


No CrossRef data available.
Article contents
Ab initio Study of the Hydrogen Molecule on ZnO Surfaces
Published online by Cambridge University Press: 18 July 2011
Abstract
We conduct first-principles total-energy density functional calculations to study the interaction of H2 on ZnO surfaces. Four surface models of Zn-terminated (0001)-, O-terminated (0001)-, 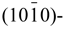 , and
, and 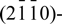 oriented ZnO planes in the presence of H2 are evaluated. The relative stability of four different surface models is examined as a function of the chemical potentials of oxygen and hydrogen. We find that only surfaces of O-terminated (0001)-oriented ZnO models exhibit active sites for the dissociation of H2, which in turn enables the formation of water from dissociative chemisorption of 2H on the O-terminated ZnO(0001) surface. The surface energy of O-terminated ZnO(0001) surface in the presence of water was found to be negative under the O-rich and H-rich condition. The findings agree with the experimental observations that ZnO epitaxial layers are easily etched by hydrogen at typical growth temperatures.
oriented ZnO planes in the presence of H2 are evaluated. The relative stability of four different surface models is examined as a function of the chemical potentials of oxygen and hydrogen. We find that only surfaces of O-terminated (0001)-oriented ZnO models exhibit active sites for the dissociation of H2, which in turn enables the formation of water from dissociative chemisorption of 2H on the O-terminated ZnO(0001) surface. The surface energy of O-terminated ZnO(0001) surface in the presence of water was found to be negative under the O-rich and H-rich condition. The findings agree with the experimental observations that ZnO epitaxial layers are easily etched by hydrogen at typical growth temperatures.
Keywords
- Type
- Research Article
- Information
- MRS Online Proceedings Library (OPL) , Volume 1327: Symposium G – Complex Oxide Materials for Emerging Energy Technologies , 2011 , mrss11-1327-g08-04
- Copyright
- Copyright © Materials Research Society 2011
References
REFERENCES
1.
Nakahara, K., Tanabe, T., Takasu, H., Fons, P., Iwatal, K., Yamadal, A., Matsubara, K., Hunger, R. and Niki, S., Jpn. J. Appl. Phys.
40, 250 (2001).Google Scholar
2.
Tsukazaki, A., Ohtomo, A., Onuma, T., Ohtani, M., Makino, T., Sumiya, M., Ohtani, K., Chichibu, S. F., Fuke, S., Segawa, Y., Ohno, H., Koinuma, H. and Kawasaki, M., Nat. Mater.
4, 42 (2005).Google Scholar
3.
Zhao, B., Yang, H., Du, G., Miao, G., Zhang, Y., Gao, Z., Yang, T., Wang, J., Li, W., Ma, Y., Yang, X., Liu, B., Liu, D., and Fang, X., J. Cryst. Growth
258, 130 (2003).Google Scholar
4.
Dai, J., Liu, H., Fang, W., Wang, L., Pu, Y., Chen, Y., and Jiang, F., J. Cryst. Growth
283, 93 (2005).Google Scholar
5.
Smith, T. P., Mecouch, W. J., Miraglia, P. Q., Roskowski, A. M., Hartlieb, P. J., and Davis, R. F., J. Cryst. Growth
257, 255 (2003).Google Scholar
6.
Pan, C.J., Tu, C.W., Tun, C.J., Lee, C.C., Chi, G.C., J. Cryst. Growth
305, 133 (2007).Google Scholar
7.
Hong, S.-K., Ko, H.-J., Chen, Y., Hanada, T., and Yao, T., J. Cryst. Growth, 214/215
81 (2000).Google Scholar
8.
Ko, H. J., Chen, Y. F., Hong, S. K., Wenisch, H., Yao, T., and Look, D. C., Appl. Phys. Lett.
77, 3761 (2000).Google Scholar
9.
Lin, C.-W., Ke, D.-J., Chao, Y.-C., Chang, L., Liang, M.-H., and Ho, Y.-T., J. Cryst. Growth
298, 472 (2007).Google Scholar
10.
Ougazzaden, A., Rogers, D.J., Hosseini Teherani, F., Moudakir, T., Gautier, S., Aggerstam, T., Ould Saad, S., Martin, J., Djebbour, Z., Durand, O., Garry, G., Lusson, A., McGrouther, D. and Chapman, J.N., J. Cryst. Growth
310, 944 (2008).Google Scholar
11.
Rogers, D. J., Hosseini Teherani, F., Ougazzaden, A., Gautier, S., Divay, L., Lusson, A., Durand, O., Wyczisk, F., Garry, G., Monteiro, T., Correira, M. R., Peres, M., Neves, A., McGrouther, D., Chapman, J. N. and Razeghi, M., Appl. Phys. Lett.
91, 071120 (2007).Google Scholar
12.
Liu, P.-L., Chizmeshya, A. V. G., Kouvetakis, J., and Tsong, I. S. T., Phys. Rev. B, 72, 245335 (2005).Google Scholar
13.
Liu, P.-L., Chizmeshya, A. V. G., and Kouvetakis, J., Phys. Rev. B, 77, 035326 (2008).Google Scholar
18.
Perdew, J. P., Chevary, J. A., Vosko, S. H.. Jackson, K. A., Petersen, M. R., and Fiolhais, C., Phys. Rev. B
46, 6671 (1992).Google Scholar