


Introduction
The term ‘non-sulfide’ is used to define a type of Zn-Pb ore deposit dominated by ‘oxidized’ Zn and Pb ore minerals, and is applied to distinguish such deposits from entirely sulfide deposits (Large, Reference Large2001; Hitzman et al., Reference Hitzman, Reynolds, Sangster, Allen and Carman2003). Non-sulfide Zn-Pb deposits can be classified into two types: supergene deposits and hypogene deposits, according to their genetic attributes and resultant mineralogies (Large, Reference Large2001; Hitzman et al., Reference Hitzman, Reynolds, Sangster, Allen and Carman2003; Boni and Mondillo, Reference Boni and Mondillo2015). Supergene deposits form after the weathering and oxidation of exhumed sulfide orebodies at, or close to, the surface, consisting commonly of Zn and Pb carbonates (smithsonite, hydrozincite and cerussite) plus a range of hydrous Zn-bearing silicates and clays (hemimorphite and sauconite), in addition to remnants of primary sulfides (sphalerite, galena and pyrite). Hypogene deposits form either by hydrothermal processes or the metamorphism of primary sulfide ores, and consist mainly of an assemblage of anhydrous Zn silicates and oxides, such as willemite, zincite, franklinite, coexisting locally with sulfides (Hitzman et al., Reference Hitzman, Reynolds, Sangster, Allen and Carman2003).
The Kabwe Zn-Pb deposit (formerly known as ‘Broken Hill’) is located in central Zambia, ~110 km north of Lusaka, in an area where several other Fe, Mn, Au and Pb-Zn-Cu-V occurrences are developed (Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009). The mineralized district forms a cluster of mixed sulfide/non-sulfide orebodies. Mineralization was discovered in 1902, and the sulfides were mined until mine closure in 1994 (Kamona and Friedrich, Reference Kamona and Friedrich2007). During its lifespan, Kabwe was the most significant Zn-Pb mine in Zambia, with production from the sulfide bodies of 1.8 Mt of Zn, 0.8 Mt Pb, 79 t Ag, 7820 t V2O5, 235 t Cd and 64 t Cu. The sulfide ores included sphalerite, galena, pyrite and chalcopyrite as well as accessory Ge-sulfides (briartite and renierite). Significant Ga and In were also recognized in the ores (Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009). Despite this, these ‘critical elements’ were never recovered from the mineralization as they were only recognized in 1991, by which time the bulk of the Zn-Pb sulfide orebody was almost completely mined out (Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009). About 1.9 Mt of Zn non-sulfide resources remain with a reported grade of 13.4 wt.% Zn and 1.5 wt.% Pb (Kamona and Friedrich, Reference Kamona and Friedrich2007). This non-sulfide resource consists of (1) Zn-silicate zones mostly containing willemite, which surround the massive Zn-Pb sulfide orebodies, and (2) distinct oxide-rich alteration bands, containing Zn-, Pb- and Cu- vanadates, phosphates and carbonates, overlying both sulfide and Zn-silicate orebodies (Kamona and Friedrich, Reference Kamona and Friedrich2007). In the oxidized zones, willemite and supergene minerals demonstrably formed at the expense of original sulfides (Kamona and Friedrich, Reference Kamona and Friedrich2007; Terracciano, Reference Terracciano2008). Although willemite is commonly considered a hydrothermal mineral (Hitzman et al., Reference Hitzman, Reynolds, Sangster, Allen and Carman2003, and references therein), a number of authors (Kamona and Friedrich, Reference Kamona and Friedrich2007; Terracciano, Reference Terracciano2008) suggest that the willemite at Kabwe may be related to either low-temperature hydrothermal oxidizing fluids or alternatively to weathering processes, particularly given that the willemite at Kabwe is associated with a ‘typical’ supergene mineral assemblage (e.g. smithsonite, cerussite, goethite).
The aim of the present study was to evaluate Ge, Ga and In related to the non-sulfide mineralization, both in willemite-dominated ore and the oxide-dominated mineral assemblage. For the present study, a reanalysis of Kabwe samples housed in the ore collection of the Natural History Museum, London (NHM) was conducted, with a specific focus on comparing the deportment of trace elements in the primary sulfides and in the oxidized mineral assemblage. In order to better constrain formation processes, oxygen isotope analyses of willemite and other secondary minerals were carried out.
Geological setting
Regional geology
The rocks underlying the Kabwe region are dominated by Neoproterozoic metasediments which unconformably cover a Paleo- to Mesoproterozoic basement consisting of granite gneiss with minor amphibolite, schist, quartzite and pegmatite (Arthurs et al., Reference Arthurs, Newman and Smith1995; Cairney and Kerr, Reference Cairney and Kerr1998; Kamona and Friedrich, Reference Kamona and Friedrich2007; Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009) (Fig. 1a). Neoproterozoic metasedimentary cover rocks are generally considered to be contemporaneous with Katangan rocks of the Copperbelt, formed between 877 and 573 Ma (Cahen et al., Reference Cahen, Snelling, Delhal and Vail1984; Cairney and Kerr, Reference Cairney and Kerr1998; Armstrong et al., Reference Armstrong, Robb, Master, Kuger and Mumba1999; Master et al., Reference Master, Rainaud, Armstrong, Phillips and Robb2005). According to various authors (Taylor, Reference Taylor1954; Whyte, Reference Whyte1966; Kortman, Reference Kortman1972; Kamona, Reference Kamona1993; Kamona and Friedrich, Reference Kamona and Friedrich2007; Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009), the Neoproterozoic succession in the Kabwe area includes, from the base to the top: (1) a basal conglomerate lying unconformably on the Paleo- to Mesoproterozoic basement complex; (2) a mixed unit known as the Kangomba Formation (Moore, Reference Moore1964; Cairney and Kerr, Reference Cairney and Kerr1998) consisting of arkoses, quartzites, conglomerates, metasiltstone, schist, phyllite and dolomite; and (3) a predominantly phyllite-dolomite unit with associated calcite-marble named the Nyama Formation (Moore, Reference Moore1964; Barr et al., Reference Barr, Cahen and Ledent1978; Intiomale, Reference Intiomale1982; Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009). In the Kabwe area (Fig. 1a), the upper part of the Kangomba Formation has been upgraded stratigraphically to a true Formation, named the Kabwe Dolomite Formation, which has been subdivided into two members (Kortman, Reference Kortman1972; Kamona and Friedrich, Reference Kamona and Friedrich2007): (1) the lower micaceous dolomite; and (2) the upper massive dolomite, which hosts the main orebodies (Kamona and Friedrich, Reference Kamona and Friedrich2007).
Fig. 1. (a) Geological sketch map of the Kabwe area (modified after Hitzman et al., Reference Hitzman, Reynolds, Sangster, Allen and Carman2003; Kamona and Friedrich, Reference Kamona and Friedrich2007). (b) Morphology and relative positions of orebodies on the 850 ft mine level (~260 m depth from the surface), underlying the surficial Main Zone (black square in (a); modified after Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009).
Main characteristics of the Kabwe mineralization
The morphology, structural setting, mineralogy and geochemistry of the Kabwe orebodies have been described previously by Kamona (Reference Kamona1993), Kamona and Friedrich (Reference Kamona and Friedrich2007, and references therein), Terracciano (Reference Terracciano2008) and Kampunzu et al. (Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009), and a summary of that work is presented here. The Kabwe primary mineralization is composed of a number of pipe-like bodies of massive Zn-Pb sulfide, containing sphalerite, galena, pyrite, minor chalcopyrite, accessory briartite and the Ge-Cu sulfide renierite, developed in massive dolomite close to the faulted contact with the micaceous dolomite (Kamona, Reference Kamona1993; Kamona and Friedrich, Reference Kamona and Friedrich2007; Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009). These sulfide cores are surrounded locally by strongly oxidized zones mostly consisting of willemite and mineralized jasperoid, with a mixture of quartz, willemite, cerussite, smithsonite, vanadates, phosphates, goethite and hematite.
In detail, (Fig. 1b) it is possible to recognize three main massive sulfide orebodies in the Kabwe region referred to as No. 1, No. 3/4 and No. 5/6, all of which occur in highly faulted massive dolomite adjacent to the Mine Club fault zone. The No. 2 body is a willemite-rich but Pb-poor massive orebody. Minor orebodies (“X”, “E”, South Orebody, No. 2 Hangingwall, No. 8) occur near the main massive sulfide orebodies. Other mineralized occurrences include the small willemite-rich Foundry and the Airfield prospects. Mineralization of the same type is also found a few kilometres from Kabwe (e.g. at Carmarnor, Chowa, Millberg; Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009). The large No. 5/6 and the No. 3/4 orebodies both have pipe-like forms, occur mainly along NE–SW trending faults, and are clearly discordant to the host dolomite (Kamona and Friedrich, Reference Kamona and Friedrich2007). The dolomitic host rock is barren a few metres from the orebodies, rarely containing finely disseminated sphalerite, galena and pyrite. Mineralization at Kabwe is also associated with breccias, which clearly cross-cut the surrounding stratified rocks (Whyte, Reference Whyte1966; Kortman, Reference Kortman1972; Samama et al., Reference Samama, Mapani and Tembo1991; Kamona and Friedrich, Reference Kamona and Friedrich2007).
Primary sulfides (pyrite, sphalerite, galena, chalcopyrite) are recrystallized and deformed (Kamona, Reference Kamona1993; Kamona and Friedrich, Reference Kamona and Friedrich2007). This deformation suggests that the mineralization was affected by a phase of low-grade metamorphism, probably related to the Lufilian orogeny at ~550 Ma., thereby fixing a minimum age for the formation of the sulfide deposit (Porada and Berhorst, Reference Porada and Berhorst2000; Kamona and Friedrich, Reference Kamona and Friedrich2007). On the basis of Pb isotopic compositions of galena, Kamona et al. (Reference Kamona, Lévêque, Friedrich and Haack1999) proposed a model age for the sulfide mineralization of 680 ± 13 Ma. Summarizing previous works, Kampunzu et al. (Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009) suggested that the Kabwe mineralization formed from basinal brines during a syntectonic hydrothermal circulation, which produced carbonate-hosted stratabound epigenetic ore bodies of MVT-type (Leach et al., Reference Leach, Sangster, Kelley, Large, Garven, Allen, Gutzmer and Walters2005). On the other hand, some authors (see e.g. Heijlen et al., Reference Heijlen, Banks, Muchez, Stensgard and Yardley2008) consider Kabwe, together with Kipushi, Dikulushi, Lombe, and Kengere, to be a vein-type Zn-Pb-Cu-Ag deposit.
Early, hypogene sulfide minerals are clearly replaced by various secondary minerals (Kamona and Friedrich, Reference Kamona and Friedrich2007; Terracciano, Reference Terracciano2008; Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009). In detail, previous work (Kamona and Friedrich Reference Kamona and Friedrich2007, and references therein, Terracciano, Reference Terracciano2008) recognized at least two willemite generations: (1) a massive willemite, replacing the sulfides directly, and (2) a crystalline willemite, precipitating in geodes. They also described a range of secondary phases including carbonates (smithsonite, cerussite, azurite, hydrozincite, aurichalcite, rosasite, and Pb- and Zn-malachite), silicates (hemimorphite, chrysocolla), oxides (magnetite, hematite, goethite, zincite, psilomelane, wulfenite and cuprite), rare phosphates (tarbuttite, parahopeite, pyromorphite, hopeite, spencerite, scholzite and zincian libethenite), vanadates (vanadinite, descloizite and mottramite), sulfates (anglesite, gypsum, linarite and goslarite), supergene sulfides (covellite, chalcocite and galena), arsenates (beudantite, mimetite) and native copper. Many of these secondary minerals are typical of a supergene environment and, in fact, occur in a weathering-related alteration profile over sulfides, persisting locally from the surface down to the deepest level of the mine (465 m) (Kamona and Friedrich, Reference Kamona and Friedrich2007; Terracciano, Reference Terracciano2008; Kampunzu et al., Reference Kampunzu, Cailteux, Kamona, Intiomale and Melcher2009). Several secondary minerals clearly formed from the mobilization of major and trace elements derived from primary minerals, i.e. arsenates, oxides, carbonates, silicates and sulfides. A limited temporal constraint to the formation of the majority of the supergene minerals at Kabwe is given by ages obtained using U/Th-He methods on descloizite yielding ages of 20–37 Ma (N.J. Evans, unpubl., in Boni et al., Reference Boni, Terracciano, Evans, Laukamp, Schneider and Bechstädt2007).
The processes leading to the formation of willemite are less clear. Willemite could have formed either during in situ weathering of the primary sulfide ore and therefore be supergene, or may have formed during hydrothermal alteration of sphalerite, and would thus be hypogene (Kamona and Friedrich, Reference Kamona and Friedrich2007; Terracciano, Reference Terracciano2008). Silica occurs partially as quartz and chalcedony in vugs, clearly of supergene origin and introduced into the oxidation zone by circulating groundwaters (Taylor, Reference Taylor1958). However, part of the silica responsible for willemite formation could have been introduced into the system during silicification of the wallrock dolomite at temperatures >150°C under oxidizing conditions (Kamona and Friedrich, Reference Kamona and Friedrich2007). Kamona and Friedrich (Reference Kamona and Friedrich2007) interpreted the first massive willemite generation, replacing the sulfides directly, as hydrothermal, whereas the second generation was supergene. On the contrary, Terracciano (Reference Terracciano2008) considered a supergene or low-temperature hydrothermal origin for both willemite generations recognized in the Kabwe deposit.
Materials and methods
For the present study, 16 specimens of Kabwe area ore bodies, housed in the NHM ore collection (Fig. 2, Table 1) were used. To define the ore textural, mineralogical and geochemical characteristics, only a limited number of diverse samples (see Tables 2 and 3) was further selected from the batch of 16 samples. One willemite specimen, belonging to the NHM Mineralogy collection (Table 1), was also used for oxygen isotopic analyses. Sample preparation and analytical methods are described in detail in Appendix 1 (Supplementary material, see below), and are summarized briefly here.
Fig. 2. Examples of Kabwe specimens (NHM ore collection). (a) BM1930-372: sphalerite and galena; (b) MI29631: silicate Zn ore; (c) OR5307: Zn-Pb-carbonate ore; and (d) OR5314: ‘Calamine’ sample: smithsonite and hemimorphite crusts.
Table 1. Specimens of the Kabwe ores used for this study (NHM ore collection).
*This specimen is housed in the NHM Mineralogy collection.
Table 2. Semi-quantitative abundances of major minerals in some of the samples analysed (data from PXRD and ‘Mineralogic' combined).
– not detected, x: <5 wt.%, xx: 5–20 wt.%, xxx: 20–40 wt.%, xxxx: 40–60 wt.%, xxxxx: >60 wt.%
Table 3. Chemical composition of the samples studied.
<, under detection limit; –, not detected.
Polished blocks were prepared for optical microscopy (OM), scanning electron microscopy with energy dispersive X-ray spectrometry (SEM-EDS) and automated mineralogy analyses; representative parts of each sample were ground to produce powder for whole-rock Powder X-ray Diffraction (PXRD) and chemical analyses.
The PXRD patterns were obtained using an Enraf Nonius PDS120 diffractometer (NHM), characterized by an asymmetric reflection geometry, with fixed angle between the X-ray tube, the sample surface and the detector. CoKα1 radiations were used at 40 kV and 40 mA. The PXRD patterns were analysed using the Highscore Plus (Panalytical) software; the PDF4 (Powder Diffraction File from the International Center for Diffraction Data – ICDD) database was used for phase identification. The instrumental configuration adopted and the sample preparation did not permit quantitative phase analysis using the Rietveld method. Mineral abundances were determined as wt.% ranges (Table 2) on the basis of the peak intensity ratio between mineral phases, the modal mineralogy of polished blocks and the whole-rock chemical analyses.
Whole-rock chemical analyses of major and minor elements (see Table 3 for the element list) were carried out at the NHM. Following 4-acid digestion, the sample solutions were analysed by inductively coupled plasma mass spectrometry (ICP-MS) using an Agilent 7700× mass spectrometer. The instrument was calibrated using multi-elements standards (Inorganic Ventures). The limits of quantification were calculated as 10 times the standard deviation of HNO3 blank solution analysed at least 10 times during the run.
The SEM-EDS analyses were carried out using a ZEISS EVO LS 15 scanning electron microscope (NHM) at 20 kV, with 8.5 mm working distance and 3 nA current mounting with X-Max detectors. Quantitative data sets of selected samples were obtained by wavelength dispersion spectrometry (WDS), using a Cameca SX100 electron microprobe operating at 20 kV, 20 nA and 10 µm spot size (NHM).
The modal mineralogy of the samples was investigated on polished blocks using the ‘Mineralogic’ system, which is an automated ZEISS EVO•50 SEM, equipped with two Bruker xFlash 5010 EDS detectors. The analyses were carried out with an acceleration voltage of 20 kV, 13 mm working distance and 0.050 s dwell time. The analyses were carried out adopting the ‘full map’ analytical mode.
The LA-ICP-MS analyses were carried out on an ESI NWR193 UV 193 nm short-pulse-width (<4 ns) laser fitted with a TwoVol2 ablation cell and coupled to an Agilent 7700× quadrupole ICP-MS configured with dual external rotary pumps for enhanced sensitivity, located in the LODE laboratory, NHM. Ablated spots were 35–50 µm in diameter, with a fluence of 3.5 J cm−2, fired at a frequency of 10 Hz. The transport gas used was He at a flow rate of 0.5 L min−1 mixed with Ar at a flow rate of 1.1 L min−1, in a signal-smoothing device.
The element menus and ICP-MS dwell time settings that were employed to obtain the compositions of the various minerals are listed in Appendix 2 (Supplementary material, see below). Element ratios to an internal standard element (29Si for willemite and hemimorphite, 57Fe for goethite and hematite, and 66Zn for smithsonite, descloizite and sphalerite) were determined by referencing background-corrected integrated intensities from mineral signals to the external calibration standard (see Appendix 1 for details). This was GSD-1g glass (USGS) for goethite and hematite, and NIST 610 for smithsonite and descloizite. Following the procedure in Choulet et al. (Reference Choulet, Barbanson, Buatier, Richard, Vennemann, Ennaciri and Zouhair2017), the external calibration standard for willemite and hemimorphite was NIST 612. More complicated was the analytical approach to the sphalerite analysis and the choice of the appropriate external standard (see Appendix 1 for details). The main limitation of the polymetallic sulfide material MASS-1 as a standard for this work is that Ge is not certified or even included as a reference value, even though Belissont et al. (Reference Belissont, Boiron, Luais and Cathelineau2014) reported a Ge concentration of 57.8 ± 2.6 ppm in the MASS-1 (Dr S. Wilson, pers. comm. in Belissont et al., Reference Belissont, Boiron, Luais and Cathelineau2014). For this reason, the external calibration standard for sphalerite was MASS-1 for 51V, 53Cr, 55Mn, 57Fe, 59Co, 60Ni, 65Cu, 71Ga, 75As, 95Mo, 107Ag, 111Cd, 115In, 118Sn, 121Sb, 137Ba, 182W, 208Pb and 209Bi, whereas NIST 610 was used for evaluating the Ge concentration (see Appendices 1 and 2 for details). Note that the Ge value in MASS-1 (as secondary standard), obtained with external calibration by NIST 610 (Table A2 in Appendix 1), was slightly more than twice the amount reported by Belissont et al. (Reference Belissont, Boiron, Luais and Cathelineau2014) as a reference value for MASS-1. Absolute element concentrations were then calculated from internal standard element concentrations (predetermined by SEM-EDS and SEM-WDS) using the program ExLAM (Zachariáš and Wilkinson, Reference Zachariáš and Wilkinson2007). Limits of detection were set at the conventional 3σ of the background signal variation (Longerich et al., Reference Longerich, Jackson and Günther1996). GSD-1g, NIST 612, or NIST 610 were used as secondary standards; NIST 2782 and BC_28 (the in-house magnetite standard of Dare et al., Reference Dare, Barnes, Beaudoin, Méric, Boutroy and Potvin-Doucet2014) were also monitored during Fe oxide or oxy-hydroxide analysis. Time-resolved raw counts-per-second signals were screened meticulously and the longest ‘clean’ integration intervals possible (up to 60 s of signal) were retained.
Carbon and oxygen isotope analyses were performed on smithsonite and cerussite. Hand-picked carbonate mineral specimens were analysed at SUERC (East Kilbride, Scotland) on an Analytical Precision AP2003 mass spectrometer equipped with a separate acid injector system, after reaction with 105% H3PO4 under He atmosphere at 70°C. Isotopic results are reported using the conventional δ‰ notation. Mean analytical reproducibility, based on replicates of the SUERC laboratory standard MAB-2 (Carrara Marble), was ~±0.2‰ for both carbon and oxygen. The δ13C data are reported relative to V-PDB (Vienna pee dee belemnite), whereas the δ18O values of carbonates are quoted relative to V-SMOW (Vienna standard mean ocean water).
The oxygen isotopic composition of willemite was analysed on hand-picked crystals, using a laser fluorination procedure, involving total sample reaction with excess ClF3 using a CO2 laser as a heat source (>1500°C; following Sharp, Reference Sharp1990). All combustions resulted in 100% release of O2 from the silica lattice. This O2 was then converted to CO2 by reaction with hot graphite, then analysed on-line by a VG Isotech SIRA II spectrometer. Reproducibility is better than ±0.3‰ (1σ), based on repeat analyses of international and internal lab standards run during these analyses. Results are reported in standard notation (δ18O) as per mil (‰) deviations from V-SMOW standard.
Results
Mineralogy, petrography and chemistry of the ores
The mineralogical and chemical bulk compositions of the samples analysed are reported in Tables 2 and 3. Samples MI32046, MI129629 1/2, MI129629 2/2 and BM1930-372 contain sulfide minerals (sphalerite, galena and pyrite), together with oxidized phases (willemite, smithsonite, cerussite and hematite) (Fig. 3). Other samples instead consist only of oxidized minerals, mostly represented by willemite, smithsonite and hemimorphite (Table 2) (Fig. 3).
Fig. 3. Examples of modal mineralogical analyses using ‘Mineralogic’ from Zeiss. (a) MI29629 2/2: sulfides replaced by willemite 1; (b) MI29631: willemite 2 overprinted by goethite; (c) MI10900: concretionary agglomerate of descloizite, hemimorphite and pyromorphite; and (d) OR5307: smithsonite-rich specimen.
From the ore textures observed, it is possible to recognize a primary galena generation (G1) interstitial to sphalerite (Fig. 4a). G1 is almost pure, whereas sphalerite can contain up to 0.9 wt.% Fe. Renierite is included locally in sphalerite (Fig. 4b). Pyrite is disseminated between the sphalerite and galena (Fig. 4c). This mineral association is overprinted by at least three different mineral assemblages. The first assemblage consists of a first generation of massive microcrystalline willemite (W1) replacing directly sphalerite and the interstitial galena (Fig. 4d). At the reaction boundaries with the Zn-silicate, sphalerite remnants are always rimmed by thin layers of a second galena generation (G2) and covellite (Fig. 4b). Other galena microdomains are finely dispersed within the massive W1, and can be associated locally with tiny Ag-sulfide inclusions. W1 seems to have been mostly developed along the fractures and the open spaces originally occurring in the sulfide association, and does not contain major elements in its structure. The second assemblage consists of a second willemite generation (W2), which is developed in well formed crystals of variable size (in some cases 0.5 mm hexagonal crystals in geodes) (Fig. 5a). W2 is zoned chemically, with alternating Pb-rich (up to 2 wt.% Pb) and Pb-poor bands (Fig. 5b). A third assemblage is formed by (Fig. 5c,d): smithsonite, cerussite, hemimorphite, sauconite, covellite, Fe-oxyhydroxides, pyromorphite, vanadinite, descloizite, mottramite, apatite, mimetite and tarbuttite. Smithsonite and hemimorphite replace willemite W1 and sphalerite directly (Fig. 5c), and also form euhedral crystals in cavities (Fig. 5d). At the reaction boundaries between sphalerite and smithsonite, covellite is observed commonly (Fig. 4c). Agglomerates of prismatic covellite crystals also occur in cavities within sphalerite remnants. Cerussite replaces galena directly (Fig. 5c). Zinc and Pb carbonates are commonly associated with goethite and hematite, which can also replace willemite directly (Fig. 6a). Hemimorphite presents two different textures: a crustiform texture (Fig. 6b), where hemimorphite crusts alternate with smithsonite crusts, and a typical crystalline texture of fan-shaped agglomerates of euhedral tabular crystals (Fig. 6c). Smithsonite crusts are commonly Mg-rich (up to 2 wt.% Mg). Minerals like pyromorphite and tarbuttite commonly fill voids between the euhedral hemimorphite (Fig. 6c,d). None of the oxidized minerals (silicates, carbonates and vanadates) show any signs of deformation.
Fig. 4. Optical microscopy - reflected light: (a) OR5304: primary sulfides assemblage: sphalerite (Sph) with interstitial galena 1 (G1); (b) MI29629 1/2: sphalerite, containing a Ge-sulfide inclusion, replaced by willemite 1 (W1); the reaction boundary between sphalerite and willemite is marked by a thin layer of galena 2 (G2); (c) MI29629 2/2: pyrite (Py), sphalerite and galena 1 remnants after willemite 1 replacement; and (d) MI29629 2/2: willemite 1 directly replacing galena 1.
Fig. 5. Backscattered electron (BSE) images of replacement textures: (a) MI29629 2/2: concretionary agglomerate of willemite 2 (W2), with alternating Pb-poor and Pb-rich layers; (b) OR5309: hexagonal prismatic crystal of willemite 2, characterized by oscillatory chemical zoning; (c) BM1930-372: an original aggregate of sphalerite (Sph) and galena 2 (G2) replaced by carbonates: galena 2 is replaced by cerussite (Cer) and sphalerite is replaced by smithsonite; and (d) OR5307: smithsonite, locally bearing Mg, forming botryoidal crust (Sm2) and replacing dolomite crystals of the host rock (Sm1).
Fig. 6. Back-scattered electron (BSE) images of typical supergene minerals: (a) MI29631: goethite (Goe) altering a Pb-bearing willemite 2 (Pb-W2) crystal; (b) OR5314: alternating crusts of hemimorphite (Hm) and smithsonite 2 (Sm2); (c) MI10900: hemimorphite platy crystals in a cavity, followed by pyromorphite (Pyrm) crystals; and (d) OR5314: tarbuttite (Tar) vein in hemimorphite. Desc = descloizite.
By correlating the mineralogy of the samples (Table 2) with whole-rock chemical compositions (Table 3), it is possible to see that largest amounts of Fe correspond to Fe-oxyhydroxide occurrences, the largest amounts of Zn coincide with sphalerite- and willemite-bearing samples, whilst Pb is associated with galena and cerussite occurrences. The critical element, Ge, is above the detection limits in willemite- and Fe-oxyhydroxide-bearing samples and in specimens containing sphalerite. Gallium reaches tens of ppm (21 ppm) only in sample BM1930-372, which contains sphalerite and willemite. The indium concentration is above minimum detection limits only in sample MI29631, which consists largely of willemite and minor goethite. Other trace elements with significant concentrations (Table 3) are V (typically associated with vanadates), Cr (1073 ppm in a goethite-rich sample), Mn, Cu and As (hundreds of ppm in samples containing sulfides, willemite and Fe-oxyhydroxides). Cadmium reaches concentrations well above 300 ppm in samples consisting of sphalerite and smithsonite. Molybdenum and U levels are ~100 ppm in samples containing Fe-oxyhydroxides. The tarbuttite-bearing sample (OR5314) is characterized by large Mn, Cu and As contents.
Chemical compositions of mineral phases
The LA-ICP-MS analyses were carried out on sphalerite, W1 and W2, hemimorphite, hematite, goethite, smithsonite and descloizite. The mean compositions of the most abundant elements revealed in the minerals analysed are given here. In Tables 4–10, maximum, minimum and median values are also reported. Zonation was registered only in willemite (Pb-zoning), whilst the other minerals analysed were remarkably un-zoned.
Table 4. Major (WDS) and trace (LA-ICP-MS) element composition of sphalerite.
LA-ICP-MS analyses: Internal standard = Zn; External standard = MASS-1. *External standard for Ge = NIST 610.
Stand. Dev. — standard deviation; Det. Lim. — detection limit.
In two analysed samples, BM1930-372 and MI29629 2/2 (Table 4), sphalerite contains minor amounts of Fe (mean 1177 and 3759 ppm), and minor to trace amounts of Cd (mean 970 and 1038 ppm), whereas all the other elements are scarce.
Willemite W1, analysed in samples BM1930-372 and MI29629 1/2 (Table 5), was found to contain discrete trace (hundreds of ppm or less) amounts of B (mean 92 and 118 ppm), Ca (mean 58 and 59 ppm) and Cd (mean 61 and 40 ppm). In the samples of W1 analysed, both As and Ge contents are variable; respective means are 8 and 586 ppm for As and 3 and 30 ppm for Ge. W1 locally has high Pb values, related to the occurrence of G2 micro-inclusions. W2 shows a chemical zoning as stated before, with alternating Pb-rich (up to 2 wt.% Pb) and Pb-poor bands. W2 was analysed by LA-ICP-MS in two samples, OR5309 and MI29631 (Table 5), where it was found to contain similar amounts of Ca and Cd as observed in W1, with minor Ge (mean 22 and 47 ppm) and Cu (mean 4 and 30 ppm), but apparently no B. W2 Pb-rich bands generally contain more Ge, As, Ca and Mg than the Pb-poor bands.
Table 5. Major (EDS) and trace (LA-ICP-MS) element composition of willemite.
–: not evaluated; <: less than the detection limit; LA-ICP-MS analyses: Internal standard = Si; External standard, LODE_NIST 612. Stand. Dev. — standard deviation; Det. Lim. — detection limit.
Hemimorphite was analysed in three samples: MI10900, OR5314 and BM1907-987 (Table 6). In these samples hemimorphite appears to have a variable trace-element composition. In sample OR5314, where hemimorphite is associated with tarbuttite, vanadium has elevated values (mean 247 ppm V). This hemimorphite also contains significant Cu (mean 92 ppm) and Cd (mean 41 ppm). In the sample MI10900, hemimorphite is associated with descloizite, and it contains significant Pb (mean 723 ppm), Cd and Cu (mean 123 ppm Cd, 61 ppm Cu), but apparently no V. In sample BM1907-987 hemimorphite is associated with cerussite, and only contains traces of Pb (mean 104 ppm). No Ge has been detected in this Zn hydro-silicate.
Table 6. Major (EDS) and trace (LA-ICP-MS) element composition of hemimorphite.
–: not evaluated; <: less than the detection limit; LA-ICP-MS analyses: Internal standard = Si; External standard = LODE_NIST 612. Stand. Dev. — standard deviation; Det. Lim. — detection limit.
Goethite and hematite have been analysed in six samples (Tables 7 and 8): BM1930-372, MI29631, OR5305, OR5309, MI10900 and MI29629 2/2. Fe-oxyhydroxides contain major (~1 wt.%) amounts of Zn, Si and Pb, and minor amounts of Al. Goethite is shown to be richer in Zn than hematite. Goethite and hematite contain hundreds of ppm of several metals (Ti, V, Cr, Mn, Co, Ni, Cu, Ge, As, Y, Mo, Cd, In, Sb, W and U), variously distributed in these minerals in the analysed samples, yet Ge appears to be very low (Tables 7 and 8). In both these Fe minerals, significant Ga was only found in sample BM1930-372 (max 138 ppm). The indium content is high in sample MI29631 (max 46 ppm in goethite and 60 ppm in hematite).
Table 7. Major (EDS) and trace (LA-ICP-MS) element composition of goethite.
–, not evaluated; <, under detection limit; LA-ICP-MS analyses: Internal standard = Fe; External standard, LODE_GSD-1G. Stand. Dev. — standard deviation; Det. Lim. — detection limit.
Table 8. Major (EDS) and trace (LA-ICP-MS) element composition of hematite.
–: element not detected; <: less than the detection limit; LA-ICP-MS analyses: Internal standard = Fe; External standard = LODE_GSD-1G. Stand. Dev. — standard deviation; Det. Lim. — detection limit.
The smithsonite trace-element composition was evaluated in sample BM1930-372 (Table 9). This Zn-carbonate contains major amounts of Mg (mean ~2 wt.%), and traces of Fe, Cd and Pb. Descloizite contains trace amounts of Ca, Cu and As (mean values in sample OR5303 of 726 ppm Ca, 2475 ppm Cu and 936 ppm As; Table 10). No Ge, In or Ga was detected in the smithsonite and descloizite analysed.
Table 9. Major (EDS) and trace (LA-ICP-MS) element composition of smithsonite.
–: not evaluated; <: less than the detection limit; LA-ICP-MS analyses: Internal standard = Zn; External standard, LODE_NIST 610. Stand. Dev. — standard deviation; Det. Lim. — detection limit.
Table 10. Major (EDS) and trace (LA-ICP-MS) element composition of descloizite.
–: not evaluated; <: less than the detection limit; LA-ICP-MS analyses: Internal standard = Zn; External standard = LODE_NIST 610. Stand. Dev. — standard deviation; Det. Lim. — detection limit.
Stable isotope geochemistry
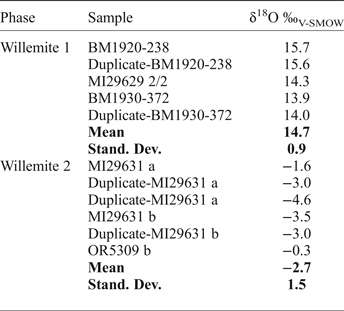
Oxygen isotope compositions have been determined for both W1 and W2 (Table 11), smithsonite (replacing sphalerite and crystals in geodes), and cerussite (replacing galena) (Table 12). W1 yields δ18O compositions of between 13.9 and 15.7‰, with a mean value of 14.7 ± 0.9‰ (1σ; n = 5). In marked contrast, W2 is characterized by negative δ18O values, ranging between −4.6‰ and −0.3‰, with a mean value of −2.7 ± 1.5‰ (1σ; n = 6). δ18O compositions of smithsonite that has replaced sphalerite, and smithsonite crystals are similar and range between 20.9‰ and 22.8‰ (all carbonate O isotope data discussed vs. the V-SMOW standard), whereas the single measurement from cerussite has a δ18O of 13.1‰. By using the cerussite-water and smithsonite-water oxygen isotope fractionation equations of Gilg et al. (Reference Gilg, Boni, Hochleitner and Struck2008), and the willemite-water oxygen isotope fractionation equation of Zheng (Reference Zheng1993):
it is possible to plot the δ18O values of the analysed phases in a temperature vs. water δ18O composition diagram (Fig. 7). Willemite W1 and smithsonite paths can be seen to cross at more than 300°C and more than 15‰ fluid δ18O (Fig. 7a), whereas smithsonite and cerussite paths cross at ~30°C and ~−8‰ fluid δ18O (Fig. 7b). W2 does not cross any W1, cerussite or smithsonite path. δ13C compositions of smithsonite and cerussite are always negative, ranging from −11.1 to −3.6‰.
Table 11. Oxygen isotope composition of willemite.
Table 12. C- and O-isotope compositions of smithsonite and cerussite.
Discussion
Mineral paragenesis and genetic processes
The petrography conducted on the selected specimens from the NHM ore collection confirmed the complex paragenesis reported in previous studies (Kamona, Reference Kamona1993; Kamona and Friedrich, Reference Kamona and Friedrich2007; Terracciano, Reference Terracciano2008). In agreement with the previous studies, two willemite generations were recognized: the first directly replaces the sulfides, and a second leading to well formed crystals in cavities. We also find that these two willemite phases were followed paragenetically by minerals consistent with a supergene alteration assemblage. Unlike previous studies (i.e. Kamona and Friedrich, Reference Kamona and Friedrich2007; Terracciano, Reference Terracciano2008), we have found no evidence in our sample suite that W2 is co-genetic with the minerals typically recognized as supergene (e.g. goethite, hematite and smithsonite). In fact, all the textures identified in the samples analysed, even though compatible with a coeval precipitation of W2 with Zn-Pb-carbonates, Fe-oxyhydroxides and vanadates, were probably the result of an ‘overprinting’ process, where smithsonite, cerussite, hemimorphite, goethite, hematite, descloizite, etc., formed together, almost at the same time, ‘above’ and ‘in the spaces between’ pre-existing minerals (i.e. the sulfides and the two willemite phases). For this reason, in the paragenetic scheme represented in Fig. 8, we suggest that the original sulfide association was altered by at least three oxidation stages: two stages that were dominated by the formation of willemite, and another characterized by weathering-related processes. The existence of these three stages is also evident when looking at the stable oxygen isotopes. When plotting the mineral-water equilibrium curves in a temperature vs. water δ18O diagram (Fig. 7), the paths of co-genetic minerals cross at points indicating their precipitation temperature and the δ18O composition of precipitating fluids. In Fig. 7, it is possible to see that smithsonite (Sm1) and cerussite paths occupy a similar area, crossing at average ambient temperatures (~30°C) as expected, confirming that these two mineral species precipitated from weathering-related fluids. In contrast, W1 and W2 fractionation curves plot in two distinct areas of the diagram. W1 curves cross smithsonite curves at >300°C, an unreasonable estimate for smithsonite precipitation (Takahashi, Reference Takahashi1960; Sangameshwar and Barnes, Reference Sangameshwar and Barnes1983; Gilg et al., Reference Gilg, Boni, Hochleitner and Struck2008), confirming that W1 and smithsonite are not co-genetic. W2 paths cover an area of the graph very far from those of the other minerals, clearly out of a possible isotopic equilibrium with any other phase. Whilst not diagnostic, these isotopic data confirm that: (1) the two willemite generations are not co-genetic; and (2) neither W1 nor W2 is co-genetic with smithsonite or cerussite. Assuming this deduction, it follows that precipitation of willemites and carbonates occurred: (1) from waters characterized by different isotopic composition; (2) from fluids having different temperatures; (3) at different times; or (4) one or more combinations of the previous three options.
Fig. 7. (a) Mineral–fluid fractionation curves based on δ18O ‰ V-SMOW of willemite 1 (red lines), willemite 2 (green lines), smithsonite 1 replacing sphalerite (magenta line), smithsonite 1 replacing dolomite (yellow lines), smithsonite 2 (light blue line), cerussite replacing galena (deep blue line). (b) Enlargement of (a).
Fig. 8. Paragenesis of the hypogene and supergene minerals detected in the samples analysed from the Kabwe ores.
Looking more in detail at the isotopic compositions of the supergene carbonate minerals, the smithsonite average composition (~21.9‰ V-SMOW) is seen to be less than that of supergene smithsonites from other mining areas of the world (~25–30‰ V-SMOW; Gilg et al., Reference Gilg, Boni, Hochleitner and Struck2008; Boni and Mondillo, Reference Boni and Mondillo2015). Cerussite (13.1‰ V-SMOW) also has smaller δ18O values than other supergene cerussite (~15–20‰ V-SMOW; Gilg et al., Reference Gilg, Boni, Hochleitner and Struck2008). Considering that smithsonite and cerussite were deposited at average ambient temperatures (~30°C), by using the cerussite-water and smithsonite-water oxygen isotope fractionation equations of Gilg et al. (Reference Gilg, Boni, Hochleitner and Struck2008), we calculated δ18O compositions for the precipitating fluid of between ~−8 and ~−10‰. These δ18O compositions for precipitating fluids, though slightly lower than the range of compositions already determined for mineralizing waters in other non-sulfide districts by Gilg et al. (Reference Gilg, Boni, Hochleitner and Struck2008), fall within the wider range of ‘unusual’ non-sulfide deposits already shown by Boni and Mondillo (2015, and references therein). In this case, the isotopic signatures of the Kabwe precipitating water must be related to the particular climatic conditions and to the location of the deposit at the time of the supergene alteration. Although a late Eocene–Oligocene age has been produced for the Kabwe descloizite (~20–37 Ma; N.J. Evans, unpubl., in Boni et al., Reference Boni, Terracciano, Evans, Laukamp, Schneider and Bechstädt2007), by analogy with other supergene deposits occurring in this region (e.g. supergene Cu-Co and manganese deposits in the Katanga region; Dewaele et al., Reference Dewaele, Muchez, Vets, Fernandez-Alonzo and Tack2006; Decrée et al., Reference Decrée, Deloule, Ruffet, Dewaele, Mees, Marignac, Yans and De Putter2010; Decrée et al., Reference Decrée, Pourret and Baele2015; De Putter et al., Reference De Putter, Ruffet, Yans and Mees2015) and, more in general, in the southern African craton (Pack et al., Reference Pack, Gutzmer, Beukes, van Niekerk and Hoernes2000; Boni et al., Reference Boni, Terracciano, Evans, Laukamp, Schneider and Bechstädt2007; Gutzmer et al., Reference Gutzmer, Du Plooy and Beukes2012; Arfè et al., Reference Arfè, Boni, Balassone, Mondillo, Hinder and Joachimski2017, and references therein), the ~300–500 m deep (Kamona and Friedrich, Reference Kamona and Friedrich2007) supergene alteration profile at Kabwe could have formed during a period longer than the single descloizite age, possibly starting in the Late Cretaceous–early Eocene and extending until the Mio–Pliocene. In this timeframe, a tropical-humid climate persisted in the region and laterite profiles developed in the Katanga region (Giresse, Reference Giresse2005). At the same time, the region experienced various uplift stages, and reached the present height (~1000 m a.s.l.) in the Miocene–Pliocene (De Putter et al., Reference De Putter, Ruffet, Yans and Mees2015, and references therein). These conditions, together with the location of the Kabwe area in the inland of the southern African craton, could have produced groundwaters characterized by very negative δ18O V-SMOW compositions (Mazor, Reference Mazor2004; Edmunds, Reference Edmunds, Aggarwal, Gat and Froehlich2005).
The nature of the fluids that precipitated the two willemite generations cannot be determined easily because the two silicates are not co-genetic and are not associated with any other oxidized mineral. Kamona and Friedrich (Reference Kamona and Friedrich2007) consider the first willemite generation to be hydrothermal, and the second generation to be supergene, whereas Terracciano (Reference Terracciano2008) considers either a supergene or low-temperature hydrothermal origin for both of the willemite generations recognized here. Comparison of the δ18O compositions of the Kabwe willemite with willemites from other deposits of the world is hampered by lack of data. At present, δ18O compositions of willemite have been only measured at Sterling Hill (New Jersey, USA) (7.4–11.4‰ V-SMOW; Johnson et al. Reference Johnson, Rye and Skinner1990), at Vazante (Minas Gerais, Brazil) (10.9–13.8‰ V-SMOW; Monteiro et al., Reference Monteiro, Bettencourt, Spiro, Graca and Oliveira1999), and at Bou Arhous (L'oriental Region, Morocco) (5.3–7.8‰ V-SMOW; Choulet et al., Reference Choulet, Barbanson, Buatier, Richard, Vennemann, Ennaciri and Zouhair2017), where willemite is considered to be precipitated from hydrothermal fluids (Johnson et al. Reference Johnson, Rye and Skinner1990; Monteiro et al., Reference Monteiro, Bettencourt, Spiro, Graca and Oliveira1999; Choulet et al., Reference Choulet, Barbanson, Buatier, Richard, Vennemann, Ennaciri and Zouhair2017). Looking at the W1-water oxygen isotope fractionation curves, we see that for a limited range of temperatures only (~0 to 40°C), the precipitating fluids would be characterized by the negative or slightly positive δ18O compositions generally indicative of a relatively unaltered surface fluid origin in tropical latitudes. At temperatures greater than this, the measured W1 paths depict distinctly positive δ18O compositions for the fluids. This, combined with the textural characteristics observed in field by Kamona (Reference Kamona1993), and with the similarity between the δ18O compositions of the Kabwe W1 and the previously reported hydrothermal willemites, strongly suggests that W1 is also of hydrothermal origin, given that the fluid O isotope compositions demand significant water–rock interaction to obtain the positive values. W2 compositions map negative fluid δ18O compositions at temperatures below ~200°C, and only slightly positive values if they were precipitated at T > 200°C. δ18O of the precipitating fluids could be very negative (from −20‰ to −15‰ V-SMOW) at ambient temperatures, and slightly less negative (−10‰ V-SMOW) for temperatures of >60°C (hydrothermal fluids). Willemite with textures similar to W2 (i.e. euhedral hexagonal habit, with chemical oscillatory zoning) has been observed at Star Zinc (willemite II; Terracciano, Reference Terracciano2008; Boni et al., Reference Boni, Terracciano, Balassone, Gleeson and Matthews2011), at Tres Marias (Saini-Eidukat et al., Reference Saini-Eidukat, Melcher and Lodziak2009, Reference Saini-Eidukat, Melcher, Göttlicher and Steininger2016), and at Bou Arhous (Choulet et al., Reference Choulet, Barbanson, Buatier, Richard, Vennemann, Ennaciri and Zouhair2017), where the mineral was considered to be of hydrothermal origin. If this was the case here, then clearly it was a distinctly different hydrothermal event compared to W1, given that the W2 precipitating fluid O isotope compositions, preserving a meteoric signature, are distinctly different from W1.
According to Kamona and Friedrich (Reference Kamona and Friedrich2007), W1 may have formed shortly after the Lufilian Orogeny (~550 Ma). Considering that the present data confirm that W1 at Kabwe is of hypogene origin, we agree with the latter authors on a possible post-‘Lufilian’ age for this phase. Boni et al. (Reference Boni, Terracciano, Balassone, Gleeson and Matthews2011) proposed the same age for another Zambian willemite mineralization, located at Star Zinc (willemite I; Terracciano, Reference Terracciano2008; Boni et al., Reference Boni, Terracciano, Balassone, Gleeson and Matthews2011), near Lusaka. The age of W2 is more difficult to constrain, but considering the ‘meteoric’ signature of the precipitating fluids, it probably formed when the sulfide deposit was already exhumed, possibly in association with, or after, major Cretaceous uplifts (De Putter et al., Reference De Putter, Ruffet, Yans and Mees2015 and references therein).
Critical-elements deportment
The critical elements Ga, In and Ge have a different deportment in the various minerals detected in the samples analysed. Gallium reaches tens of ppm only in sample BM1930-372, where the element has been detected directly in sphalerite, W1, goethite and hematite using LA-ICP-MS. Gallium concentrations in these minerals are not very high, but account for Ga levels measured in the bulk sample. Indium concentrations are above the detection limits only in sample MI29631, where it is found mostly in goethite and hematite, and in trace amounts in willemite. In this case, there is a slight disagreement between the bulk-rock composition and In levels calculated using a combination of the PXRD mineral abundances and direct data from LA-ICP-MS of Fe-oxyhydroxides and willemite. This mismatch is probably due to errors in evaluating the average In concentration in willemite (which is the most abundant mineral in the sample). No In was detected in the sphalerite analysed. Germanium is above the detection limits in willemite- and Fe-oxyhydroxide-bearing samples, and in specimens containing sphalerite and willemite; moreover, it has been detected as discrete Ge-sulfides. Through LA-ICP-MS analysis, significant Ge ppm levels have been found in W1 and W2, and small traces of it in the supergene minerals (hemimorphite, goethite and hematite). The LA-ICP-MS data from these minerals together with mineralogical abundance account for the measured bulk-rock Ge contents, but note that Ge discrete minerals (e.g. sulfides or Ge-Fe-oxyhydroxides; see Melcher, Reference Melcher2003), may have been missed during our microanalytical study and could have created a slight mismatch between the measured bulk-rock Ge concentrations and mineral-phase analysis coupled with LA-ICP-MS data.
From these data, it is possible to infer that a remobilization of the elements occurred during the various stages of oxidation of the primary sulfides. Assuming that Ga, Ge and In originally occurred in sulfides, it appears that, when sulfides were initially altered to form the first willemite generation, W1 was only able to incorporate Ge and limited Ga. During the second oxidation stage, only Ge and limited In were incorporated into W2. Finally, during supergene oxide formation, Ga, In and Ge were comprehensively incorporated in different amounts into the Fe-oxyhydroxides. These different deportments among the various phases are probably controlled by three main factors: (1) differences in initial element abundance in the mineralization (Ge was probably more abundant than Ga and In in the primary sulfides); (2) variability in element solubilities at different temperatures and pH; and (3) the diverse affinity of the elements to the range of minerals formed.
At 25 and 100°C, the solubility of α-GaOOH has a minimum between pH = 2 and pH = 6 and increases at pH <2 and pH >6, solubility of GeO2 is independent of pH at pH <8 and increases at pH <8, whereas the solubility of various In compounds has a broad minimum from pH ~4 to pH ~9 and increases for smaller and greater pH values (Wood and Samson, Reference Wood and Samson2006). In addition, note that the most common oxidation states of Ga, In and Ge in aqueous solution are Ga3+, In3+ and Ge4+, and that at neutral pH, Ge is more soluble than both Ga and In (Wood and Samson, Reference Wood and Samson2006). It follows that under certain conditions, the alteration of Ga-In-Ge-bearing sulfides can produce Ge-rich Ga-In-poor solutions, with Ga and In probably forming residual compounds in sulfides or oxyhydroxides. Germanium incorporation in willemite is not unusual, having already been observed in various Zn-silicate deposits, e.g. Franklin (Sheffer, Reference Sheffer1966), Tsumeb (Lombaard et al., Reference Lombaard, Günzel, Innes, Krüger, Anhaeusser and Maske1986), Beltana (Groves et al., Reference Groves, Carman and Dunlap2003), Bou Arhous (Choulet et al., Reference Choulet, Barbanson, Buatier, Richard, Vennemann, Ennaciri and Zouhair2017) and Tres Marias (Saini-Eidukat et al., Reference Saini-Eidukat, Melcher and Lodziak2009, Reference Saini-Eidukat, Melcher, Göttlicher and Steininger2016). In particular, Saini-Eidukat et al. (Reference Saini-Eidukat, Melcher, Göttlicher and Steininger2016) have described the structural occurrence of Ge in the Tres Marias willemite in detail, and have shown that “Ge in willemite occurs as Ge4+ and is four-fold coordinated with oxygen, as expected because the ionic radius of Ge4+ (0.44 Å) is close to that of Si4+ (0.39 Å) and isomorphism of Ge4+ with Si4+ is most probable”. The fact that W1 and W2 contains more Ge than Ga and In could be related to the strong geochemical affinity between Ge and Si, but could also indicate that during the formation of W1 and W2, the Ge concentration in the precipitating fluids was greater than that of Ga and In. Willemite is stable at pH >7 in oxidizing environments (McPhail et al. Reference McPhail, Summerhayes, Welch, Brugger and Roach2003), and if solutions were particularly rich in Ge, Ge-bearing willemite could form in those conditions. Associated Fe-oxyhydroxides could also scavenge a range of elements having chemical affinities with Fe (Wood and Samson, Reference Wood and Samson2006, and references therein). However, the present results suggest that Ge-bearing goethite is absent from the samples analysed, and both goethite and hematite have greater average Ga concentrations than either Ge or In. Considering the solubilities of the three elements, and assuming that the Fe-oxyhydroxides formed at ambient temperature and near-neutral pH (Wood and Samson, Reference Wood and Samson2006), one explanation could be that during the last stage of supergene alteration of sulfides, Ge was leached progressively from the sulfides, whilst the more immobile Ga (under those pH and T conditions), was incorporated residually in newly formed Fe-oxyhydroxides, similar to the process which enriches Ga passively into bauxites (Wood and Samson, Reference Wood and Samson2006). The same process could also explain the In concentration in the Fe-oxyhydroxides.
The present study revealed that at Kabwe, Fe-oxyhydroxides also host variable amounts of a range of other elements (Ca, Ti, V, Cr, Mn, Co, Ni, Cu, Ge, As, Y, Mo, Cd, In, Sb, W and U), which could derive from weathering of the host rocks or the primary sulfide assemblage. Note that the elements Co, Ni, Cu, As, Mo, Cd, Sb and W, which have been not detected in the sphalerites or willemites analysed (and are unlikely to be present in the host rock at Kabwe or in other deposits of similar type, e.g. Tsumeb and Khusib Springs), are normally contained in Ge-bearing sulfides (Melcher, Reference Melcher2003).
Conclusions
Petrography has confirmed the complex paragenesis reported in previous studies on the Kabwe deposit. In the newly analysed samples, the relict sulfides consist mostly of sphalerite, containing Ge-sulfide inclusions, associated with ‘interstitial’ galena. The present authors suggest that this original sulfide association was altered by at least three oxidation stages: two stages that were dominated by the formation of willemite, and another characterized by deep weathering-related processes. The existence of these three stages is suggested by the oxygen isotope data, consistent with textural observations. δ18O compositions of smithsonite (Sm1) and cerussite (replacing galena) suggest that both mineral species precipitated from weathering-related fluids. W1 and W2 have markedly different δ18O compositions suggesting that the two willemite generations are not co-genetic. The δ18O compositions and the textural characteristics of W1, resonating with those of previously reported hydrothermal willemites, strongly indicate a hydrothermal origin for W1. The distinct δ18O composition of W2 indicates that this silicate precipitated from isotopically light fluids, probably of meteoric origin. There is no evidence to support either a supergene or a low-T hydrothermal origin for W2.
The critical elements Ga, In and Ge have a different deportment in the various minerals. Gallium has been detected at low ppm levels in sphalerite, W1, goethite and hematite, detectable but low values of In are found in goethite and hematite, whereas Ge forms proper sulfides, and occurs at low ppm levels in both W1 and W2, and in small traces in clearly supergene minerals (hemimorphite, goethite, hematite). The diverse deportments among the various phases are probably due to different initial Ga, In and Ge abundances in the sulfide mineralization, the different solubilities of the three elements at different temperatures and pH values, and finally to their different affinity for the neoformed mineral phases.
Acknowledgments
N. Mondillo and co-authors are indebted to the former curator of the Natural History Museum ore collection, Helena Toman, for granting access to the Kabwe specimens, and for help during sample selection. The authors are also grateful to the staff of the Core Research Laboratories at the NHM and to the staff of SUERC Stable Isotope Laboratory for support during the analytical work. The authors are indebted to Peter Williams and Roger Mitchell, Principal Editors and to John Bowles (Guest Principal Editor), Mineralogical Magazine, for their handling of the manuscript, and to Frank Melcher and an anonymous reviewer for comments and suggestions, which greatly enhanced the quality of the paper. This project has received funding from the European Union's Horizon 2020 research and innovation program, in the form of a Marie Skłodowska-Curie Individual Fellowship (Project Number 660885) awarded to R. Herrington, supporting the fellowship of N. Mondillo.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/minmag.2017.081.038




















 Open access
Open access