


In the past two decades, companion diagnostic tests (CDx) have emerged due to the increasing understanding of molecular drivers of disease, discovery of many biomarkers, and the subsequent development of targeted pharmaceutical treatments.
CDx measure an individual's protein or gene expression or detect genetic variation (biomarkers) (Reference Byron, Crabb, George, Marlow and Newland1). The use of genetic variation to determine individual response to drug therapy is also known as pharmacogenomics. In the clinical context, CDx can be used to guide the choice and/or dose of a particular drug therapy and improve patient outcomes (Reference Byron, Crabb, George, Marlow and Newland1).
Although the potential benefits of personalized medicine are substantial, from increased therapeutic efficacy and safety to lower medical costs, the uptake by health systems remains limited (Reference Meckley and Neumann2). Rapid expansion of this field over the next decade is anticipated as scientific understanding of disease at the molecular level becomes more prevalent. Such technological progress may also require regulatory agencies to adapt their terminology and frameworks for approval. In addition, as the number of drugs requiring CDx is expected to rise; challenges may be posed for reimbursement processes. In Canada, examples of such challenges include the need to revise the health technology assessment (HTA) framework to evaluate co-dependent technologies (i.e., drug and associated CDx) and the development of reimbursement strategies for this new technology, particularly in the area of oncology.
Other related challenges noted in the literature include the reluctance of some pharmaceutical manufacturers to restrict the use of their drug products through biomarker tests and the hurdles inherent to the development of CDx from discovery to clinical validation. In addition, there seems to be difficulties for regulatory bodies in developing effective mechanisms to synchronize the review of submissions for pharmaceuticals with submissions for diagnostic devices (Reference Philip, Carrington and Chan3).
To better understand the magnitude of this issue, an environmental scan was conducted by the Canadian Agency for Drugs and Technologies in Health (CADTH) to inform Canadian policy makers. The objective of this report is to provide an overview of the current and projected use of CDx “required” for the prescription of targeted drug therapies, as well as the regulatory and reimbursement processes for these devices and drugs in Canada and internationally. Of note, the terms “efficacy” and “effectiveness,” although semantically different, are interchangeable in this article as their use reflects terminology in documents consulted. This may be explained by the fact that, although the efficacy measures from clinical trials are required for regulatory approval, clinical effectiveness (whether the benefits are maintained across varied clinical practice and laboratory settings) (Reference Singal, Higgins and Waljee4) measures are not often required or reported for the purposes of regulatory or reimbursement approval (5).
That being said, given the anticipated significant market expansion of co-dependent technologies in the future, there will be an increasing need to demonstrate the real world effectiveness of such technologies, particularly for informing policy decisions. Of particular interest is whether the anticipated benefits of co-dependent technologies will materialize once deployed in clinical practice. For example, it may be expected that benefits such as reduced treatment failure rates and decreased treatment costs may only be seen with the optimal evidence-based use of co-dependent technologies. There may, however, be clinical situations where such optimal use may not occur, for example, administering a given drug to a patient not displaying the targeted genetic variance due to lack of alternative treatment options and the genuine desire of the clinician to help the patient. As a consequence, assessing real world effectiveness of co-dependent technologies will be important.
METHODS
The results of this environmental scan are based on a focused literature search of systematic reviews, high-level reviews and economic studies (Table 1) as well as feedback solicited from targeted stakeholders in 2014 (thirty-nine pharmaceutical manufacturers, forty-two diagnostic manufacturers, two genetic testing centers, fifteen professional associations, five patient groups). Stakeholders who responded included two pharmaceutical companies and three professional associations.
Table 1. Literature Search Methodology

CDx, companion diagnostics.
Of note, the scope of this scan is limited to diagnostic tests that are listed as “required” by the regulator, as opposed to recommended, for the prescription of the associated drugs; these are formally recognized as CDx (Reference Mansfield6;7).
FINDINGS
Current Landscape of Pharmaceuticals That Require a CDx
The number of available CDx on the market is rapidly growing (Reference Agarwal, Ressler and Snyder8). Outside of oncology, examples of other approved indications with CDx tests include cystic fibrosis, human immunodeficiency virus (HIV), and severe growth failure (Reference Agarwal, Ressler and Snyder8). Likewise, while the majority of CDx are being developed for oncology (Reference Agarwal, Ressler and Snyder8;Reference Das9) CDx development is growing for other therapeutic areas (e.g., neurology, cardiology, gastroenterology, and musculoskeletal diseases) (Reference Das9). Examples of therapies and their associated biomarkers required for the CDx, can be found in Table 2.
Table 2. Examples of Therapies and Associated Biomarkers (47)
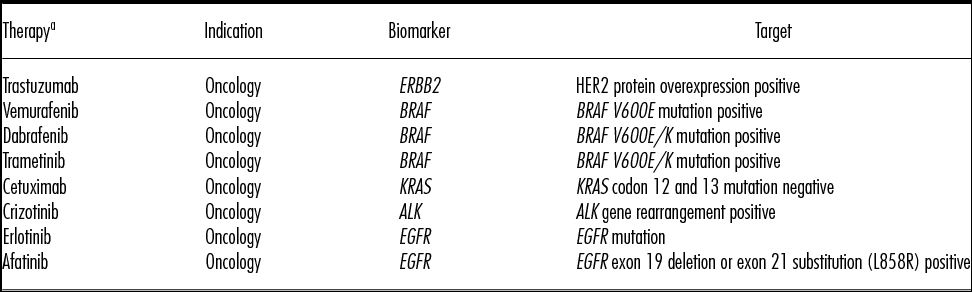
a All of the therapies listed in the table are approved for use in Canada and the United States.
ALK, anaplastic lymphoma kinase; BRAF, B-Raf proto-oncogene, serine/threonine kinase; EGFR, epidermal growth factor receptor; ERBB2, erb-b2 receptor tyrosine kinase 2; HER2, human epidermal growth factor receptor; KRAS, Kirsten rat sarcoma viral oncogene.
As a reflection of the increased attention given by the healthcare industry toward personalized medicine and targeted pharmacological therapies with CDx, several health market research companies now conduct evaluations and surveys in this domain. Advertisements for reports published by these companies provide an idea of the market trends and confirm the expected importance of this form of treatment in the future. Examples follow.
-
• The global market for CDx is expected to significantly grow in the future, with compound annual growth rate (CAGR) varying between 18.1 percent and 23.9 percent. Of note, potential differences in the definition used for market values may explain differences reported, as follows:
-
o One market research company reports that the value of the global market for CDx is projected to increase from US$1.8 billion (in 2013) to US$5.6 billion by 2019; this would represent a CAGR of 18.1 percent for the forecast period (10).
-
o Another market research company indicates that the global CDx market reached US$1.1 billion in 2012 and US$1.2 billion in 2013, respectively, and will progress to US$3.5 billion in 2018. This would represent a CAGR of 23.9 percent for the period (11).
-
o A third company projects that the global market value of CDx is expected to grow from US$3.1 billion in 2014 to US$8.7 billion in 2019, representing a CAGR of 22.7 percent for the forecast period (12).
-
-
• The global market composed of targeted pharmacological therapies and CDx, that is, co-dependent technologies, is projected to grow from a current value of US$42 billion to more than US$60 billion by 2019 (13;14). Key segments of this market include oncology, cardiovascular disease, as well as infectious disease treatment and diagnostics (13;14).
Co-development and Collaboration for CDx
With respect to a collaborative model, the availability of internal diagnostics divisions within pharmaceutical manufacturers, such as at Roche, Abbott, and Novartis, does not as yet appear to represent a more favorable structure than the use of external diagnostics providers, such as at GlaxoSmithKline, Pfizer, or AstraZeneca (Reference Blair, Stratton and Kaufmann15). Of note, the number of co-development and partnership agreements between pharmaceutical and diagnostic manufacturers increased from seven in 2008 to twenty-five in 2010; thirty-four of the forty-four (77 percent) deals made in 2009–10 were for oncology indications (Reference Leamon and Sherman16).
The limited stakeholder feedback received suggested that as knowledge of biomarkers and molecular drivers of disease increases, and new research challenges are encountered (e.g., the need to screen large amounts of patients to identify those with a specific genetic mutation), there may be a shift toward smaller and alternative trial designs in personalized medicine (e.g., adaptive trial designs, enrichment, and stratified clinical studies).
Current and Emerging Regulatory Practices
Regulatory practices for CDx are complex. The regulatory practices for CDx from Health Canada, U.S. Food and Drug Administration (FDA), and European Medicines Agency (EMA) are described below; key differences are highlighted in Table 3.
Table 3. Comparison of Key Market Requirement for Companion Diagnostics (Adapted from Ansari 2013 [20])
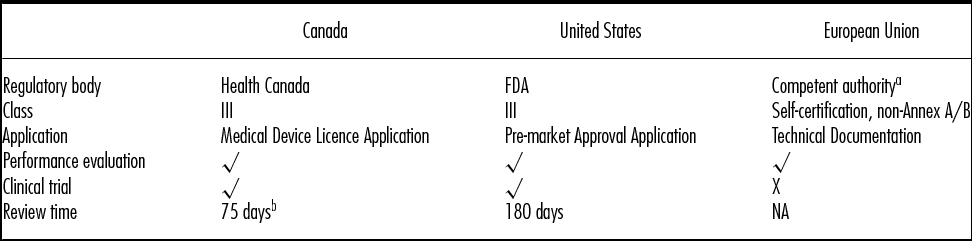
a Based on legal manufacturer's physical location.
b Performance review target to first decision (60 days) + submission screening phase (15 days) (48).
Source: Ansari, M., Therapeutic Innovation & Regulatory Science Journal (July 2013, vol. 47, no. 4: 405–415)
Table 1, p. 411, copyright © 2013 by (The Authors(s)) Reprinted by Permission of SAGE Publications.
FDA = Food and Drug Administration; NA = information not available.
Health Canada
In Canada, pharmaceutical drugs are regulated by the Therapeutic Products Directorate and biologic drugs are regulated by the Biologics and Genetic Therapies Directorate. In vitro diagnostic (IVD) devices are regulated by the Therapeutic Products Directorate's Medical Devices Bureau. All devices intended to be used for pharmacogenomics testing are classified as Class III medical devices and require a pre-market scientific assessment of the safety and effectiveness by the Medical Devices Bureau (17). Medical device classification is based on the risk associated with their use; Class III devices are associated with moderate risk (18).
For new drug submissions, or a supplement to a new drug submission, involving pharmacogenomics tests to support a therapeutic decision (e.g., the choice of a drug or dose), Health Canada encourages manufacturers to apply for a medical devices license as they progress through their drug development program. If the pharmaceutical manufacturer, in its clinical trials, used a companion test that is already licensed in Canada, it is recommended that the manufacturer indicates in its submission the name, description, and license number of the IVD device that was used; however, there is no provision for joint application and review processes for the drug and the companion test (17). Stakeholder feedback indicated that it may be a challenge for manufacturers to coordinate and align the review processes (so that the drug and the associated CDx receive marketing authorization at the same time), given that the regulatory review timelines for each of these components are different.
Quality assurance of CDx is also important, although not directly regulated by Health Canada. As the number of drug treatments for which genetic testing is required increases, access to reliable high-quality testing must be ensured to maximize the benefit that can be derived from personalized medicine. Reliability and quality of testing can be assured through establishing an effective framework for clinical laboratory operations, medical testing, and diagnostic devices. Hospitals and private laboratories offering genetic testing are subject to provincial regulations related to laboratory operations, accreditation, and quality control. Concerns have recently been expressed regarding the significant variation in the regulatory frameworks across the provinces and the lack of national oversight or guidelines to facilitate harmonization and good practice in laboratories throughout Canada (Reference Butts, Kamel-Reid and Batist19). Stakeholder feedback was received concerning the potential need to provide information on the legal implications to Canadian healthcare institutions and their laboratories of using proprietary CDx, or equivalent laboratory developed tests (LDTs), including on- and off-label uses of such tests.
Food and Drug Administration — United States
The FDA typically classifies CDx as Class III medical devices as the agency maintains that CDx carry the same risk profile as the companion drug. As such, proper use of the diagnostic device is critical to the proper use of the drug (Reference Philip, Carrington and Chan3;Reference Ansari20). Class III medical devices require pre-market approval and carry the highest risk in the U.S. regulatory system (Reference Philip, Carrington and Chan3;7;Reference Ansari20).
The FDA assesses IVD CDx testing products for safety and effectiveness. FDA guidance includes a provision for the approval of CDx devices alongside novel therapeutic products and recommends that the CDx be developed and approved at the same time to ensure safe and effective use (21). In some instances, the FDA may approve a drug product even though the CDx is not being approved simultaneously. Under these circumstances, if the therapy demonstrates benefits so pronounced as to outweigh the risks from an unapproved CDx, the FDA does not delay the therapeutic product's approval until the CDx is approved. However, the FDA expects the CDx will be subsequently approved through an appropriate IVD device submission, and the therapeutic product label will be revised accordingly (21). Moreover, the FDA may consider additional measures (e.g., a risk evaluation and mitigation strategy, or a post-marketing requirement) to address any potential safety issues related to the use of the companion drug without an approved CDx. In cases where the manufacturer intends to market an already approved IVD diagnostic device for another therapeutic product, the FDA requires a pre-market submission for the new use (7).
The FDA released new guidance to industry for IVD CDx devices in August 2014 (7). The FDA requires labelling on how, when, or whether a product should be used for all prescription therapeutic and device products (7;21). In the labeling of IVD CDx, a device intended for use with a therapeutic product must specify the therapeutic product(s) (or class of therapeutic products) with which it has been approved for use. The August 2014 guidance also notes that these devices, when used for informing treatment decisions in clinical trials, will generally be considered investigational IVD devices, unless the intended use of the device is already approved or cleared (7).
The FDA also intends to regulate CDx developed in private and institutional laboratories. In October 2014, the FDA issued draft guidance titled “Framework for Regulatory Oversight of Laboratory Developed Tests” which would update the Medical Devices Amendment which has regulated LDTs since 1976 (22). The proposed framework defines an LDT as an IVD that is intended for clinical use and designed, manufactured, and used within a single laboratory. Although the proposed framework would apply to most LDTs, it is mentioned that CDx will be considered high-risk Class III LDTs, which means that laboratories manufacturing these tests will be subjected to submit a pre-market submission within 12 months from issuance of the final FDA guidance (22;Reference Thorton23). Of interest, similarly to the FDA's intention, the Australian government recently began to regulate LDT's (24). Of note, as regulatory requirements expand for CDx to include not only commercially available products but also LDTs, and regulation scope expands beyond demonstrating assay validity, for example, demonstrating clinical utility, it may be anticipated that such changes will add complexity to the regulatory process for the diagnostic industry.
European Medicines Agency — Europe
In Europe, the regulatory framework consists of three directives: active implantable medical devices (Directive 90/395/EEC); medical devices (Directive 93/42/EEC); and IVD (Directive 98/79/EC). Currently, the regulation of IVD medical devices falls under the Directive of the European Parliament and of the Council (“the IVD Directive”) (25). Under this regulatory framework, IVD devices are not subject to pre-market authorization by a regulatory authority, but rather, are subject to a conformity assessment, which is the sole responsibility of the manufacturer for the majority of medical devices (25). For high-risk devices and devices for self-testing, an independent third party is required for the conformity assessment. Once certified, medical devices are allowed to circulate freely in the European Union or European Free Trade Association countries and Turkey (25).
Proposed regulations will follow guidance put forth by the Global Harmonization Task Force, in which CDx will be classified as high-risk, or Class C medical devices. As such, CDx devices must be assessed by an independent third party before certification is issued (Reference Le Gledic and Malek26). The proposed regulations also provide some clarification for the regulation of new therapeutic products requiring a CDx; including consultation with the EMA (or a competent authority) in the case of CDx devices, concerning the “suitability of the companion diagnostic in relation to the safe and effective use of the medicinal product in question” (25). The proposal is “awaiting council 1st reading position/budgetary conciliation convocation” (27). The proposed regulations stipulate that the independent third parties assessing CDx must consult the EMA or an individual national competent authority for their evaluation of the suitability of the CDx in relation to the therapy concerned. The EMA consultation includes a review of the draft summary of clinical safety and efficacy, as well as instructions for use; however, the purpose or benefit of the EMA review is unclear, and any steps to resolve discrepancies between the EMA and third-party assessments are also lacking (Reference Le Gledic and Malek26).
Current and Emerging Reimbursement Practices
Reimbursement policies and practices for CDx are not well defined. Both government and private payers in the countries listed below typically use HTA to inform their evaluation of clinical and economic benefits associated with pharmaceuticals. Standardized methods for determining clinical utility (i.e., how the novel diagnostic will impact clinical practice leading to improvement in patient outcomes) and reimbursement rates for diagnostic tests are lacking compared with the more established systematic and evidence-based HTA approaches used to guide reimbursement decisions for pharmaceuticals. At this time, individual payers seem to determine what evidence is required for CDx to show improvement in patient outcomes and cost-effectiveness (Reference Conway28).
Canada
Once a pharmaceutical product has been authorized for sale through a Health Canada review, it will be reviewed by CADTH Common Drug Review (CDR) or pan Canadian Oncology Drug Review (pCODR) process, depending on whether it is a non-cancer drug or a cancer drug, respectively. These programs assess the drug by reviewing scientific evidence on its comparative clinical- and cost-effectiveness. Expert committees [pCODR Expert Review Committee (pERC) and Canadian Drug Expert Committee (CDEC)] then develop recommendations to inform reimbursement decisions at the jurisdictional level (29). No specific reimbursement policies for companion drugs and associated diagnostics have been identified from any of the provincial bodies. In addition to these publicly funded drug plans, private insurance is also available in Canada to assist with costs related to prescription drugs (30;Reference Paris and Docteur31).
Funding decisions for genetic tests are made at the provincial level and decisions may vary across jurisdictions. Some provinces may not have dedicated processes in place to review, fund, and implement such tests which may result in increased pressure on individual hospitals. Hospital-based decisions may also result in considerable duplication of effort and prevent standardization of policies across institutions. Sometimes, when jurisdictional funding is lacking, pharmaceutical companies may offer to cover the cost of CDx. This approach is based on the anticipated return on investment when new patients become candidates for the drugs sold by the company (Reference Butts, Kamel-Reid and Batist19).
While such an approach allows health ministries and hospitals to save money on genetic testing, funding from industry may be limited in time or associated with conditions, where funding for a CDx associated with a particular drug may be limited to patients qualifying for therapy with this specific drug as the latter is sold by the manufacturer providing funding for the test. These situations may lead to confusion for patients and clinicians regarding availability of genetic tests and may result in inequities regarding the availability of such tests (Reference Butts, Kamel-Reid and Batist19). Stakeholder feedback regarding the latter point indicated support for a centralized structure for funding of CDx to ensure the quality of tests offered and consistency in patient access to this technology across Canada.
United States
Both government and private payers in the United States use HTA to inform their evaluation of the clinical and economic benefits associated with pharmaceuticals, which provides the basis for reimbursement and coverage decisions. Reimbursement for diagnostics can be slightly different in that pricing is often benchmarked to the Medicare Clinical Laboratory Fee Schedule, although other approaches (e.g., direct payment negotiation with payers) sometimes occur. In the case of IVDs, in addition to HTA findings regarding efficacy, payers also consider “the clinical utility of novel diagnostics attempting to understand (through data) how the novel diagnostic will impact clinical pathways, treatment decisions, prognosis, and ultimately outcomes. Lack of evidence related to clinical impact can limit coverage” (32). The HTA process is decentralized for both private and public payers, and varies across payers, who typically have a committee with an HTA or similar evidence-based review function. In addition to these committees, payers may also use third-party HTA organizations to provide evidence-based reviews and rate the evidence to help inform decision making by the payer (32).
No reimbursement policies for CDx were identified from either the Medicare or the Medicaid Web site. Of interest, Medicare “can create national or local coverage policies; however, most diagnostics are paid without explicit policies” (32). With respect to Medicaid, it appears that “each state sets its own guidelines regarding eligibility, services, and reimbursement” (32). The Veterans Affairs (VA) Health Services Research & Development Program is a national program tasked with “identifying and evaluating innovative strategies that lead to accessible, high quality, cost-effective care for Veterans and the nation” (33). CDx would fall under the “genomic medicine” assessed (34). The VA also has some personalized medicine research initiatives such as the Precision Oncology Program; however, no explicit policies were identified from the Web site (Reference Fiore, Brophy and Turek35).
In the United States, there are also many private payers who provide specific coverage and payment to their health plan members. CDx would be covered under the medical benefit of these health plans and the coverage and reimbursement of these devices or therapies would be reviewed within each organization (32). For example, in January 2016, BlueCross BlueShield Technology Evaluation Centre posted an announcement to the effect that one of the Association's members (Independence Blue Cross) will become the first major insurer in the United States to cover next-generation whole genome sequencing for the treatment of patients with cancer (36).
United Kingdom
In the United Kingdom, the National Institute for Health and Care Excellence (NICE) defines CDx as a diagnostic technology that identifies people who are likely to benefit from a specific therapy for their condition. These tests may also help in stratifying disease status, selecting the proper medication, and tailoring dosages to patients’ needs. In some cases, the use of CDx technologies may be necessary to comply with the licensed indications of pharmaceuticals (37).
Reflecting the two clinical development paths for such tests, NICE provides guidance on CDx through one of the two following programs (37;Reference George38): (i) Tests linked to new drugs are appraised through the Technology Appraisal Programme (TAP) as part of the appraisal of the new drugs, (ii) Tests linked to established drugs follow the Diagnostics Assessment Programme (DAP). Both of these programs use quality-adjusted life-years (QALYs) to compare the clinical and cost-effectiveness across technologies (Reference Byron, Crabb, George, Marlow and Newland1).
Australia
In late 2010, acknowledging the need to improve current models for the assessment of personalized medicine, to inform reimbursement decisions and to provide clarity to industry regarding policy-makers’ expectations, the Australian Government Department of Health and Ageing released the first integrated national framework for reviewing co-dependent technologies (defined as biomarker, test, and drug packages) (Reference Merlin, Farah and Schubert39). The framework allows for direct evidence, or linked evidence of the impact on patient health outcomes to be used in the submission process. This framework (Reference Merlin, Farah and Schubert39;40) includes five components: (i) Section A: Context for the submission, (ii) Section B: Clinical benefit, effectiveness, and safety of the pair of co-dependent technologies, (iii) Section C: Can the test-drug evidence of effectiveness be translated to an economic model for the Australian clinical setting? (iv) Section D: Is the proposed use of the pair of co-dependent technologies cost-effective? (v) Section E: What is the financial impact of the proposed listing of the pair of co-dependent technologies?
The framework also includes a checklist of seventy-nine items as well as consideration of the type of evidence supporting the efficacy of the co-dependent technologies (Reference Merlin, Farah and Schubert39;40). Although this framework was developed for the Australian health system, it is anticipated that it may be a suitable model for other health systems (Reference Merlin, Farah and Schubert39). Of note, a review of the Pharmaceutical Benefits Advisory Committee Guidelines is currently under way, with a final draft expected to be completed in November 2016 (41). This review includes an updated chapter on co-dependent technologies, which has been shared for public consultation but not yet formally approved.
New Zealand
Similar to Canada, no formal policies or processes were identified for the reimbursement of companion drugs and associated diagnostics.
Potential Financial Impact, Particularly on Public Payers
By demanding increased evidence of cost-effectiveness before reimbursing treatments, payers may actually contribute to the drive toward developing required CDx (Reference Byron, Crabb, George, Marlow and Newland1). For example, a recent estimate suggests that for patients with metastatic colorectal cancer, approximately $600 million could be saved annually if panitumumab or cetuximab were limited to patients with the wild-type KRAS gene (Reference Byron, Crabb, George, Marlow and Newland1). Other examples of CDx associated with either cost-saving or reasonable cost-effectiveness ratios (e.g., < $50,000/QALY) are presented in Table 4 (Reference Meckley and Neumann2). CDx may also reduce adverse drug reactions by preventing the prescription of certain treatments to those individuals with specific biomarkers, in turn, reducing the potential costs for medical attention or hospitalization (Reference Byron, Crabb, George, Marlow and Newland1).
Table 4: Examples of Cost-Attractive Companion Diagnostic Tests (Adapted from Meckley and Neumann, 2010 [2])

Source: Reprinted from Health Policy, Volume 94, Issue 2, Meckley LM, Neumann PJ. Personalized medicine: Factors influencing reimbursement, pages 91–100, 2010 Feb (© 2009), Table 3, p. 95, with permission from Elsevier.
FDA, Food and Drug Administration; HER2, human epidermal growth factor receptor 2; UGT1A1, UDP glucuronosyltransferase family 1 member A1; QALY, quality-adjusted life-year.
As indicated earlier, further evidence and practice-based data of the benefits, harms, and costs associated with the broader deployment of CDx are required to confirm whether their anticipated advantages will materialize. Should co-dependent technologies not be used optimally in routine clinical practice, that is, used outside the targeted populations for which they were initially designed for, the expected clinical utility may be decreased. This situation would also result in reduced cost-effectiveness and unproductive spending.
From a budgetary perspective, the introduction of a new drug with a CDx affects both laboratory and pharmacy services. Stakeholder feedback indicated some concerns regarding the proprietary nature of the CDx and the potentially higher costs to laboratories and funding agencies. It was mentioned that laboratories should have the opportunity to choose how they will test for a genetic variation after demonstrating equivalent sensitivities with the test used in clinical trials. This practice could lead to laboratories developing more tests in-house; alternatively, laboratories could choose other commercial molecular assay kits that test for the same mutation as the testing kit used in the clinical trials, should other manufacturers commercialize such tests at a lower price.
IMPLICATIONS
In Canada, pCODR reports that as of December 31, 2013, there were potentially fifteen individual drugs and thirty-one drug-indications linked to twelve different CDx on the horizon for cancer treatment (42). Of note, pCODR had completed thirteen reviews of targeted therapies for cancer treatment as of December 2015; these required five different CDx (43). Similarly in Europe and the United States, the development of therapeutic products that are paired with diagnostic tests is becoming increasingly common (21;Reference Garfield44). Currently available CDx aim to detect a single biomarker or genetic variance in a single gene; in the future, whole genome sequencing and genomic profiling of tumors or individuals may be possible (Reference Payne and Annemans45).
Of interest, as the number of available CDx increases and the clinical validation of their use expands, some have started to argue that the era of precision medicine may in fact lead to greater uncertainty. Indeed, the use of CDx will add to, and not replace, the use of currently established diagnostic tools. Therefore, clinicians will be faced with an increasing amount of information, the latter also being more complex, leading to challenges in interpreting such data to inform clinical decision making. This personalized information will range from estimates of disease risk based on the patient genetic profile to, once a particular disease is diagnosed, estimates of prognostic and therapeutic options with precision medicine. In addition, new tools will need to be developed to help patients absorb large amounts of complex information so that they can make choices about their care (Reference Hunter46).
LIMITATIONS
A limitation of this environmental scan is the low response rate for stakeholder feedback. Of the 103 organizations approached for feedback, only five responded including two pharmaceutical companies and three professional associations. Although a higher rate of feedback from industry and public payers may have generated more specific information on co-development opportunities and potential financial impact, respectively, we believe the feedback received is aligned with information available from the grey literature and do not expect any opposing views. As such, despite the low response rate, findings from the stakeholder survey validate and complement the information retrieved from the literature.
Another limitation resides in the relative scarcity of information available. This may be explained, in part, by the limited information available in the public domain as well as the wide spread of pharmaceutical and diagnostic test manufacturers, which makes information identification and retrieval more challenging. Another factor to consider is the confidential nature of technologies in development for which information may not be publically available to protect their commercial operations and marketing strategy, this in turn may also add to information retrieval challenges. Lastly, the scope of this work is in itself limited. Indeed, this environmental scan was not aiming at providing in-depth analysis of the comparative effectiveness of co-dependent technologies. Rather, its main purpose was to gain a better understanding of the importance of this relatively new technology and the stage of development of associated regulatory and policy contexts. Findings from this environmental scan may, however, inform future policy work and HTA methodology development activities.
CONCLUSION
The global market for CDx is expected to grow considerably until the end of this decade, with CAGR around 20 percent. The co-development of CDx and new drugs, as well as the partnership between diagnostic and pharmaceutical companies, may be strategies that will be increasingly adopted to capture part of this growing market. This growth will be driven by increasing scientific knowledge of biomarkers and molecular drivers of disease, which will likely result in more co-development of CDx with associated drug therapies. Other growth drivers include the increased interest by some regulatory authorities for simultaneous approval of co-dependent technologies and the potential for cost-savings associated with lower rates of treatment failures and adverse drug reactions.
However, until the clinical development and deployment of pharmaceuticals with CDx expand, and further scientific evidence and practice-based data are available, it remains uncertain whether the expanded use of co-dependent technologies will lead to more cost-effective drug therapy. Anticipating the expansion in the availability of pharmaceuticals with CDx, countries such as the United States, the United Kingdom, and Australia are adapting their regulatory and/or reimbursement frameworks; however, several countries, including Canada, do not yet have published policies for co-dependent technology. As availability of CDx increases, and the clinical use of these tests expands, HTA will likely play an important role in interpreting and assessing developments in co-dependent technologies in ways that will provide more evidence-based information to guide reimbursement decisions, clinical practice guidelines, as well as other decisions and policies. However, to do so, current HTA methods may need to be refined to evaluate co-dependent technologies.
ACKNOWLEDGMENTS
The authors would like to sincerely thank Hayley Fitzsimmons, MLIS, as well as Kelly Farrah, MLIS, for their valuable assistance with information retrieval and reference management.
CONFLICTS OF INTEREST
T.C. is the founder and principal consultant of Medlior Health Outcomes Research Ltd., an independent private research organization with public sector and industry clients; M.B. is employed by CADTH. The authors declared no conflict of interest.




