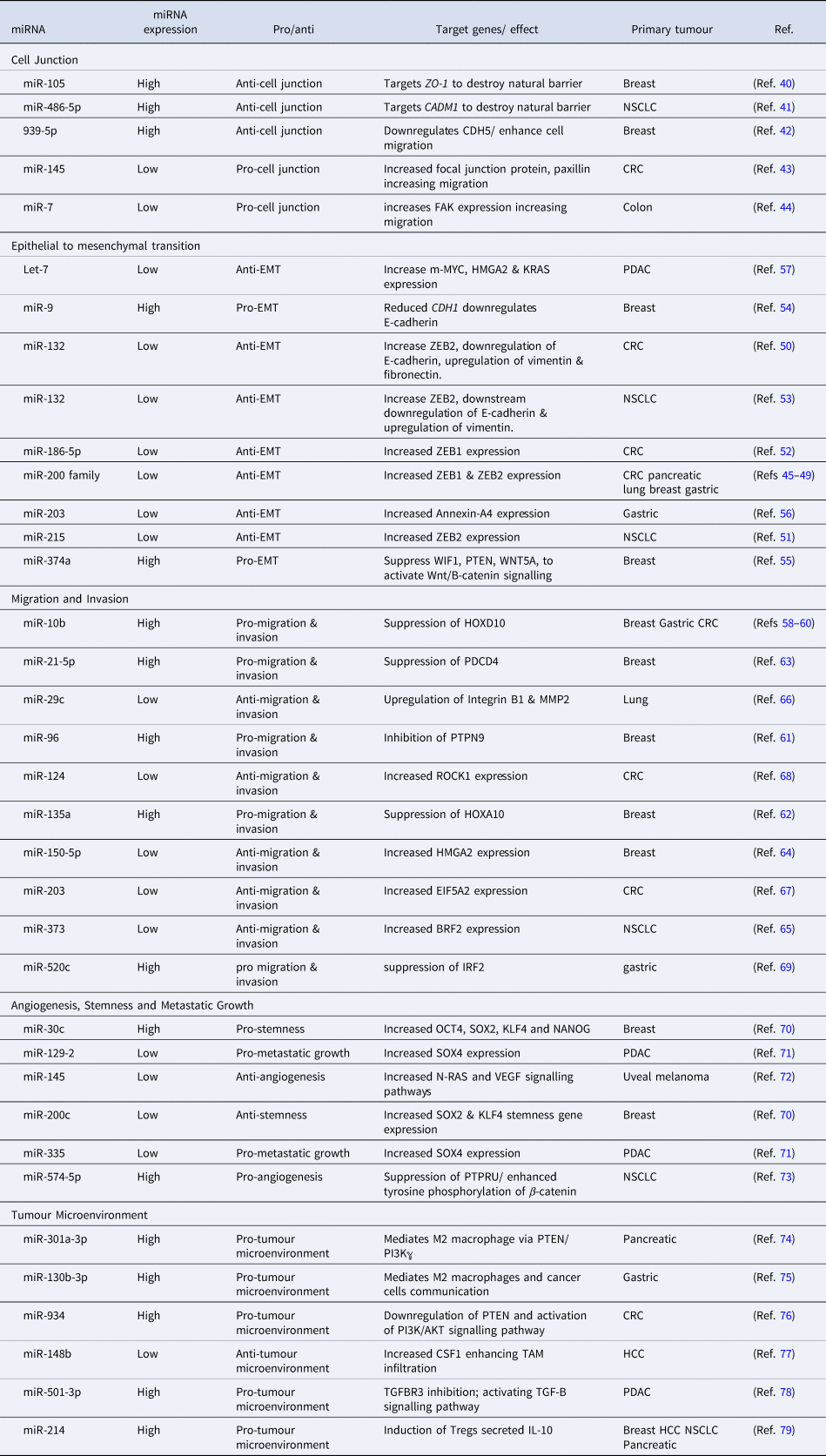
MiRNAs are regulators of virtually every cellular process, including the ones that lead to the development of metastasis (Ref. Reference Solé and Lawrie35). MiRNA can target the mRNA of tumour suppressors or oncogenes implicated at different phases of the developing metastatic tumour (Table 1) (Ref. Reference Peng and Croce36). Genetic mutations in cellular pathways resulting from the dysregulation of miRNA have been widely identified in the metastatic pathways (Ref. Reference Iorio and Croce37). Here we discuss miRNA dysregulation and how it impacts key stages of metastasis.
Table 1. A list of microRNAs involved at different stages of cancer metastasis
MiRNAs and cell junctions
MiRNAs regulate the transcription of cellular junction proteins critical for signalling communication, growth and migration (Ref. Reference Zhuang38). For metastasis to occur, the disruption of cellular junctions is essential (Ref. Reference Martin39). Several miRNAs regulate the expression of zonula occluden-1 (ZO-1), a major component of tight junctions (Ref. Reference Zhou40), e.g. miR-105 regulates the metastasis of breast cancer by inhibiting ZO-1 (Ref. Reference Zhou40). Furthermore, overexpression of miR-105 induces the metastasis of cancer cells to distant organs including the liver (Ref. Reference Zhou40). The cell-cell adhesion for the adaption of the premetastatic niche is promoted by oncogenic miR-105 through the tight junction protein ZO-1 (Ref. Reference Zhou40).
A highly elevated expression of miR-486-5p in CD31+ vascular endothelial (VE) cells increases permeability and promotes non-small cell lung cancer (NSCLC) metastasis (Ref. Reference Sun, Han and Shi41). The transfection of human VE cells, with miR-486-5p antagomirs, targets CADM1 and destroys the tight junctions of VE cells. Similarly, the downregulation of adherens junction protein, VE cadherin (CDH5), by miRNA, enhances breast cancer cell migration (Ref. Reference Di Modica42). MDA-MB-231-GFP breast cancer cells transfected with miR-939 mimics, show higher migration through the endothelial barrier which is mediated by a downregulation of CDH5 (Ref. Reference Di Modica42). MiR-145 negatively correlates with the focal junction protein paxillin in colorectal cancer (CRC) (Ref. Reference Qin43). Delivery of miR-145 mimics downregulates paxillin and inhibits cell proliferation, migration and invasion (Ref. Reference Qin43). Similarly, miR-7 targets focal adhesion kinase (FAK) expression to suppress colon cancer proliferation, migration and invasion (Ref. Reference Zeng44). HCT-8 and Caco-2 colon cancer cell lines transfected with miR-7 mimics have been shown to have a negative correlation with colon cancer metastasis (Ref. Reference Zeng44).
MiRNAs in epithelial to mesenchymal transition (EMT)
Epithelial to mesenchymal transition (EMT) of cells is mostly controlled by zinc finger E-box binding protein transcription factors (TFs) ZEB1 and ZEB2 and leads to an upregulation of vimentin and N-cadherin and repression of E-cadherin (Ref. Reference Vu and Datta45). The miR-200 family (miR-200a, miR-200b, miR-200c, miR-124, miR-429) has been extensively investigated as a regulator of ZEB1 and ZEB2 in cancer that mostly metastasises to the liver, including colorectal (Ref. Reference Vu and Datta45), pancreatic (Ref. Reference Gui46), lung (Ref. Reference Liu47), breast (Ref. Reference Ji48) and gastric cancers (Ref. Reference Yu49). ZEB1 and ZEB2 are direct targets of several miRNAs in CRC and NSCLC. Zheng et al. reported a significant downregulation of miR-132 in CRC and furthermore used a luciferase activity assay to demonstrate miR-132-mediated regulation of ZEB2 (Ref. Reference Zheng50). MiR-215-mediated regulation of ZEB2 expression has been demonstrated in CRC while low levels of miR-186-5p in CRC inhibit EMT by targeting ZEB1 (Refs Reference Chen51, Reference Li52). Additionally, it was reported that miR-132 directly targets ZEB2 in NSCLC driving EMT (Ref. Reference You53). There are other miRNAs that promote EMT in breast cancer. MiR-9 repression of E-cadherin, increases EMT in breast cancer (Ref. Reference Ma54) while oncogene miR374a targets negative regulators of Wnt/B-catenin pathway including WIF1, PTEN and WNT5A and activates the Wnt/B-catenin pathway to promote EMT in breast cancer (Ref. Reference Cai55). The downregulation of miR-203 promotes EMT in gastric cancer by releasing the repression of its target gene Annexin A4 (Ref. Reference Li56). In pancreatic ductal adenocarcinoma (PDAC), EMT is driven by an increase in the expression of c-MYC, HMGA2 and KRAS mediated by a reduction in Let-7 miRNA expression (Ref. Reference Wang57).
MiRNAs in cancer cell migration and invasion
MiR-10b was the first miRNA to be associated with metastasis in patients with advanced breast cancer (Ref. Reference Kim58). MiR-10b promotes the migration and invasion in different cancer types including colorectal, breast and gastric cancers by regulating HOXD10 (Refs Reference Kim58–Reference Wang60). In breast cancer, migration and invasion is enhanced by an upregulation of miR-96, miR-135a and miR-21 and the downregulation of miR-150. Similarly, in breast cancer, oncogenic miR-96 targets PTPN9 (Ref. Reference Hong61) and miR-135a represses HOXA10 to drive the migration and invasion (Ref. Reference Chen62). Another oncogene miR-21-5p upregulates the expression of programmed cell death protein 4 (PDCD4) to increase breast cancer invasion and migration (Ref. Reference Li63). Tumour suppressor miR-150, which targets HMGA2, is aberrantly expressed in breast cancer and promotes migration and invasion (Ref. Reference Wang64). In NSCLC, a downregulation of miR-373 which regulates TFIIB-related factor 2 (BRF2), promotes cell migration and invasion (Ref. Reference Wang65). Multiple investigations of cancers metastasising to the liver including pancreatic cancer and lung cancer, demonstrate that cell migration and invasion is controlled by miR-29c-mediated regulation of MMP2 (Ref. Reference Wang66). Low levels of miR-124 and miR-203 promote migration and invasion in CRC by targeting ROCK1 and eukaryotic initiation factor 5A2, respectively (Refs Reference Deng67, Reference Zhou68). Similarly, gastric cancer migration and invasion is driven by the dysregulation in miR-520c-mediated regulation of IRF2 (Ref. Reference Li69).
MiRNAs in stemness, angiogenesis and metastatic growth
Key events of metastatic growth and tumour mediated angiogenesis are regulated by miRNAs. Downregulation of miR-200c and overexpression of miR-30c targets stemness-related genes in breast cancer influencing secondary tumour growth (Ref. Reference Rahimi70). Oncogene miR-9-mediated repression of E-Cadherin contributes to an overexpression of vascular endothelial growth factor (VEGF) leading to an increase in angiogenesis which is essential for the growth of secondary tumour (Ref. Reference Ma54). The co-repression of miR-129-2 and miR-335 significantly upregulates oncogenic SOX4 driving metastatic growth in PDAC (Ref. Reference Huang71). Likewise, VEGF overexpression leading to angiogenesis, tumour growth and invasion in uveal melanoma was significantly suppressed by miR-145 mimics directly targeting N-RAS and VEGF signalling pathways (Ref. Reference Yang72). Upregulation of miR-574-5p promotes metastatic growth in NSCLC enhancing tyrosine phosphorylation of B-catenin via the repression of protein tyrosine phosphate receptor type U (PTPRU) (Ref. Reference Zhou73).
MiRNAs in tumour microenvironment
MiRNA is an important mediator of the crosstalk between the tumour microenvironment and tumour cells, playing an important role in metastasis progression. Tumour-associated macrophages (TAM) are key components of the tumour microenvironment, regulated by miRNAs, to exhibit pro-tumour activity in the microenvironment. For example, highly expressed miR-301a in pancreatic cancer cells induces M2 macrophage polarisation via the PTEN/PI13Kℽ signalling pathway to promote pancreatic cancer cell metastasis (Ref. Reference Wang74). Similarly, miR-130-3p upregulated in gastric cancer mediates communication between M2 macrophages and cancer cells in the tumour microenvironment by promoting the expression of mixed lineage leukaemia 3 (MLL3) gene and grainyhead-like 2 (GRHL2) gene (Ref. Reference Zhang75). Tumour-derived exosomal miR-934 promotes liver metastasis of CRC by regulating the interaction between TAMs and the metastatic microenvironment (Ref. Reference Zhao76). Downregulated miR-148b expression negatively correlates with the upregulation of colony-stimulating factor-1 (CSF1), promoting CSF1 signalling and inducing TAM infiltration to promote hepatocellular carcinoma (HCC) metastasis (Ref. Reference Ke77). Furthermore, M2 macrophage-derived exosomal miR-501-3p downregulated TGFRR3 to promote liver and lung metastasis of PDAC in nude mice by activating the TGF-β signalling pathway (Ref. Reference Yin78). Upregulated miR-214 negatively correlates with PTEN in several cancers including breast, HCC, NSCLC and pancreatic cancer. The upregulated miR-214 promotes regulatory T-cells (Tregs) which secret high levels of IL-10 and enhance immune suppression for metastatic progression (Ref. Reference Yin79).
Regulation of metastatic miRNAs
The inter-regulation between miRNAs and different TFs lead to a finely tuned and spatio-temporally regulated transcriptional and post-transcriptional gene regulation system which gets perturbed during metastasis (Ref. Reference Liu80). Evidences suggest that the dysregulation of miRNAs in cancer can occur at the genomic level. An example is the frequently lost genomic locus of miR-146a in acute myeloid leukaemia (Ref. Reference Zhao and Starczynowski81). However, aberrations at the transcription level are widely studied and thought to be more impactful. For instance, tumour suppressive TF p53 regulates the expression of the miR-16, miR-145 and miR-34 family (Ref. Reference He82). While miR-145 is repressed by the oncogenic RAS-responsive element-binding protein 1 (RREB1) (Ref. Reference Kent, Fox-Talbot and Halushka83). Other reported TFs that regulate miR-145 include CCAAT/enhancer-binding protein beta, beta-catenin/T cell factor 4 and forkhead TFs FOXO1 and FOXO3 (Refs Reference Zeinali84, Reference Gan85). The oncogenic c-Myc TF suppresses expression of miRNAs 29, 30 and let-7 family (Refs Reference Chang86–Reference Zhang88). ZEB1 and ZEB2, key activators of EMT repress the expression of miR-200 family of genes (Ref. Reference Guan89) including miR-200c (Ref. Reference Chen90). Similarly, studies have demonstrated that nuclear receptors, especially estrogen receptor (ER) and androgen receptor (AR) can directly regulate the transcriptional activity of miRNAs in cancer by binding to promoter or repressor regions. For instance, ER binds to the promoter region of the miR-221 and inhibits its expression in breast cancer (Ref. Reference Di Leva91). Interestingly in prostate cancer, a negative feedback loop that regulates miR-135a and AR protein expression in an androgen-dependent manner was identified. Here, androgen stimulates the expression of miR-135a which inhibits AR expression. In turn, AR binds to the miR-135a locus and controls its expression (Ref. Reference Coarfa92). Some studies have also indicated roles of epigenetic factors like DNA methylation in the regulation of metastatic miRNAs like miR-200c (Refs Reference Davalos93, Reference Ceppi94). Similarly, the promoter of miR-34a is hypermethylated in ovarian cancer (Ref. Reference Schmid95). Other factors that modulate miRNA activity like regulation and post-translational modifications of AGO proteins (Ref. Reference Cheng, Li and Han96), miRNA transport to the cytoplasm and regulation of miRNA–mRNA interactions need to be further explored in the context of metastasis.
Several recent studies have also compared changes in miRNA expression in primary and secondary tumours to identify potential drivers of metastasis. For instance, a study involving 33 CRC patients with metastasis and 14 patients without metastasis revealed differential expression of 17 miRNAs and their 198 predicted targets. There was a strong association of the target genes with cancer progression and metastasis (Ref. Reference Lee97). In another study involving metastatic breast cancer patients, the upregulation of miR-342-3p and miR-187-3p was associated with an increased progression-free survival (PFS) and overall survival (OS); while, the downregulation of miR-301a-3p was associated with a higher PFS and OS (Ref. Reference Martinez-Gutierrez98). In addition to being therapeutic targets, studies in several different cancer types have identified differences in expression levels of several miRNA indicating that they may serve as diagnostic or prognostic markers (Refs Reference McGuire, Brown and Kerin99–Reference Song106).
MiRNA-based therapies
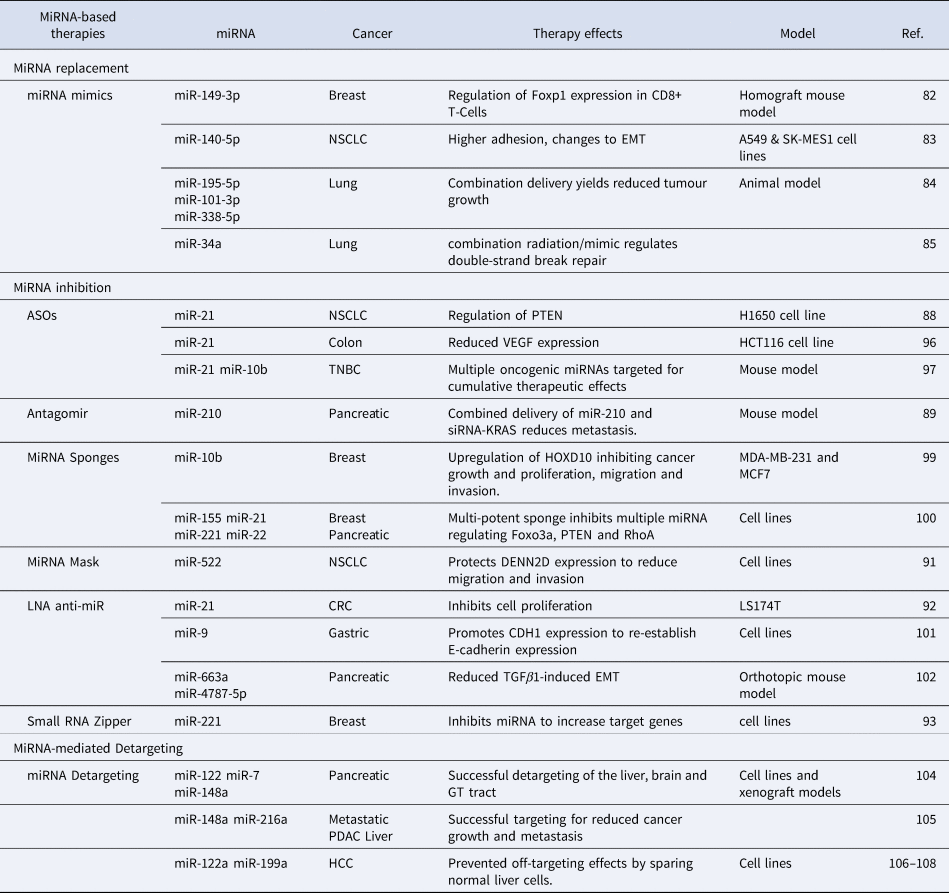
Endogenous miRNAs play a crucial role in maintaining cellular homoeostasis (Ref. Reference Rupaimoole107). The genomic and transcriptomic alterations in cancer cells can perturb the global miRNA expression profile causing genome-wide transcriptional changes (Ref. Reference Bartel108). These changes can lead to an upregulation of oncogenes and/or a downregulation of tumour suppressors which is critical for metastasis (Ref. Reference Bartel108). Most miRNA-targeted cancer therapies focus on restoration or inhibition of dysregulated miRNAs (Ref. Reference Raue109) but recently, miRNA-based detargeting strategies have been utilised for cell/tissue-specific targeted therapies (Ref. Reference Dhungel, Ramlogan-Steel and Steel110). Table 2 provides a comprehensive list of miRNA-based therapeutic strategies which will be discussed in this section in detail.
Table 2. MiRNA-based therapies for cancer metastasis: Dysregulated miRNAs are potential therapeutic targets to treat metastatic cancer
MiRNA replacement
The restoration of miRNAs that are downregulated in cancer is one approach to target metastasis (Ref. Reference To4). MiRNAs in metastatic cells can be restored using miRNA mimics (Ref. Reference Hosseinahli111) which are small synthetic RNA duplexes containing an antisense strand with the same sequence as the endogenous miRNA (Ref. Reference van Rooij and Kauppinen112). To increase stability of the duplex and to enhance cellular uptake, the sense strand can be chemically modified. The sense strand may also contain several mismatches to minimise off-target effects (Ref. Reference van Rooij and Kauppinen112). Like the naturally occurring miRNA, these miRNA mimics are loaded into the RISC complex and inhibit downstream targets (Fig. 3a) (Ref. Reference van Rooij and Kauppinen112). MiRNA mimics have been widely studied for therapeutic purposes in both in vitro and in vivo cancer models (Ref. Reference Zhang113). For instance, miR-149-3p mimics suppress breast cancer growth and metastasis by regulating inhibitory receptors and Foxp1 gene expression in CD8+ T cells in a homograft mouse model (Ref. Reference Zhang113). Treatment with miR-149-3p mimics reduced the apoptosis of CD8+ T cells which mediate the immune surveillance of cancer cells (Ref. Reference Zhang113). The promotion of CD8+ T cells resulted in the death of 4T1 mouse breast cancer cells (Ref. Reference Flamini, Jiang and Cui114). Similarly, a reduction of migration and invasion in A549 and SK-MES1 squamous carcinoma NSCLC cell lines was observed after the transfection of miR-140-5p mimics (Ref. Reference Flamini, Jiang and Cui114). The authors also reported a higher adhesion to an artificial extracellular matrix (ECM), indicating a change in EMT (Ref. Reference Flamini, Jiang and Cui114). A combined delivery of three miRNA mimics, miR-195-5p, miR-101-3p and miR-338-5p, is more effective in reducing tumour growth and the number of metastatic nodules in animal models of lung cancer (Ref. Reference Liu115). Similarly, delivering miR-34a mimic sensitises primary and metastatic derived lung cancer cell lines to radiotherapy, in vitro and in vivo (Ref. Reference Cortez116). Several other miRNA replacement therapies are currently being tested in both clinical and pre-clinical settings.

Fig. 3. MiRNA-based therapies. (a) MiRNA replacement with mimics function like an overexpression of endogenous miRNA and increase the degradation or repression of target mRNAs. (b) The miRNA inhibitor approach minimises the binding of miRNA-induced silencing complex (miRISC) to target mRNAs. Different strategies used for miRNA inhibition includes antisense oligonucleotides (ASOs), antagomir antisense oligonucleotides, locked nucleic acid (LNA), antisense oligonucleotide and small RNA zippers. (c) MiRNA sponge binds to the miRISC complex reducing its binding to the target mRNA. (d) MiRNA mask prevents the miRISC from binding to the mRNA by ‘masking’ the miRNA binding site.
MiRNA inhibition
Another approach to target metastasis is to inhibit upregulated oncogenic miRNAs (Ref. Reference Nguyen and Chang117). The inhibition of oncogenic miRNAs overexpressed during metastasis can restore silenced tumour suppressors (Ref. Reference Nguyen and Chang117). MiRNA inhibitors are single-stranded oligonucleotides complimentary to an endogenous miRNA (Ref. Reference To4). These inhibitors can bind to endogenous miRNAs and inhibit their incorporation into the RISC complex (Fig. 3b) (Ref. Reference Rupaimoole and Slack118). Several types of miRNA inhibitors have shown therapeutic advantages both in vitro and in vivo including antisense oligonucleotides (ASOs) (Ref. Reference Ge119), antagomirs (Ref. Reference Xie120), miRNA sponges (Ref. Reference Tay121), miRNA masks (Ref. Reference Zhang122), locked nucleic acid (LNA) anti-miRNAs (Ref. Reference Nedaeinia123) and small miRNA Zippers (Ref. Reference Meng124).
Synthetic Antisense Oligonucleotides (ASOs)
ASOs are single-stranded, chemically modified DNA molecules, 20-25 nucleotides in length, with a full complementarity to a target miRNA (Ref. Reference Bajan and Hutvagner125). ASOs inhibit the binding of mature miRNA to its target mRNA by producing an ASO-miRNA duplex which can lead to the cleavage of the miRNA and the upregulation of the target mRNA (Ref. Reference Bajan and Hutvagner125). ASOs have already been approved by the Food and Drug Administration for the treatment of Duchenne muscular dystrophy and spinal muscular atrophy whereby exon skipping strategies are utilised to restore the dystrophin expression (Ref. Reference Rinaldi and Wood126). For cancer therapy and metastasis inhibition, some pre-clinical studies have been reported with ASOs. For example, Ge et al. designed an ASO to target miR-21 which is overexpressed in NSCLC. MiR-21 regulates the activity of PTEN, a regulator of invasion and a metastasis promoter (Ref. Reference Ge119). The ASO-based drug was successful in reducing miR-21 expression and induced apoptosis in H1650 NSCLC cell line (Ref. Reference Ge119). Likewise, the transfection of synthetic ASOs targeting miR-21 significantly reduced migration and invasion of HCT116 human colon carcinoma cell line accompanied by a reduction in the expression of VEGF which is critical for colon cancer metastasis (Ref. Reference Tao127). Multiple oncogenic miRNAs can also be targeted simultaneously for additive therapeutic effects (Ref. Reference Devulapally128). ASOs targeting miR-21 and miR-10b have been successfully delivered in cell lines and tumour xenografts for triple-negative breast cancer (TNBC) (Ref. Reference Devulapally128). The simultaneous delivery of these miRNAs induces cancer apoptosis and inhibits tumour growth and metastasis in a mouse model of TNBC (Ref. Reference Devulapally128).
Antagomir antisense oligonucleotides
Antagomirs are artificially synthesised single stranded RNA of 23 nucleotides length complementary to a miRNA. Antagomirs can be chemically modified with a cholesterol moiety for greater stability (Ref. Reference Xie120). In a mouse model of pancreatic cancer, a cholesterol-modified polymetric CXCR4 antagonist was delivered with nanoparticles via an intraperitoneal delivery to localise efficacy and limit systemic side (Ref. Reference Xie120). The co-delivery of antagomirs against miR-210 and siRNA against KRAS to this model demonstrated a reduced metastatic activity (Ref. Reference Xie120). Of particular importance was the complete inhibition of liver metastasis, the primary metastatic site of PDAC (Ref. Reference Xie120).
MiRNA sponges antisense oligonucleotide
MiRNA sponges are short, synthetic transcripts with the same sequence as the 3′UTR of mRNAs targeted by the miRNA. Acting as a decoy, sponges inhibit the ability of miRNAs to regulate their target mRNAs (Fig. 3c) (Ref. Reference Tay121). There are some reports where miRNA sponging has been successfully performed for a single miRNA (Ref. Reference Ebert and Sharp129). Liang, Zhang, Zhou, Wu, Lin and Liu (Ref. Reference Liang130) designed a miRNA sponge plasmid to target miR-10b in metastatic breast cancer cell lines, MDA-MB-231 and MCF-7, demonstrating an inhibition of miR-10b and upregulation of its target HOXD10. This resulted in an inhibition of cancer growth and proliferation as well as a reduction in migration and invasion (Ref. Reference Liang130). Additionally, a multi-potent miRNA sponge that simultaneously inhibits 4 oncogenic miRNAs, miR-155, miR-21, miR-221 and miR-222 was developed (Ref. Reference Jung131). This multi-potent miRNA sponge was successful in inhibiting multiple oncogenic miRNAs, thus promoting anti-tumour effects in human breast cancer and pancreatic cancer cells (Ref. Reference Jung131). Results demonstrated the multi-potent miRNA sponge to be more effective in inhibiting proliferation when compared to single miRNA-targeted sponges and demonstrated a 1.3-2.3-fold change in the protein levels of Foxo3a, PTEN and RhoA which are associated with an increased metastatic potential (Ref. Reference Jung131).
MiRNA-masking antisense oligonucleotide
MiRNA-Masking (miR-Mask) is an inverted approach to protect mRNAs from miRNA-mediated repression (Ref. Reference Zhang122). In this approach, the miR-Masking oligonucleotides shield the miRNA binding sites of the mRNA to be protected (Fig. 3d) (Ref. Reference Zhang122). A full complementarity is required for better specificity (Ref. Reference Zhang122). This approach inhibits miRNA-mediated repression of targeted mRNAs without effecting the expression and potentially important functions of a miRNA (Ref. Reference Zhang122). Zhang et al. studied the effects of a miR-mask designed to complement the miR-522 binding site within DENND2D for the treatment of NSCLC and observed a reduced cell migration and invasion in NSCLC cells (Ref. Reference Zhang122).
Locked nucleic acid (LNA) antisense oligonucleotide
Another alternative oligonucleotide designed to inhibit miRNA oncogenic function are locked nucleic acid anti-miRs (LNA-i-miR) (Ref. Reference Nedaeinia123). LNAs are chemically modified by connecting the 2′ oxygen and 4′ carbon to form an extra methylene bridge locking the ribose ring (Ref. Reference Nedaeinia123). This leads to a higher thermal and in vivo stability and a greater binding affinity with mRNA targets (Ref. Reference Nedaeinia123). LNA against miR-21 was effective in reducing the invasiveness and inhibited the proliferation of human colorectal adenocarcinoma cells (Ref. Reference Nedaeinia123). Likewise, Lima et al. devised a strategy whereby LNA was efficient even when delivered at a low dose. In their study, miR-9 was targeted with LNAs to promote the expression of CDH1 for the reestablishment of E-cadherin in human gastric cancer cells (Ref. Reference Lima132). In another study, the delivery of LNA-i-miRs against miR-663a and miR-4787-5p reduced tumour burden and metastasis in an orthotopic mouse model of pancreatic cancer by decreasing TGFβ1-induced EMT (Ref. Reference Mody133).
Small RNA zippers
In this approach, oligonucleotides complementary to the second and the first half of a miRNA are synthesised and delivered into the cells (Ref. Reference Meng124). Small RNA zippers connect multiple copies of a miRNA end-to-end by forming a duplex of multiple miRNA copies and inhibit the function of the target miRNA (Ref. Reference Meng124). Like chemically modified LNAs, small RNA zippers have increased affinity, specificity and stability (Ref. Reference Meng124). A 70–90% inhibition of miR-221 and miR-17 and rescue of their target genes was observed in breast cancer cell lines using miRNA zippers (Ref. Reference Meng124). Further, the oncogenic effects of miR-221 were reversed by miR-221 zippers as demonstrated by the cell migration assay. However, the in vivo applications of miRNA zippers are yet to be tested (Ref. Reference Meng124).
MiRNA-mediated detargeting
Unlike miRNA replacement or inhibition therapies, this approach utilises the binding sites of miRNAs that are downregulated in cancer for detargeting the therapy from the normal cells and thereby reduce off-target effects. This approach is mostly useful for genetic therapies which utilise therapeutic gene transfer (Ref. Reference Kopp134). For instance, Baertsch et al. utilised three miRNAs; miR-122, miR-7, miR-148a, expressed at high levels in the liver, brain and the gastrointestinal tract, respectively, and demonstrated successful detargeting of these organs for a measles virus-mediated oncolytic virotherapy of pancreatic cancer in cell lines and murine xenograft models (Ref. Reference Baertsch135). In another example, the binding sites of miRNAs downregulated in PDAC, miR-148a and miR-216a, were used for detargeting in locally advanced and metastatic pancreatic and liver cancer (Ref. Reference Bofill-De Ros, Gironella and Fillat136). 8-miR148aT demonstrated detargeting effects by repressing all miR-148/152 family members in the pancreas and liver (Ref. Reference Bofill-De Ros, Gironella and Fillat136). This study demonstrated that this method was highly efficient for targeted therapies as a significant decrease in cancer growth and metastasis was observed (Ref. Reference Bofill-De Ros, Gironella and Fillat136). To prevent off-targets effects in the liver after suicide gene therapy, the binding sites of miR-122a and miR-199a, which are significantly downregulated in HCC, were used in multiple studies. Adeno-associated virus-based vectors were used to deliver miRNA122a and/or miRNA199a-regulated the suicide gene therapy system cytosine deaminase (CD)/ 5-fluorocytosine (Refs Reference Dhungel137–Reference Dhungel, Ramlogan-Steel and Steel139). Limited killing of normal liver cells with this system demonstrated an efficient liver detargeting using the binding sites of these miRNAs (Refs Reference Dhungel137–Reference Dhungel, Ramlogan-Steel and Steel139).
MiRNA-based therapies in clinical trials
MiRNA-based strategies have demonstrated therapeutic potential in a range of conditions including advanced cancers (Ref. Reference Fortunato and Iorio140). In fact, several miRNA-targeted therapeutics are at different phases of clinical development for the treatment of advanced cancers and metastases (Ref. Reference Ivkovic141). For cancer therapy, miRNA therapeutics are injected directly into the site of the tumour which can increase the specificity, efficacy and reduce off-target effects (Ref. Reference van Rooij and Kauppinen112). Below we summarise clinically applied miRNA-targeted therapies (Table 3). Although not exclusive to metastasis, these therapies have shown promise in treating advanced cancers including those with metastasis to secondary organs.
Table 3. A list of cancer therapy clinical trials utilising miRNA-based strategies

Clinical miRNA replacement
Tumour suppressor miR-15/16 family is downregulated in various cancers including lung and colon cancer that metastasise to the liver (Ref. Reference Reid142). Downregulation of miR15/16 can increase drivers of metastasis including tumour growth, angiogenesis, EMT and stemness (Ref. Reference Ivkovic141). In pre-clinical studies, miR-16 mimic replacement safely inhibited growth and metastasis of Malignant Pleural Mesothelioma (MPM) and NSCLC xenograft tumours (Ref. Reference Reid143). TargomiRs are minicells coated with an anti-EGFR-specific antibody carrying miR-16 mimics for a cancer-targeted delivery (Ref. Reference Reid143). A Phase 1 clinical trial of TargomiR initiated in 2014 demonstrated the safety of the approach in 26 patients. Of the 22 patients who were assessed for response, one showed a partial response and 15 had stable disease (Ref. Reference van Zandwijk144).
Tumour suppressor miR-34a downregulates the expression of oncogenes including MET, MYC, PDGFR-α, CDK4/6 and BCL2 (Ref. Reference Hong145). Both in vitro and in vivo studies have reported that miR-34a mimics can reduce tumour growth, migration and invasion and metastasis (Ref. Reference Reda El Sayed146). Anti-tumour activity of co-injecting let-7 and miR-34 was demonstrated in multiple NSCLC cell lines (Ref. Reference Kasinski147). The anti-tumour activity of this combinatorial therapy was also tested in KrasLSD−G12D/+; p53flx, flx mouse model of NSCLC (Ref. Reference Kasinski147). A second in vivo study was then initiated using lipid-based delivery agent (NOV340) to deliver miR-34a mimics (Ref. Reference Kasinski147). A Phase I clinical trial with MRX34, a liposomal formulation of a synthetic, double-stranded miR-34a mimic, was initiated for patients with HCC and unresectable liver metastasis. (Refs Reference Kasinski147, Reference Beg148). Unfortunately, adverse immune-mediated toxicities precluded the trial advancing to phase II (Clinical Trial identifiers: NCT01829971, NCT02862145) (Ref. Reference Beg148).
Tumour suppressor miR-193a-3p is downregulated in a range of cancers including HCC (Ref. Reference Grossi149), NSCLC (Ref. Reference Gao150) and TNBC (Ref. Reference Yu151). The repression of miR-193a-3p in these cancers decreases apoptosis, increases cell proliferation and migration tumour growth and metastasis (Ref. Reference van den Bosch152). Targets of miR-193a-3p play an important role in malignant cell behaviour including KRAS (Ref. Reference Fan153), ERBB (Ref. Reference Liang154), and S6K2 (Ref. Reference Yu155) in lung cancer, PLAU in bladder cancer (Ref. Reference Lv156), MCL-1 in glioma (Ref. Reference Kwon157), CCND1 in prostate cancer (Ref. Reference Liu158), RAB27B in osteosarcoma (Ref. Reference Pu159) and SRSF2 in HCC (Ref. Reference Bader160). Telford et al. reported that miR-193a-3p mimics reduce cancer cell proliferation/survival by inducing cell cycle arrest, apoptosis, increased cell senescence, DNA damage and inhibit migration (Ref. Reference Telford161). INT-1B3, a 193a-3p mimic replacement drug, consists of a lipid nanoparticle-based delivery system (Ref. Reference van den Bosch152). In preclinical studies with tumour bearing mice, systemic injection of INT-1B3 shows significant anti-tumour activity (Ref. Reference van den Bosch152). A phase 1/1b clinical trial to investigate the safety, preliminary efficacy, pharmacokinetics and pharmacodynamics of INT-1B3 is currently ongoing (Clinical Trial identifier: NCT04675996) (Ref. Reference van den Bosch152).
Clinical miRNA inhibition
Oncogenic miR-221-222 cluster located on the X chromosome is highly expressed in several solid tumours such as lung cancer (Ref. Reference Garofalo162), breast cancer (Ref. Reference Miller163), HCC (Ref. Reference Callegari164), and glioblastoma (Ref. Reference Zhang165) as well as haematological malignancies including myeloma (Ref. Reference Di Martino166). In advanced cancers, upregulation of miR-221 interferes with the expression of its targets p27, p57, PUMA and PTEN promoting tumour growth (Ref. Reference van den Bosch152). Di Martino et al. reported anticancer effects of anti-miR221-targeted LNAs both in vitro and in vivo. LNA-i-miR-221 is a 13-mer antisense oligonucleotide that uses LNA technology and phosphorothioate backbone chemistry for increased affinity for miR221 targeting (Ref. Reference Di Martino166). A phase I clinical trial of LNA-i-miR-221 will administer the drug via an intravenous injection to patients with multiple myeloma and advanced solid tumour (Clinical Trial identifier: NCT04811898).
Oncogene miR-155 regulates immune cell function and its overexpression affects multiple genes associated with the promotion of solid tumours including breast cancer, lung cancer, liver cancer as well as haematological malignancies including leukaemia (Ref. Reference Higgs and Slack167). Upregulation of miR-155 has been linked to JAK/STST, NK-KB and PI3K/AKT survival pathways stimulating T-cell receptors (Ref. Reference Seto168). MiR-155 inhibition reduces proliferation and increases apoptosis in T-cell lymphoma cell lines (Ref. Reference Seto168). In xenografts of B-cell lymphoma, miR-155 silencing with a LNA delivered systemically, reduced tumour burden and metastasis (Ref. Reference Zhang169). In another preclinical study, an anti-miR-155 molecule with a peptide nucleic acid backbone was used for greater sensitivity and efficient delivery to treat haematological malignancies (Ref. Reference Cheng170). These preclinical studies lead to the development of Cobomarsen which is a single-stranded, chemically modified miR-155-targeting molecule with chemical modifications for increased stability (Ref. Reference Seto168). A Phase 1 trial of Cobomarsen in patients with cutaneous T-cell lymphoma (CTCL) [mycosis fungoides (MF) subtype] was initiated in 2016 and reported some therapeutic benefit for 95% of all enrolled patients with increased benefits reported for subjects who underwent more than one cycle (Ref. Reference Seto168). Encouraging early data was followed by phase 2 clinical trial which started in 2019 but was terminated in 2020 due to commercial reasons (Clinical Trial identifiers: NCT03603431, NCT03713320, NCT03837457).
Challenges for miRNA-based cancer therapies
There are several challenges in using mi-RNA based therapies including insufficient delivery to the target tissue (cancer), stability in the biological system, immune responses and unwanted off-targeting. Arguably the primary challenge for miRNA-based cancer therapies is their efficient delivery to target tissues. Tumours can have poor blood perfusion and the complexity of ECM often hinder the delivery of miRNA-based therapies. In addition, scavenging cells like TAMs, neutrophils and monocytes can prevent the miRNA carrying vehicle from reaching cancer cells (Ref. Reference Yu, Huang and Li171). To overcome these challenges, different modes of delivery are being investigated, including both viral and non-viral vector systems (Refs Reference Bulcha172, Reference Santana-Armas and Tros de Ilarduya173). The more common viral vectors are derived from adeno-associated viruses, adenoviruses and lentiviruses, while non-viral vectors can include exosomes, polymers and liposomes (Refs Reference Bulcha172, Reference Santana-Armas and Tros de Ilarduya173). Viral vectors have a high efficiency and have been successfully used in several clinical trials. They also form the basis of a number of FDA approved therapies. (Ref. Reference Lundstrom174). Non-viral approaches may also be beneficial. The biocompatibility and biodegradability of polymers and liposomes are some of their advantages (Ref. Reference Karlsen and Brinchmann175). The conjugate vehicles have selective targeting and high stability due to the use of lipid or receptor binding molecules (Ref. Reference Biscans176). Naturally occurring exosomes are an advantage due to their immune compatibility (Ref. Reference Li177). Both viral and non-viral systems have inherent advantages and shortcomings, therefore the choice of the delivery system should be based on the overall design of a study.
Once delivered to the target tissue, the effect of miRNA therapeutics in non-cancer cells needs to be prevented. Similarly, effects on off-target mRNAs within cancer cells is another concern as miRNAs can target multiple transcripts simultaneously (Ref. Reference Bandi and Vassella178). MiRNA imperfect complementarity to the targeted mRNA 3′UTR has the capacity to indiscriminately silence off-target genes (Ref. Reference Kara, Calin and Ozpolat179). Another cause of off-targeting is the possibility of artificial exogenous miRNA competing with endogenous miRNA creating a dysregulation in gene expression (Ref. Reference Kara, Calin and Ozpolat179). Off-target effects can induce the silencing of tumour suppressors or activation of oncogenes in normal cells (Ref. Reference Seok180). For instance, miR-15/16 cluster regulate a large proportion of the whole transcriptome in leukaemia cells (Ref. Reference Pepe181). Thus, these miRNAs would not likely be used as therapeutic targets. The use of cancer targeted viral and non-viral delivery vectors can also reduce unwanted off-target effects (Refs Reference Santana-Armas and Tros de Ilarduya173, Reference Segal and Slack182). Furthermore, cancer cell-specific regulatory elements like tumour specific promoters can be used in the delivery system (Ref. Reference Dhungel183). Vigilant bioinformatic and wet-lab studies need to be performed with proposed inhibitors or mimics to identify any potential off-target effects in pre-clinical studies.
Both the miRNA and the delivery vector can elicit an immune response (Ref. Reference Ceppi184). MiRNA duplexes can trigger toll like receptor response (Refs Reference Yu, Huang and Li171, Reference Ceppi184) leading to an interferon response against miRNA therapeutics (Ref. Reference Yu, Huang and Li171). MiRNA therapeutics designed with certain chemical modifications can mitigate these immune responses (Ref. Reference Yu, Huang and Li171). Chemical modifications can enhance the miRNA stability in vivo. For instance, miRNAs without chemical modification of the ribose 2′-OH are prone to nuclease-mediated degradation and have a short half-life when injected systemically. Three generations of ASOs modification techniques developed (Ref. Reference Rawat1) First-generation modifications substitute phosphodiester backbone with phosphothiorate to increase in vivo stability; (Ref. Reference Kozomara, Birgaoanu and Griffiths-Jones2) Second-generation modifications substitute the 2′-O-alkyl group of the sugar moieties with 2′-OMe, 2′-O-methoxyethyl (2′MOE) or 2′-Fluoro to enhance efficacy and bioavailability and to reduce the immune stimulation and toxicity; (Ref. Reference Mishra, Yadav and Rani3) Third-generation modifications are the chemical alteration to the furanose ring with 2′4′-methylene producing LNAs to reduce nuclease degradation and increase membrane penetration (Ref. Reference Rinaldi and Wood126).
Concluding remarks
The inability of current therapies to effectively treat advanced or metastatic cancers stems from an incomplete understanding of the molecular mechanisms governing metastasis. Understanding molecular drivers of cancer metastasis can provide opportunities to develop novel therapeutic approaches. MiRNAs play a central role in regulating gene expression, thus, the dysregulation of miRNAs in metastasising cells warrants special attention. The dysregulation of several miRNAs is observed at every step of the metastatic process and restoring their levels is an attractive therapeutic avenue. Depending on the cancer type, dysregulated miRNAs can function as an oncogene or tumour suppressor. These dysregulations can lead to significant alterations in the expression of downstream target genes. Therapeutic approaches that utilise miRNAs aim to restore the normal levels of dysregulated miRNAs using miRNA mimics and inhibitors. There are several studies which report a successful use of miRNA mimics and inhibitors in pre-clinical in vitro and in vivo studies for targeting both primary tumours and metastasis for different cancer types. This has led to the initiation of human clinical trials using miRNA-based therapies for both solid and blood cancers. However, there are both technical and practical limitations for delivering miRNA-based therapies to patients. The in vivo stability and delivery of these miRNAs at therapeutic levels to the target tissue is a major issue for clinical applications. Similarly, therapy-induced toxicity and potential off-target effects are major concerns. There are several ongoing developments in this area to increase the stability of miRNAs mostly involving chemical modifications. Similarly, developments in the field of both viral and non-viral vector-based delivery can make the therapy cancer-specific and reduce off-target effects. Further research needs to be performed in order to identify novel miRNAs which control metastasis and potential therapeutic target; only then will the full potential of miRNA-based therapies for cancer metastasis be realised.
Abbreviations: BRF2, TFIIB-related factor 2, CADM1, Cell Adhesion Molecule 1, CDH1, Cadherin-1 or Epithelial cadherin, CDH5, Cadherin 5 or VE-Cadherin, CSF1, Colony-stimulating factor-1, EIF5A2, Eukaryotic Translation Initiation Factor 5A2, FAK, Focal adhesion kinase, HMGA2, High-mobility group AT-hook 2, HOXA10, Homeobox A10, HOXD10, Homeobox D10, IRF2, Interferon regulatory factor 2, IL-10, Interleukin 10, KLF4, Krüppel-like factor 4, KRAS, Kirsten rat sarcoma viral oncogene homologue, MMP2, Matrix metalloproteinase-2, MYC, MYC proto-oncogene, bHLH transcription factor, NANOG, Nanog Homeobox, N-RAS, Neuroblastoma RAS viral oncogene homologue, Oct-4, Octamer-binding transcription factor 4, PDCD4, Programmed Cell Death 4, PTEN, Phosphatase and tensin homologue, PTPN9, Tyrosine-protein phosphatase non-receptor type 9, PTPRU, Protein Tyrosine Phosphatase Receptor Type U, ROCK1, Rho Associated Coiled-Coil Containing Protein Kinase 1, SOX2, SRY-Box Transcription Factor 2, SOX4, SRY-Box Transcription Factor 4, TGF-β, Transforming growth factor beta, TGFBR3, Transforming growth factor beta receptor 3, VEGF, Vascular Endothelial Growth Factor, WIF1, WNT Inhibitory Factor 1, Wnt5a, Wnt Family Member 5A, ZEB1, Zinc finger E-box binding homeobox 1, ZEB2, Zinc finger E-box-binding homeobox 2, ZO-1, Zonula occludens-1. Cancers: Breast, Colon, CRC, Colorectal cancer, Gastric, HCC, Hepatocellular carcinoma, NSCLC, non-small cell lung cancer, pancreatic, PDAC, Pancreatic ductal adenocarcinoma, uveal melanoma.
Acknowledgements
The figures in this manuscript were drawn with Biorender.
Conflict of interest
The authors declare no competing interests.







 Open access
Open access