Science is typically slow work. It can take years or even decades for exploratory work on, say, a new concept in materials to become a product ready for the market place. But advances in artificial intelligence (AI) have the potential to greatly accelerate that tortuous process. Computer algorithms are increasingly helping with the exploring and understanding, and the direction of experimentation, modeling, and simulation. They are working in parallel with human creativity and ingenuity to find and refine new materials for tomorrow’s technologies. Launching one effort to harness computational and data-driven resources, the Materials Genome Initiative, in 2011, US President Barack Obama laid out the objective. “The invention of silicon circuits and lithium-ion batteries made computers and iPods and iPads possible,” he said, “but it took years to get those technologies from the drawing board to the market place. We can do it faster.”
Yet sometimes the choices available to materials scientists are enough to make you despair. Take so-called “high-entropy alloys,”Reference Tsai and Yeh1 which have high strength and are mixtures of five or more metallic elements; some may contain up to 20 elements. How can one ever hope to probe all the possible permutations and phases? Or take the exotic quantum mechanical properties discovered during the past decade or so in complex materials with compositions such as Ca10Cr7O28 and YbMgGaO4.Reference Balz, Lake, Reuther, Luetkens, Schönemann, Herrmannsdörfer, Singh, Nazmul Islam, Wheeler, Rodriguez-Rivera, Guidi, Simeoni, Baines and Ryll2,Reference Paddison, Daum, Dun, Ehlers, Liu, Stone, Zhou and Mourigal3 How can we find out in any comprehensive, systematic way what other new and potentially useful behaviors might exist in combinations of elements that no one has thought to look at before?
It is unfeasible to trawl blindly through all the options experimentally. Previously, the options were narrowed down largely through intuition. But human intuition becomes severely tested by both the range and complexity of the possible choices.
Yet computer algorithms can now develop a kind of intuition too through the same process that we tend to use: looking for patterns and regularities in what we already know. This is machine learning (ML), an aspect of AI that aims to digest and generalize existing knowledge to find new solutions to problems. It is used today in all manner of applications in which a large amount of data exceeds human capability to assimilate it all—from genomics and drug design to analysis of financial markets and the development of game-playing algorithms. It seems increasingly likely that some of the outstanding challenges in materials design will be solved this way too.
The potential effect of AI in materials science, however, extends well beyond the discovery of new substances and compositions. Far from being merely a tool for automated materials exploration, said Benji Maruyama of the Air Force Research Laboratory in Dayton, Ohio, AI might supply nothing less than a new way of doing science, helping to improve, streamline, and guide the process of acquiring new knowledge about the materials universe (see the Materials Research Society OnDemand® Webinar at mrs.org/ondemand-ai). “We are on a long-term trend towards more AI being integrated into the research process,” said Patrick Riley, an AI researcher at Google.
“Machine-learning algorithms could help eliminate bottlenecks that appear in the research process,” said Kristofer Reyes, a computational materials scientist at the University at Buffalo, The State University of New York. Algorithms that learn from experience might help researchers to choose and design experiments, analyze the results, and generalize the knowledge extracted. And to judge from experience in other areas, such as the autonomous game-playing capabilities of AI, it is possible that such systems could take genuinely creative leaps beyond the bounds of human intuition. In the years to come, the relationship between humans and computers might be realigned: defined no longer in terms of users and tools, but as a collaboration. “Certainly, ML can predict materials with desirable properties,” said Bryce Meredig of Citrine Informatics in Redwood City, Calif., a company that has developed a commercially available AI platform for materials informatics. “But if we think about a future in which all materials scientists have ML-based co-pilots, just like in our daily lives with Alexa and Google Assistant, the possibilities are much broader.”
“We’re still at the beginning of this journey,” cautioned Riley. But he feels that if the technical challenges can be met, AI could become an “amazing research assistant.”
The next generation of materials discovery
The most familiar and, in some ways, currently the most advanced applications of AI are in discovery science, where computational resources are used to analyze and learn from volumes of data that far exceed the ability of humans to process.
For thousands of years, new materials were discovered by random trial and error—essentially by accident. That was probably how glass and metals such as iron were first prepared in anti quity, and little had changed up until the 19th century, when Thomas Edison sought materials for his incandescent bulb filaments by trying out anything he could lay his hands on.
As the properties of materials became better understood and quantified in mathematical models and equations, it became possible to bring to bear some element of design to this quest, for example, by extrapolating observed relationships between composition and function. Materials such as advanced composites could be planned based on known determinants of crack formation and propagation, leading to the advent of fiber-reinforced materials in the mid-20th century.
An ability to calculate properties from first principles, for example from classical approximations of interatomic forces or, more recently, from ab initio quantum mechanics, ushered in the modern era of materials rationally designed with the aid of computer calculations. In principle, vast swathes of materials space can be mapped out this way without having to synthesize and purify anything in the lab.
But there are limits to what even super-computers can achieve in such a prospecting venture, and what’s more, there are limits to how reliably some materials properties can be predicted this way. All the same, materials discovery and design informed by computation of properties such as electronic band structure and cohesive energies have given rise to materials such as advanced alloys and electrically or optically active polymers.
Now the challenge of making new materials is progressing to what some researchers see as a fourth stage.Reference Agrawal and Choudhary4 It is being driven by three factors: access to larger sets of data on materials structure–function relationships than in the past; improvements in the scope and reliability of computer modeling and simulation; and algorithms that permit ML, which can analyze big data sets to extract trends, laws, and principles beyond the reach of human intuition. The use of ML in materials discovery makes no claims yet to mimic the processes of human intelligence: on the whole this is merely a matter of using rather literal-minded algorithms to generalize knowledge gleaned from existing data sets in order to make predictions about how new substances will behave. All the same, the approach is already proving fertile for finding new materials with useful properties. And, ultimately, it might go beyond number-crunching interpolation and start to seek out general principles governing the relationships between composition, structure, and function.
From correlation to prediction: Supervised and unsupervised learning
The use of ML techniques acknowledges that the range of possible substances—the materials universe—is much vaster than the human mind can encompass or than the experimentalist can hope to explore empirically. In this regard, materials scientists face the same challenge as chemists, who can’t hope to explore experimentally more than a tiny corner of the space of all possible molecules, and so need help with navigating the options and focusing on the regions that show the greatest promise for a given application, such as drug action.Reference Butler, Davies, Cartwright, Isayev and Walsh5
The aim here is to learn from experience—to try to deduce which materials might be promising based on those that have proved successful already. Take thermoelectric materials, which can either convert heat into electricity or use electricity to absorb heat. One of the most efficient thermoelectric materials known so far is lead telluride, but the presence of toxic lead limits potential applications. Many other candidate thermoelectrics have been identified, but the criteria for a commercially viable product—adequate performance from constituents that are neither expensive nor toxic—are demanding. Rather than using quantum mechanical calculations to blindly crunch out the electronic properties of potential materials, seeking the characteristics needed for a good thermo-electric, ML algorithms would look for correlations between the composition, structure, and properties of those currently known to make inferences about other materials that might work. These empirical correlations between microscopic and macroscopic properties are called descriptors. Once they have been deduced and tested by algorithms for materials known to have the desired properties, they can be applied to other materials in a database to find new candidates.
In this way, ML can fill in gaps, making deductions from known data that can be extended to unknown materials and compositions. “It is best used for situations where we have no understanding or predictive algorithms, but for which we have a lot of data available,” said Gerbrand Ceder of the University of California at Berkeley. “These are the kind of problems where ML can, in principle, shine.”
In general, ML algorithms use part of the available data set for learning to look for relationships between the input data and the desired properties. The other known data are then used for model validation and optimization to see how well the ML model predicts the target property for cases where the answer is known. Once the appropriate descriptors are identified, they can then be used to seek new candidate materials among those known but not tested for the property of interest, whether that be performance as a battery cathode materialReference Ceder6 or topological insulator behavior.Reference Yang, Setyawan, Wang, Buongiorno Nardelli and Curtarolo7
The most common approach is called “supervised learning.” This assumes that you know what you are looking for—the value of Young’s modulus or simply a classification of materials into metals, semiconductors, and insulators. You identify the necessary parameters (e.g., valence states or atomic radii of the constituent atoms) for that property. Then the algorithm searches for some functional relationship between the inputs and the outputs—the descriptor appropriate to the problem at hand. Once this relationship has been refined to the required degree of fidelity, it can be tested for making predictions on known materials.
Supervised learning can be strengthened in an approach called deep learning, in which the training data include not only many examples of the thing to be recognized (e.g., a cat), but also negative examples (e.g., things that are not cats). “Deep learning is essentially function fitting to training data ‘on steroids,’ ” said computer scientist Carla Gomes of Cornell University in Ithaca, N.Y.
Supervised learning compels you to make some choices about which factors you think matter to the target property. “You tend to put in things that you think are important—you’re testing a hypothesis,” said Kristin Persson of the University of California, Berkeley. To check that you’re getting sensible results, you might also put in parameters that you think absolutely should not correlate; to take an absurd example, the alphabetical order of the compound names, so that if the ML model finds there is a correlation, then you’d have to get suspicious.
“Once we were machine-learning Young’s modulus with about three thousand data points,” said Persson. “An initial set of descriptors included the space-group label of the structures. That has zero importance—it’s an arbitrary assignment.” But it was found to be the second most important descriptor. “This suggests there is an artificial correlation through something else,” said Persson. “You sometimes get these pathological correlations.” That’s why you need intuition at both ends of the learning process—to choose the parameters and to decide what to look for.
Sometimes, though, there can be a benefit in standing back and letting the machine decide what matters. So-called unsupervised learning doesn’t make any assumptions about what we’re looking for. Given a bunch of materials with particular compositions or other input parameters and with a range of observed properties, the algorithm looks for trends or clusters between the two, with no particular preconceptions of what you’re looking for or what matters for a given property. “Unsupervised algorithms are typically when you have no labels and you are just looking for patterns in the data,” said Olexandr Isayev of The University of North Carolina at Chapel Hill. “Arguably, unsupervised methods are much more powerful. This is, after all, closer to how the human brain works.”
For example, Shou-Cheng Zhang of Stanford University in California and co-workers applied unsupervised learning to existing experimental data on approximately 60,000 inorganic 2-, 3-, and 4-component compounds.Reference Zhou, Tang, Liu, Pan, Yan and Zhang8 The algorithm produced a vast, multidimensional but sparse matrix of atom–environment pairs encoding similarities in composition between the compounds formed by different types of atoms. In effect, each atom has an associated vector for which the dimensions are abstract quantities learned from scratch by the algorithm. Some of these vector dimensions loosely correlate with known properties of the elements concerned—one, for example, strongly predicts nonmetal behavior and another metallic behavior. But the algorithm decides which vector components to heed. The algorithm accurately identifies the family groupings familiar from the periodic table, for example, the kinship of halogens or alkali metals. The atom-wise properties turn out to effectively predict the characteristics of compounds, such as whether they are metals or insulators.
“When dealing with complex materials, I believe unsupervised ML works best, said Zhang, whose recent death at the age of 55 shocked and saddened many in the community of condensed-matter physics. His team is now trying the same approach to look for structure–property relationships in antibody proteins, which might reveal useful information for biomolecular recognition and drug design.
Once you have conducted your search for promising materials, selecting the best candidate for further exploration and attempted synthesis isn’t always an easy task. Sometimes it involves a compromise between properties, which might factor in cost. Veronique Van Speybroeck of Ghent University in Belgium and her co-workers have developed an algorithm for ranking materials in computational screening in which a “win fraction” for each candidate quantifies the fraction of the tradeoff with another candidate that favors the one under consideration, summed over all the design criteria.Reference Lejaeghere, Cottenier and Van Speybroeck9 A ranking can then be created by finding the minimum of the win fraction for each candidate with respect to all of the others: the larger this minimum, the better the tradeoff of desirable properties. Van Speybroeck and her colleagues showed that their approach produces intuitively sensible results in a computational search for economical metals and alloys with high mass density and for finding candidate materials with an optimal balance of hardness, thermal resistance, and cost.
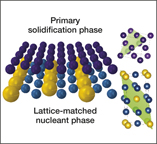
Resources for accelerating materials science are also being developed in the private sector. Citrine Informatics hopes that their materials database will assist not just in the discovery but also in the commercialization of materials in areas ranging from batteries and photovoltaics to aerospace superalloys and screen coatings for personal electronics. This AI resource has already been used to identify nucleating agents for producing crack-free microstructures in three-dimensional (3D) printed aluminum alloys (see Figure 1),Reference Martin, Yahata, Hundley, Mayer, Schaedler and Pollock10 and industrial customers, ranging from BASF and Boeing to Panasonic, are using Citrine’s database to seek materials for applications such as catalysis, aerospace engineering, and electronics.
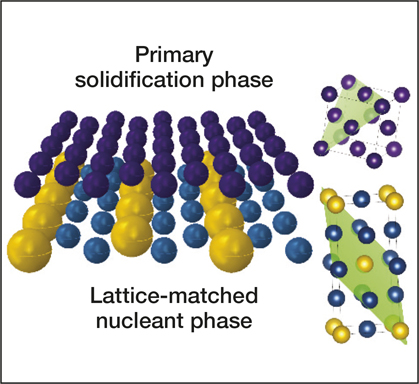
Figure 1. Schematic representation of how lattice-matched nanoparticles (bottom phase in blue and yellow) induce low-energy-barrier epitaxial growth of solidifying metals (top phase in purple), with lattice-matched planes in the unit cells, indicated in green on the right. Reprinted with permission from Reference Reference Martin, Yahata, Hundley, Mayer, Schaedler and Pollock10. © 2017 Macmillan Publishers Ltd.
Searching for structure
One of the most basic problems in materials science is the prediction of structure. In 1988, the editor of Nature pronounced it a “scandal” that a priori computational prediction of crystal structures was not possible. That situation is somewhat better today, but it is still far from routine. A typical computational approach is to take a given material composition (e.g., the ratios of elements), and calculate the energies of all possible unit cells to see which one has the lowest energy, which takes a lot of time and effort.
But what if there is some relationship between composition and structure, so that, for example, materials with particular types of valence states, stoichiometries, and ratios of atomic or ionic radii tend to favor specific structures? Then ML might be able to make a good guess at the structure from the features of the constituents.
Indeed, “this is a perfect problem for ML,” according to Ceder. Crystal structure databases have more than 100,000 structure assignments, so, in principle, a machine should be able to predict structure by learning from these data. In 2006, Ceder and his colleagues described a ML algorithm that enabled structure prediction for a wide range of binary-metal alloys.Reference Fischer, Tibbets, Morgan and Ceder11 They found that not only were particular structures correlated with the size ratio of the component atoms, but the structure adopted by a binary combination at one stoichiometry was a good predictor of the structure of the same pair at another stoichiometry (e.g., AB2 and AB3).
By training their model on learning data taken from a large database of binary crystal structures, they found that they could make good predictions of the structures of nearly 4000 others in the data set, even though no alloys with the same pair of elements was included in the training set. The researchers used the ML algorithm to narrow the set of candidate structures for a given alloy to approximately five, so that it then became practical to use quantum mechanical calculations for just these candidates to pick out the one likely to be the most stable. In this way, they were able to correctly predict the actual structure 90% of the time.
Even if you have structural information on a new material, such as x-ray diffraction data, it can be challenging to extract a structure from it. Gomes and her co-workers are developing methods that seek to use prior knowledge and reasoning in AI algorithms to assist that process.Reference Bai, Xue, Bjorck, Le Bras, Rappazzo, Bernstein, Suram, van Dover, Gregoire and Gomes12 Mapping back from the diffraction data to a structure is a computational problem that is “NP-hard,” which means that finding the solution can take a long time and increases exponentially with the complexity of the system being studied. What’s more, the possible solutions for a given x-ray diffraction pattern are not unique, and so finding the best one requires judgment and comparison with prior data.
Nicola Marzari of the École Polytech-nique Fédérale de Lausanne and his co-workers have explored ML for finding two-dimensional (2D) materials with interesting electronic properties (see Figure 2).Reference Mounet, Gibertini, Schwaller, Campi, Merkys, Marrazzo, Sohier, Castelli, Cepellotti, Pizzi and Marzari13The strength and exotic electronic structure of graphene—2D layers of graphite-like carbon—and hexagonal boron nitride have made it clear that similar materials could be useful for electronics and other applications. Marzari and colleagues used ML to search for 2D candidate materials among more than 100,000 3D crystal structures, finding around 6000 of them. They then used electronic-structure calculations to whittle this list to ones that seemed stable enough for the individual layers to be separable in the process called exfoliation, which can produce graphenefrom graphite. They ended up with approximately 1000 candidates that they screened computationally for potentially useful electronic and magnetic properties.
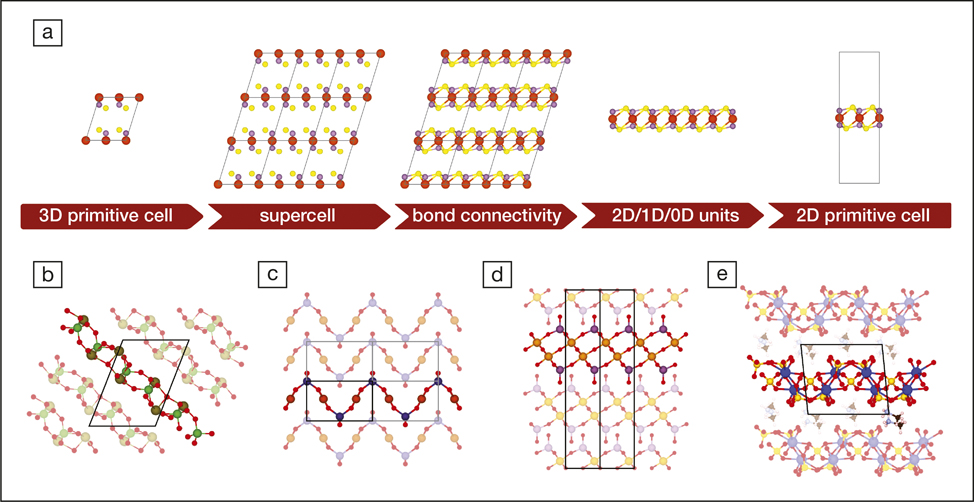
Figure 2. (a) Schematic representation of the fundamental steps needed to find low-dimensional units of a parent 3D crystal (here MgPS3). (b–e) Examples illustrating non-trivial layered structures that can be identified in (b) triclinic or monoclinic structures that are not layered along the [001] crystallographic direction (As2Te3O11). (c) Layered compounds whose constitutive layers extend over multiple unit cells and thus require the use of supercells to be identified (CuGeO3). (d) Layers that have partial overlap of the atomic projections along the stacking direction, with no manifest vacuum separation between them (Mo2Ta2O11). (e) Composite structures that contain units with different dimensionality [(CH6N)2(UO2)2(SO4)3, where 2D layers of uranyl sulfate are intercalated with 0D methylammonium molecules]. Reprinted with permission from Reference Reference Mounet, Gibertini, Schwaller, Campi, Merkys, Marrazzo, Sohier, Castelli, Cepellotti, Pizzi and Marzari13.© 2018 Nature Publishing Group.
One of these corresponded to a so-called quantum spin Hall insulator, a material in which the quantum spin Hall effect, which can create metallic, electrically conducting edge states in otherwise insulating material, is suppressed by the opening of a bandgap between the conducting and localized electron states. Marzari and colleagues predicted that this behavior would be seen in a 2D version of a natural mineral called jacutingaite, a compound of platinum, mercury, and selenium. They were able to verify, using detailed quantum calculations, that this material should have the predicted properties.Reference Marrazzo, Gibertini, Campi, Mounet and Marzari14
This approach of following up a ML sweep of the materials database with first-principles calculations of the most promising candidates is common. Isayev said that in their searches for new materials, they would typically perform first-principles electronic-structure calculations on the best candidates found by their ML algorithms, using the standard method called density functional theory (DFT) before trying to make them experimentally, to check if there are any possible surprises.
But “the first-principles theories we have are not that accuratefor a number of things,” said Marzari. Their ability to provide good thermodynamic predictions is not great, which means it’s hard to be sure if the material is stable or if it will be easy to make in preference to alternative materials. It’s also hard to anticipate how amenable metastable materials might be; after all, based on energy minimization alone, one wouldn’t expect diamond to persist under ambient conditions.
Yet ML can help with these theoretical shortcomings. It can circumvent the need for computationally intensive calculation of a property, such as a bandgap or electronic structure, by learning to predict those things as well.Reference Brockherde, Vogt, Li, Tuckerman, Burke and Müller15 “If you have large databases of DFT calculation/simulation results,” said Gomes, “it is possible for deep-learning models to learn and speed up DFT calculations; the models predict the outcome of a DFT simulation without running the actual simulation.” In this way, said Marzari, “one can use machine learning to predict the results of calculations that would be very expensive, either because they require large systems or long simulation times, or because they are very complex.”
The Materials Project, an initiative being developed at the Lawrence Berkeley National Laboratory and directed by Persson, currently has approximately 60,000 band structures in its database. These are calculated using the standard approach of DFT, and the data set has been used, for example, to look for novel thermoelectric materials or electrode materials for lithium batteries.Reference Jain, Hautier, Ong and Persson16 “We can now predict the bandgap for any material that is within the circle of the data set to an accuracy equal to that of DFT today,” Persson said. All the same, without adding more experimental data, such predictions still can only tell you the result that DFT would deliver; they avoid the need to carry out the calculations themselves, but can’t escape the approximations that the theory makes.
Not enough data
All this sounds very promising, but there is a big obstacle to the successful prediction of all manner of materials properties using ML. No algorithm, however good, can be expected to generate knowledge in regions of materials space where none currently exists. “ML techniques essentially rely on predictions based on what was seen in the known training data,” said Gomes. “However, to discover truly new materials, we want to go essentially outside what is known so far.” In other words, the learning process is only as good, and as broad, as the data set that informs it. What’s more, ML algorithms cannot anticipate anomalies and unexpected behaviors within the available data. It creates a continuous landscape in a discrete variable space, assuming that the path between two points is always smooth – which might not be true.
The sheer lack of data, though, is currently a limiting factor. Right now, there are 200,000 to 500,000 known inorganic materials, which might sound like a lot, but it is a relatively small subset of all those possible. “You can’t machine-learn something that isn’t supported by your input data,” said Persson. So ML is very good at finding hidden correlations in large data sets that are difficult for human beings to see, but is not particularly good at finding materials that are not supported by the data set that you give it. “Google asked us recently how we could machine learn a better battery,” she said, “and I told them it is very difficult because we don’t have a large enough data set to address a complex problem like that.”
“Modern algorithms really excel in modeling of very complex data relationships, but vast amounts of data will be needed to train such models,” said John Gregoire of the California Institute of Technology in Pasadena, who leads a project searching for new electrocatalysts for artificial photosynthesis. “What remains to be seen is whether we could ever generate enough materials data to take advantage of this approach.”
All the same, ML might spot trends and commonalities in existing data, such as the fact that the ions like to move between an octahedral coordination site in one place and a tetrahedral one in another. If you now give it a huge number of crystal structures, it might be able to find such structures. You cannot just give it cycling curves for all the batteries made to date and expect it to figure out what is needed, Persson said, because there are a lot of bad data in there—it is not a clean, robust data set, but is full of noise. “There has been a focus on collecting a lot of data, but what we need is quality, not just quantity,” said Marzari.
Producing and harnessing data is essential to this project, and journals could help by encouraging the presentation of data in standardized ways. “We write papers the way we have done historically, as a kind of story,” said Persson. “But if we want to use natural language processing to extract information, that’s not the best way.” She thinks there will need to be some changes to how papers are written for ease of data extraction, perhaps as a tabular version with the metrics of the synthesis, the outcomes, and so forth. “In academia, we are notoriously bad at data capture,” said Isayev. “In addition, most data is locked in PDF files and must be extracted. We really need to establish best practices and publish negative data too.” The problem is there is no single format for data that are well suited to all applications, he added. “Ultimately, those features are dependent on what specific question is to be answered.”
The issue here is not simply about collecting more data, but about integrating different types of data. It is one thing to collect vast amounts of crystal structures, for example, but how might that be usefully combined with theories or simulations of structure–property relationships, or with experimental measurements that might depend on issues such as impurities or higher-order microstructure? ML might deduce correlations between such multi-model data sets that are invisible to human researchers.
Citrine is making concerted efforts to gather and integrate diverse kinds of data sets, said Meredig. “Being able to understand multi-modal data sets is essential to generating value from materials data. In our field, data are inherently multi-modal, hierarchical, heterogeneous, and generally complex.” Meredig said that Citrine aims to let users store any kind of materials data on its platform: simulations, experiments, sample processing histories, and so forth.
The need for more data also highlights the neglected value of negative results. If a material does not have the properties you are looking for, that could still be a valuable piece of information for the global materials knowledge set, but such results are generally hard to publish. “It’s a real problem that we only publish successes,” said Persson. “And even then, there’s often some fudging and massaging of the data. We need complete data sets, not just the good ones. We can be smarter about how we use the knowledge that’s out there.”
But it can be hard to find good metrics for inputting data. “We don’t even have a good metric for local coordination,” said Persson. We can look at a crystal structure and be unsure if we are looking at fivefold or sixfold coordination, for example. There is not always a simple, discrete number or category to describe the parameter we want to give the algorithm. This problem of finding good input metrics becomes even harder for noncrystalline structures, or those with a high degree of disorder or heterogeneity. For example, the electrical behavior of semiconductors can depend sensitively on the presence of dopants, which may be irregularly distributed throughout a crystal lattice. The failure or fracture of many materials is initiated at flaws that may be rare, but nonetheless critical, to the overall performance. Of course, some materials are not crystalline at all, but amorphous–particularly organic and polymeric materials. It is a challenge then to find metrics appropriate for training ML algorithms and anticipating trends in behavior. Still, it can be done. For example, a team led by researchers at the Stanford Synchrotron Radiation Lightsource in California, has trained a ML model to find new ternary metallic glasses.Reference Ren, Ward, Williams, Laws, Wolverton, Hattrick-Simpers and Mehta17 And materials scientist John Mauro of The Pennsylvania State University has argued that by applying ML to composition–property relationships in glasses, it should be possible to “decode the glass genome”—the biological analogy refers to the idea that there might be a kind of code that links composition, structure, and properties—to find faster, cheaper routes to new glassy materials.Reference Mauro18
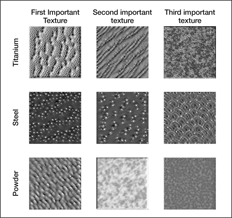
Isayev said that one of the most useful features to be able to predict with ML would be a material’s microstructure: the level beyond the crystal structure itself, for example, in terms of grain boundaries or compositional gradients. Bulk properties such as mechanical behavior can be very sensitive to such factors. But he says that getting this information experimentally is typically slow, and there are no good general computational methods for predicting it. Nonetheless, there are already efforts under way to use ML to classify and interpret microstructures from imaging studies (see Figure 3).Reference Ling, Hutchinson, Antono, DeCost, Holm and Meredig19

Figure 3. The most important textures for a set of microstructures in various materials. From left to right, the columns represent the first, second, and third most important texture features for each case. From top to bottom, each row represents a different case: titanium, steel, and powder. Reprinted with permission from Reference Reference Ling, Hutchinson, Antono, DeCost, Holm and Meredig19. © 2017 Elsevier.
The challenge of making predicted materials
Aside from the need for more and better data, there is another big obstacle to turning ML into a method that actually delivers tangible, useful materials: you have to be able to make them. Even if a material is stable, in principle, that does not mean you will be able to figure out how to make it. “A specialty of my lab is trying to rapidly synthesize new materials, and I have had collaborators give me machine-predicted materials from their AI algorithms,” said Gregoire. “But we have yet to successfully synthesize a single one. Most models are focused on the functionality of the materials, but synthesizability also needs to be incorporated.” It is for this reason, said Marzari, that “I prefer to explore the unknown properties of materials that are known to exist,” such as jacutingaite.
“Synthesis is currently the bottleneck,” agreed Persson. “It can take me minutes to predict a novel piezoelectric. I know this because we did it a couple of years ago. And then it sat for two years in the project,” because no one could see how to make it. Finally, one brave researcher decided to try, “and it took her another two years.” It did, however, have the properties predicted.
But Persson believes that synthesis too can be machine-learned. “We need to know what drives synthesis. Some people think of it as an entirely unpredictable kinetic landscape, but I do not believe that. I’m sure there are rules; nature always goes by rules.”
It will not be easy to find them though. It is one thing to use ML or traditional ab initio methods to calculate phase diagrams that will tell you if a material is thermodynamically stable in principle. It’s quite another to predict how surface effects, defects, or heterogeneities in real materials might modify their stability, let alone to figure out the roles of kinetic factors and reactive intermediate states in the nucleation of new phases.
“Today, there is no predictive theory of which compounds can be synthesized and how,” said Ceder.
Guiding experiments
AI can help researchers figure out where to look in the materials universe to stand a good chance of finding substances with specified target properties. It could even help to devise ways of making candidate materials. But that is still only a part of the process of getting some useful product from the research process. “I think we’ll gradually see greater use of AI for a broader set of questions,” said Riley. “Most of the uses of AI in materials today ask very specific questions, like ‘Will this hypothetical material have the property I want?’ But we already have AI systems that try to answer more subtle questions, like ‘What’s most unusual about this set of examples?’ or ‘Can you find a simple equation that explains this data?’ ”
The philosophy of ML is that anything can be learned in an automated way if there are enough data to train the algorithm. So why not use it to assist in devising a research strategy? (See the 2018 MRS Spring Meeting interview with Maruyama at mrs.org/interview-maruyama.)
That strategy is generally iterative: you conduct a set of experiments, whether real or computational, refine your aims based on what you learned, and conduct some more. “It’s often useful in the discovery process to have lots of iterations, not just lots of runs of the same basic experiment,” said Maruyama. Each iterative step needs thoughtful experimental design, and sometimes that design process is the rate-limiting step toward genuine innovation. “Most people are using AI to assess what’s going on in their experiments, but not to make the decisions,” said Maruyama. In general, a “closed-loop” system deploys robotics to synthesize and characterize materials products, and then feeds back the gained information into the decisions made by ML about the next cycle. This creates a much more directed search than traditional rapid-throughput methods that explore large materials spaces in a blind, scattershot manner: the best materials can be identified much more quickly.
Maruyama and his co-workers have devised a prototype system to create a closed-loop automated process for growing carbon nanotubes (see Figure 4).Reference Nikolaev, Hooper, Webber, Rao, Decker, Krain, Poleski, Barto and Maruyama20 These carbon nanostructures are typically grown by chemical vapor deposition from a carbon-rich source onto microstructured particles of a catalytic metal. The challenge is that there are so many experimental parameters one could vary (e.g., temperature pressure, feedstock composition). Exploring the entire high-dimensional parameter space is impractical, and it is preferable to gradually improve the outcome by making decisions about the next experiment that incorporates knowledge gleaned from the last.
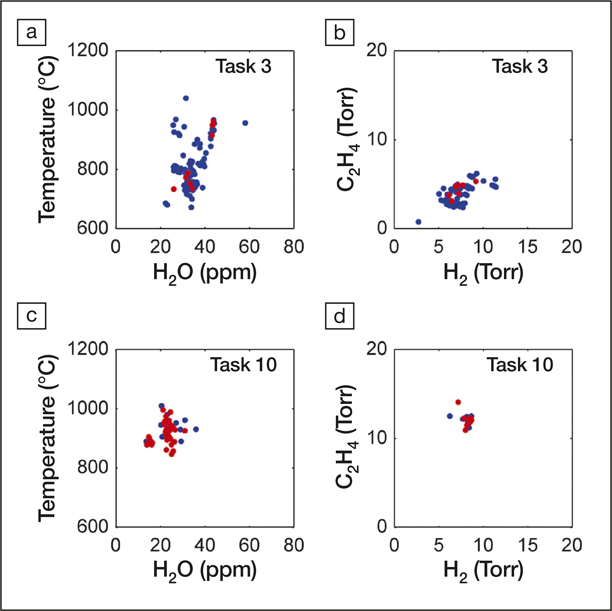
Figure 4. Variability in the experimental parameter space with learning. Experimental conditions chosen by the autonomous research system (ARES) in Task 3, before convergence (a, b), and in Task 10, after convergence (c, d), are compared over four experimental parameters (temperature, water concentration, and H2 and C2H4 partial pressures). Red dots represent successful, on-target experiments. (a, b) Before convergence, ARES sampled a wide range of growth conditions, and only 8% of experiments were on target. (c, d) After convergence, ARES sampled a narrow range of growth conditions, with 68% on-target experiments, demonstrating its ability to autonomously optimize multiple experimental parameters. Reprinted with permission from Reference Reference Nikolaev, Hooper, Webber, Rao, Decker, Krain, Poleski, Barto and Maruyama20. © 2016 Nature.
The researchers first carried out 84 experiments, spanning much of the available parameter space, as a training set for their automated research system. In each case, the nanotube growth rate was measured using Raman spectroscopy. The AI software was then used to plan and execute 600 further experiments that aimed to target specific growth rates, refining each iteration using the previously collected data. The system learned to achieve target rates of 500, 3000, and 16,000 nanotubes per second, allowing it to develop well-defined nanotube surface densities in some specified time.
In a similar way, Turab Lookman of the Los Alamos National Laboratory in New Mexico and co-workers have used an automated iterative process to search experimentally for new shape-memory alloys with minimum thermal hysteresis, a factor that leads to fatigue in repeated deformation cycles. They identified 14 compositions based on the nickel-titanium system that had smaller hysteresis than any of those in the training set. The best performance was achieved by an alloy containing small and precise amounts of copper, iron, and palladium that would have been difficult to spot from an exhaustive search of the ∼800,000 potential compositions that their system could generate.Reference Xue, Balachandran, Hogden, Theiler, Xue and Lookman21
As well as assisting in experimental selection and design, AI techniques can speed up the analysis of results. Right now, said Reyes, a graduate student might spend several days painstakingly extracting quantities such as particle-size distributions from microscope images, whereas image-analysis and trained deep-learning algorithms could complete a task like this almost instantaneously.
ML can also help to condense massive, high-dimensional data sets into a meaningful form. “As researchers, we often take a large amount of effort in reducing and summarizing data into something we can visualize and plot,” said Reyes. “ML algorithms don’t have this limitation and can see structure in hundreds to thousands of dimensions.”
Reyes is also developing algorithms to plan experiments that work on similar lines to the AlphaGo AI system, which has defeated the best human players at the game Go. However, “unlike a Go game, experimental results are noisy and have inherent randomness, and we must incorporate this variability,” he explained. It is a bit like having to decide on a move without perfect knowledge of the state of the board and where pieces are liable to move somewhat at random. To deal with this imperfect knowledge of a materials system, Reyes and colleagues incorporate Bayesian reasoning into their algorithms, where prior beliefs about the best outcome are updated as new information becomes available.Reference Wang, Reyes, Brown, Mirkin and Powell22
Gomes, meanwhile, is a member of a multi-institutional project to develop a system called the Scientific Autonomous Reasoning Agent (SARA), which will“integrate first-principles quantum physics, experimental materials synthesis, processing, and characterization, and AI-based algorithms for reasoning and conducting science, including the representation, planning, optimization, and learning of materials knowledge.” The project has been recently launched and has not yet led to publications, but Gomes said the aim is to develop a closed-loop iterative process that will formulate hypotheses about making materials with given structures and properties, to plan and execute experiments (such as x-ray diffraction), to interpret the results, and to use that knowledge to devise the next round of hypothesis testing. “We have an ambitious plan for SARA,” she said.
Ultimately, AI systems might turn data into full-blown theories. “Long term, I absolutely think that AI will be suggesting novel research directions and theories,” said Riley. “True machine super-intelligence is still a way off—our machine learning often has trouble understanding the context of information or dealing with very disparate types of data, something that humans can do quite well. But in narrow domains, we are already seeing machines produce creativity of a sort.” Riley added that in his work at Google on small-molecule chemistry, “our algorithms have produced ideas that caused our collaborators to say, ‘I didn’t think of that, but it’s a good idea!’ ”
Does this mean that research itself might become fully automated? “The idea that soon we will have AI algorithms that can perform scientific discovery completely autonomously is a bit far-fetched,” said Gomes. “Nevertheless, I do believe that AI algorithms can dramatically speed up discovery by several orders of magnitude.” Maruyama said that this kind of approach could permit researchers to be more ambitious in tackling targets that might seem too dauntingly complex and multidimensional to be attempted by conventional human-led experimental design. He hopes that approaches like this could create a kind of Moore’s Law for the speed of research: a steady acceleration as computer power expands.
If so, the social ramifications could be enormous. Faster and more efficient research should ultimately translate into cheaper products—ones that work better and require less investment of time and resources in R&D. In a sector such as materials for energy conversion—photovoltaic, thermoelectric, and battery materials—this could help governments to reduce their reliance on fossil fuels,Reference Tabor, Roch, Saikin, Kreisbeck, Sheberla, Montoya, Dwaraknath, Aykol, Ortiz, Tribukait, Amador-Bedolla, Brabec, Maruyama, Persson and Aspuru-Guzik23 a vital concern in the light of the urgency expressed by the 2018 report of the Intergovernmental Panel on Climate Change.
Such acceleration of materials discovery is the objective of the Materials Genome Initiative, which is supported by the US Departments of Energy, Defense, Commerce, and others. The program aims to “discover, manufacture, and deploy advanced materials twice as fast, at a fraction of the cost,” and AI should be vital to that goal. In the coming years, “AI-driven autonomous materials research is going to fundamentally change how we do materials science,” said James Warren, the technical program director for the initiative at the National Institute of Standards and Technology in Gaithersburg, Md. (see Figures 5 and 6).
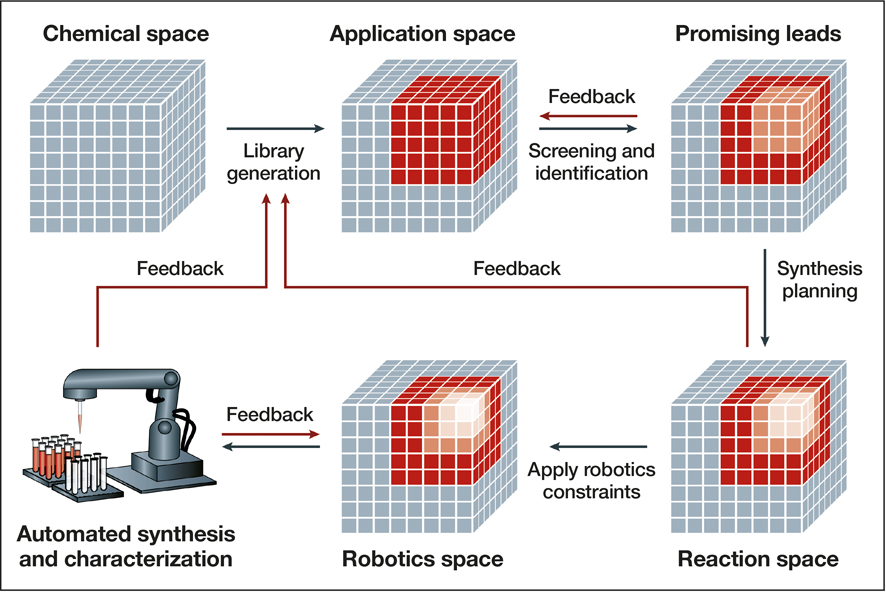
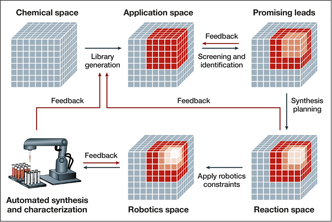
Figure 5. Workflow of a closed-loop approach to autonomous materials discovery. The procedure begins with identifying an application space of candidates for a given problem. The promising leads from this library are identified, potentially through computational screening, and are further narrowed by identifying the synthetically accessible molecules. Finally, the constraints of available robotics systems are taken into consideration before starting automated synthesis and characterization. Feedback from in situ experimentation is used to adjust the model, building the application space for the next iteration of this loop. Other feedback mechanisms at various stages of the loop aid in ensuring the candidates are compatible with all stages of the loop and reduce trial and error in the long term. Reprinted with permission from Reference Reference Tabor, Roch, Saikin, Kreisbeck, Sheberla, Montoya, Dwaraknath, Aykol, Ortiz, Tribukait, Amador-Bedolla, Brabec, Maruyama, Persson and Aspuru-Guzik23. © 2018 Macmillan Publishers Ltd.
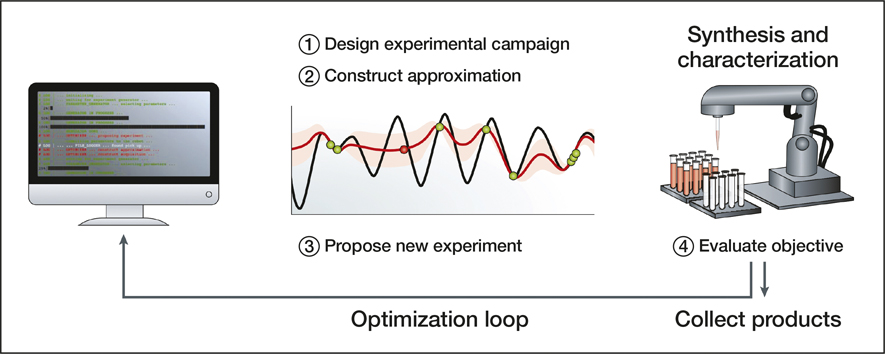
Figure 6. Autonomous experimentation bridges computational power and robotics solutions to create a virtuous cycle. The approximation function (red) models how a particular set of experimental conditions will perform in terms of, for example, yield and reaction time. Once constructed, this function is used to propose the next experiment. The model is updated and moves toward maximizing the performance of the reaction based on the predefined conditions. Reprinted with permission from Reference Reference Tabor, Roch, Saikin, Kreisbeck, Sheberla, Montoya, Dwaraknath, Aykol, Ortiz, Tribukait, Amador-Bedolla, Brabec, Maruyama, Persson and Aspuru-Guzik23. © 2018 Macmillan Publishers Ltd.
AI as collaboration, not competition
Despite such bold predictions, it is not yet clear where ML can take us in the materials universe. “I don’t think we know what properties will be reliably predicted, and I don’t think I’ve seen any convincing prediction of properties of new materials or even any materials beyond the scope of the training set,” said Gregoire. “Generative models that provide a material that meets user specifications is an ultimate goal, but there’s been limited progress to date for materials.” The most-immediate impact of ML in materialsscience, he thinks, will be in automation and acceleration of tasks we already know how to do.
All the same, exploring the physical world via relationships extracted empirically from vast data sets by ML is becoming an increasingly common way to do science. Although the results can be predictively useful, some argue that they barely qualify as science, because they amount to just finding correlations, without any mechanistic understanding of what controls them. “What we really want is to understand the underlying physics and chemistry,” said Maruyama, ideally without having to collect immense data sets to do so.
Some researchers worry that AI and ML, as they become more pervasive in science, might come to supplant human ingenuity, and science itself might then become an exercise in blind number-crunching, lacking the spark of true creativity. However, it would be a mistake to regard this as an either/or situation. The skill sets of AI and human researchers are complementary, as is obvious from the way it has proved so difficult to make AI systems that can process visual or audio information with the ease of the human mind, or that can apply sheer “common sense” to extract meaning from ambiguous sentences. So maybe the ideal is to work together. “I think it is better to view an AI system as a collaborator, where we should teach that system everything we know about the problem,” said Gregoire. “If the high-level goal of a project is to assess the statistical significance of some relationship, then one might be concerned with human bias. But when the goal is to discover useful materials, human bias is not really a problem. Given that practically all discoveries to date have been made by human reasoning as opposed to machine reasoning, we want to keep the experts involved in the process.”
Maruyama said that by freeing human researchers from routine data-crunching and experimental optimization, AI could allow them more time for aspects of research that require the creativity and imagination in which humans excel. For Reyes, optimal use of the technology involves finding ways to blend the“domain knowledge” acquired by ML from massive data sets with human expert opinion. A major challenge here, he said, is for human experts to handle the high-dimensional quantities that ML tends to generate. “We have thought a lot about visualization and are building interactive tools to help the expert codify their knowledge,” he added.
AI could also help them to cope with the increasingly overwhelming scale of the literature. “It’s quite difficult to keep up with the expanding rate of research output,” said Riley. “We’re likely to see some combination of AI methods and researchers producing more structured data and documents so that machines can more effectively assist researchers in understanding the research output of their fields.”
Properly applied, said Persson, ML will be an empowering tool, not a competitor. “We’re not going to make ourselves obsolete,” she said. “We are just going to ask different questions.”