Introduction
In less than 50 years obesity has become a worldwide epidemic and is estimated to affect over one billion individuals(Reference Caballero1). One of the most concerning aspects related to obesity is the increased risk of developing the metabolic syndrome and associated co-morbidities(Reference Rezaianzadeh, Namayandeh and Sadr2). Currently the leading cause of liver disease in North America, non-alcoholic fatty liver disease (NAFLD) is considered the hepatic manifestation of the metabolic syndrome and is characterised as an increase in intrahepatic fat that may be accompanied by inflammation in the absence of alcohol abuse(Reference Boyle, Thompson and Gregg3, Reference Hamaguchi, Takeda and Kojima4).
NAFLD includes a spectrum of liver diseases that includes simple steatosis as well as the progression of non-alcoholic steatohepatitis (NASH) that can develop into cirrhosis and hepatocellular carcinoma (HCC)(Reference Levene and Goldin5, Reference Michelotti, Machado and Diehl6) (Fig. 1). Although it is estimated that up to 90 % of individuals with obesity (BMI >30 kg/m2) may have hepatic steatosis, this condition represents a relatively benign and asymptomatic accumulation of intrahepatic fat(Reference Bellentani and Tiribelli7). However, approximately 30 % of patients will develop steatohepatitis characterised by inflammation leading to hepatocyte damage and potentially fibrosis(Reference Levene and Goldin5). Given time, NASH can progress to liver cirrhosis and HCC(Reference Michelotti, Machado and Diehl6, Reference Yasui, Hashimoto and Komorizono8). While pharmacological treatments are being actively studied they are in the early stages of development; therefore current treatment strategies for NASH are limited to intensive dietary and lifestyle modification(Reference Engl, Sturm and Sandhofer9, Reference Milic and Stimac10). Due to the severe complications associated with NASH, identification of the underlying mechanisms driving its pathogenesis is required.

Fig. 1 A current model demonstrating the progression of non-alcoholic fatty liver disease (NAFLD) in humans. While animal models can recapitulate certain aspects of NAFLD, non-alcoholic steatohepatitis (NASH) and even fibrosis, not one model exists that covers all stages of the liver disease. Further development of animal models is needed to continue the study of disease mechanisms and evaluation of possible treatment strategies.
The factors contributing to the development of NAFLD are under extensive study. NAFLD is associated with obesity, insulin resistance, diabetes, hyperlipidaemia and increased risk of CVD and cancer(Reference Michelotti, Machado and Diehl6, Reference Pacana and Fuchs11). The accumulation of central adiposity along with insulin resistance has been linked to the development of liver steatosis and probably contributes to the further progression to NASH(Reference Bugianesi, Moscatiello and Ciaravella12, Reference Tilg and Moschen13). Genetic susceptibility and environmental factors such as diet have also been implicated in disease progression of NASH(Reference Tilg and Moschen13). Although excessive adiposity and the metabolic syndrome are linked to the majority of cases of NAFLD, steatohepatitis has also been reported as a bystander effect due to pharmacological therapies most commonly associated with oxidative stress and aberrant changes in lipid metabolism(Reference Larrain and Rinella14). Individuals receiving total parenteral nutrition (TPN) are also at increased risk of developing NASH(Reference Larrain and Rinella14, Reference Buchman15). Recently it was shown that patients with chronic hepatitis C carrying the PNPLA3 rs738409 G variant is associated with more severe steatosis and steatohepatitis in the absence of obesity(Reference Petta, Vanni and Bugianesi16). Caution should be taken when interpreting results based on the characterisation of non-obese patients with steatosis or steatohepatitis as many studies use BMI for the classification of obesity; however, this has been shown to underestimate the prevalence of obesity in a number of different racial/ethnic populations(Reference Richmond, Thurston and Sonneville17–Reference Jitnarin, Poston and Haddock19).
The use of animal models has proven to be a useful tool to provide insight into the development of NAFLD and progression of NASH. However, research in this field has been hampered by the lack of an animal model that can fully recapitulate the disease phenotype in a consistent and timely manner. Due to the number of factors that are associated with the progression of NAFLD and still unclear pathogenesis, rodent models must be carefully chosen to answer the research question. Highlighting this fact is a recent publication directly comparing two widely used dietary murine models of NAFLD demonstrating the critical impact of diet on the pathogenesis of these diseases(Reference Machado, Michelotti and Xie20). In addition, innovative murine models are still needed to facilitate evaluation of novel treatments for NAFLD and NASH.
Pathogenesis of non-alcoholic fatty liver disease
We have learned a considerable amount about NAFLD since it was first described over three decades ago(Reference Ludwig, Viggiano and McGill21). The development of NAFLD in patients has been closely linked to adipose tissue insulin resistance(Reference Milic and Stimac10). During obesity, loss of peripheral insulin sensitivity may lead to the aberrant release of NEFA from adipose tissue as well as increased de novo lipogenesis promoting hepatic steatosis(Reference Kong, Zhu and Bai22). It is thought that an influx of NEFA from adipose tissue stores may contribute up to 80 % of intrahepatic fat(Reference Donnelly, Smith and Schwarzenberg23). Diets higher in saturated fat and cholesterol have been linked to patients with NAFLD and may influence the development of hepatic insulin resistance(Reference Donnelly, Smith and Schwarzenberg23, Reference Lottenberg, Afonso Mda and Lavrador24). In addition, levels of proinflammatory cytokines including TNF-α and IL-6 as well as elevated levels of leptin and reduced adiponectin have been detected during NAFLD(Reference Milic and Stimac10, Reference Das and Balakrishnan25, Reference Handa, Maliken and Nelson26). Recent advances in the understanding of autophagy put into question the exact role of insulin resistance during NAFLD with reports suggesting the accumulation of excess lipid within the liver may in fact initiate insulin resistance(Reference Kim, Jeong and Oh27). Considerable research is required to further understand the contribution of autophagy to the pathogenesis of obesity, the metabolic syndrome and NAFLD(Reference Kwanten, Martinet and Michielsen28).
The diagnosis of NAFLD often occurs in asymptomatic patients following the detection of elevated serum aminotransferases and/or ultrasound findings of steatosis during routine screening(Reference Tuyama and Chang29). A positive diagnosis of NAFLD usually requires the exclusion of all other causes of liver steatosis including viral infection and autoimmunity as well as the assessment of liver function, lipid profiles and insulin sensitivity measured by the homeostatic model assessment of insulin resistance (HOMA-IR) index(Reference Neuschwander-Tetri, Clark and Bass30). Alcohol abuse causing alcoholic liver disease must also be separately identified(Reference Ludwig, Viggiano and McGill21). Interestingly, moderate ethanol consumption (2·5 g/kg body weight per d) in leptin-deficient (ob/ob) mice was recently associated with reduced steatosis and improved liver function possibly due to increased adiponectin levels and enhanced activation of the sirtuin 1 (SIRT1) pathway and Akt phosphorylation(Reference Kanuri, Landmann and Priebs31). This demonstrates that the degree of alcohol consumption and NAFLD should be considered although this is currently beyond the scope of the present review. Liver biopsy remains the ‘gold standard’ for diagnosis; however, the use of biomarkers is currently being developed as a less invasive diagnostic tool(Reference Wieckowska and Feldstein32).
Development of non-alcoholic steatohepatitis from obesity and non-alcoholic fatty liver disease
The development of NASH was previously associated with a ‘two-hit’ model where liver steatosis is accompanied by the metabolic syndrome that progresses into hepatitis following additional stresses(Reference Day and James33). However, this theory has now been extended to include multiple factors that are considered to promote the development of inflammation and fibrosis including increased mitochondrial dysfunction, endoplasmic reticulum stress, changes in lipid oxidation and increased endotoxin levels in the liver leading to immune cell activation and infiltration contributing to a heightened proinflammatory microenvironment leading to hepatocyte cell stress and death(Reference Musso, Gambino and Cassader34–Reference Farrell, van Rooyen and Gan38).
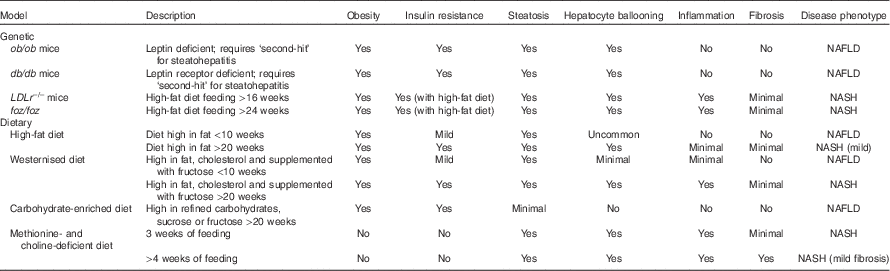
The histopathology of NASH includes evidence of steatosis, both macro- and microvesicular as well as ballooning hepatocytes, lobular inflammation and the development of pericellular fibrosis(Reference Pagadala and McCullough39). Elevated aminotransferase levels including alanine aminotransferase (ALT) and aspartate aminotransferase may signal changes in liver biochemistry but are inconsistent whereas liver biopsy remains the ‘gold standard’ diagnostic procedure for NASH(Reference Neuschwander-Tetri, Clark and Bass30). Given the heterogeneity inherent to the development of NASH, treatment of this disease poses a challenge. Selecting an appropriate model that captures the underlying chronic, low-grade inflammation due to the metabolic syndrome along with the active inflammation process is key. Although a single animal model has yet to capture the complete human disease pathology a number of models are able to recapitulate certain disease pathologies (Table 1).
Table 1 Murine models commonly used to study non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH)
Murine models with increased genetic susceptibility to non-alcoholic fatty liver disease
When choosing an animal model to study obesity, type 2 diabetes and fatty liver disease the genotype x environment interactions should be considered(Reference Waller-Evans, Hue and Fearnside40–Reference Nagao, Asai and Inaba42). A distinct difference was detected when comparing the disease susceptibility to high-fat diet (HFD) treatment between two commonly used mouse strains C57BL/6J and BALB/c(Reference Waller-Evans, Hue and Fearnside40). Following 23 weeks of HFD C57BL/6J mice gained more weight and had higher adiposity compared with BALB/c mice(Reference Waller-Evans, Hue and Fearnside40). Analysis of gene transcription in the liver between the mouse strains demonstrated that each strain differentially regulated genes associated with fatty acid uptake and efflux when fed HFD affording increased disease susceptibility to C57BL/6J mice and relative resistance to BALB/c mice fed a HFD(Reference Waller-Evans, Hue and Fearnside40). To further support this finding, 8 weeks of HFD treatment in C57BL/6J, 129X1/SvJ, DBA/2 and FVB/N mice resulted in similar body-weight gain and impaired glucose tolerance whereas BALB/c mice had normal glucose tolerance and resisted hepatic lipid accumulation(Reference Montgomery, Hallahan and Brown41). It has been suggested that while C57BL/6J mice are the most susceptible to diet-induced obesity, BALB/c mice might be a good model of ‘healthy obese humans’ since BALB/c mice have lower hepatic lipid accumulation and normal glucose tolerance(Reference Montgomery, Hallahan and Brown41). Within the C57 sub-strains, C57BLKS/J mice have been reported to gain less weight over 12 weeks of HFD treatment and have improved glucose tolerance compared with C57BL/6J mice although the strains share 70 % genetic homology(Reference Sims, Hatanaka and Morris43). Genetic differences between C57Bl/6N and C57BL/6J mice have also been linked to differences in disease susceptibility in HFD-fed mice(Reference Heiker, Kunath and Kosacka44, Reference Podrini, Cambridge and Lelliott45).
Numerous genetic models of both NAFLD and NASH have been recently described(Reference Nagarajan, Mahesh Kumar and Venkatesan46). These models may develop obesity, impaired insulin sensitivity, glucose intolerance and hepatic steatosis over time independent of dietary challenge(Reference Nagarajan, Mahesh Kumar and Venkatesan46). In other cases dietary manipulation is required to expose the development of the metabolic syndrome(Reference Nagarajan, Mahesh Kumar and Venkatesan46). Some of the more widely used models have been highlighted in greater depth below.
LDLr–/–
Often mice with a genetic alteration will be treated with a HFD to exacerbate an underlying phenotype. This is particularly important for the LDL receptor-deficient mouse. Due to a lack of LDL receptors and an inability to clear intermediate-density lipoproteins and LDL these mice develop hypercholesterolaemia and hypertriacylglycerolaemia when fed a HFD(Reference d Oliveira, Carvalho and Polo47). One of the mechanisms that has been proposed for the development of the insulin-resistant phenotype observed in the LDLr –/– mouse is due to a lack of β-cell hypertrophy to compensate for reduced insulin release(Reference d Oliveira, Carvalho and Polo47). Treatment of a high-fat high-cholesterol diet in LDLr –/– mice for 12 weeks leads to hyperlipidaemia as well as glucose and insulin intolerance(Reference Bojic, Burke and Chhoker48). LDLr –/– mice fed a Western diet supplemented with olive oil for 16 weeks resulted in a robust steatohepatitis phenotype including elevated ALT levels, inflammation and the development of early fibrosis(Reference Depner, Traber and Bobe49). A high-fat, high-carbohydrate diet with added cholesterol has also been shown to induce a NASH-like phenotype in LDLr –/– mice after 24 weeks of treatment(Reference Subramanian, Goodspeed and Wang50).
Leptin deficient (ob/ob) and leptin receptor deficient (db/db)
Numerous transgenic mouse models have been used to study obesity and related co-morbidities including the leptin-deficient ob/ob mouse and the db/db mouse that lacks functional leptin receptor signalling(Reference Nagarajan, Mahesh Kumar and Venkatesan46, Reference Trak-Smayra, Paradis and Massart51, Reference Tamura, Sugimoto and Murayama52). Shortly following weaning these mice become obese, insulin resistant and develop steatosis on a chow diet which can become exacerbated on a HFD(Reference Trak-Smayra, Paradis and Massart51) (Fig. 2(a)). Although these mice readily develop steatosis they do not consistently progress to steatohepatitis without additional metabolic challenge(Reference Handa, Maliken and Nelson26) (Fig. 2(b)). Following 10 weeks of high-saturated fat diet feeding in db/db mice, half of the cohort developed NAFLD while the remaining mice progressed to NASH(Reference Handa, Maliken and Nelson26). There was an inverse relationship between db/db mice with NASH and levels of adiponectin, suggesting that hypoadiponectinaemia may be a critical component of the metabolic syndrome and the progression to NASH(Reference Handa, Maliken and Nelson26). A thorough review of the variety of genetic models available to study NAFLD has been recently published(Reference Nagarajan, Mahesh Kumar and Venkatesan46).
Fig. 2 Formalin-fixed, paraffin-embedded liver sections stained with Gomori’s Trichrome from db/db mice fed either a high-fat, high-sucrose diet or a methionine- and choline-deficient (MCD) diet. All images are at 20X magnification with insets at 40X magnification. (a) db/db mice fed a high-fat, high-sucrose diet for 12 weeks developed microvesicular steatosis. (b) db/db mice fed a MCD diet for 3 weeks had macrovesicular steatosis and lobular inflammation. CV, central vein; PT, portal tract.
foz/foz
Similar to Alström syndrome in humans, foz/foz mice with a mutation in the Alms1 gene manifest a disease phenotype characterised by obesity and type 2 diabetes(Reference Arsov, Silva and O'Bryan53). After 250 d of feeding a regular chow diet both male and female foz/foz mice develop significantly increased body fat mass compared with wild-type controls(Reference Arsov, Silva and O'Bryan53). Switching foz/foz mice onto a HFD for 24 weeks leads to an accelerated obesity phenotype and the development of steatohepatitis including elevated ALT levels, lobular inflammation, ballooning hepatocytes and increased expression of fibrosis genes type 1 collagen, α-smooth muscle actin (α-SMA) and matrix metallopeptidase 9 (MMP-9)(Reference Larter, Yeh and Haigh54, Reference Van Rooyen, Larter and Haigh55). Interestingly, when foz/foz mice are switched back to a chow diet after 12 weeks of HFD feeding they have improved liver function, reduced blood glucose levels and increased adiponectin compared with foz/foz mice fed on HFD(Reference Larter, Yeh and Haigh54). The reduced progression of steatohepatitis was linked to a shift in lipid metabolism including the redistribution of TAG storage in adipose tissue over the liver, reduced hepatic fatty acid uptake and macrophage activation favouring tissue recovery over fibrosis, demonstrating a key link between diet and the progression of NASH(Reference Larter, Yeh and Haigh54).
Murine models best for diet-induced non-alcoholic fatty liver disease
When choosing an animal model to study NAFLD there are a number of characteristics that should be considered. Due to the association between adipose tissue inflammation, insulin resistance and steatosis, diets high in saturated fat are often used to model obesity, glucose intolerance and NAFLD in mice(Reference Carpenter, Strohacker and McFarlin56). Depending on the primary aim of the study it is key to identify the differences in dietary models as HFD alone have been shown to lead to alterations in weight gain and insulin sensitivity compared with high-sucrose and high-fat diets, and high-sucrose diets(Reference Omar, Pacini and Ahrén57, Reference Pyndt Jorgensen, Hansen and Krych58). Comparison of diets ranging in fat from 30 to 60 % demonstrated that a diet containing >45 % of energy from fat results in the greatest weight gain after 18 weeks of treatment in CD-1 mice(Reference Carpenter, Strohacker and McFarlin56). Extrapolation of findings from HFD-fed mice must be used with caution as certain obesity-related diseases are more representative of the human condition than others(Reference Brainard, Watson and Demartino59).
High-fat diets
Dietary treatment with a diet high in fat is one of the most common animal models to study obesity, insulin resistance and liver steatosis(Reference Xu, Kim and Bai60–Reference Galloway, Lee and Brookes65). Often the diets are composed of 40–60 % energy from fat that can be a derived from a combination of saturated fat, probably from lard and unsaturated fat in maize oil(Reference Lafarge, Pini and Pelloux66). One of the most critical factors when employing the HFD model is that a long duration of feeding, usually greater than 10 weeks, is required for the full effects of the diet to manifest into obesity, glucose intolerance and NAFLD(Reference Xu, Kim and Bai60–Reference Galloway, Lee and Brookes65). While mice fed a HFD for over 10 weeks develop steatosis, a much longer duration exceeding 40 weeks of HFD feeding is required to initiate a mild degree of inflammation more closely representing NASH(Reference Hennig, Mikula and Goryca67).
In addition to increased fat mass and impaired glucose tolerance, long-term HFD feeding has also been linked to a number of defects including delayed fracture healing in mice(Reference Brown, Yukata and Farnsworth68). At 5 weeks following injury, mice on a HFD had reduced bone volume and biomechanically weaker repair compared with chow-fed mice(Reference Brown, Yukata and Farnsworth68). It was proposed that this difference was due to an increased number of adipocytes at the fracture location and a subsequent loss of mesenchymal-derived osteoblasts(Reference Brown, Yukata and Farnsworth68). The mutually exclusive relationship between adipocyte formation and bone formation from mesenchymal stem cells continues to be evaluated(Reference Brown, Yukata and Farnsworth68). Recently, HFD feeding for 8 weeks was also associated with reduced metabolic activity at the blood–brain barrier(Reference Ouyang, Hsuchou and Kastin69). Analysis of HFD feeding for 10 weeks on the lymphatic system demonstrated a reduction in inguinal lymph node size, dysregulation of T cell migration, increased infiltration of B cells and macrophages and impaired lymphatic fluid transport(Reference Weitman, Aschen and Farias-Eisner70).
Sex and age differences between male and female C57B/Bl6 mice on HFD feeding have been reported(Reference Yang, Smith and Keating71). Male mice fed HFD on average consume more energy and therefore weigh more than their chow-fed male controls as well as female mice fed HFD(Reference Yang, Smith and Keating71). There was an increase in fat mass in both male and female groups fed HFD whereas no difference in fat-free mass(Reference Yang, Smith and Keating71). In addition, older-aged mice (>23 weeks of age) were found to have greater variability in fat mass than young mice (8 weeks of age)(Reference Yang, Smith and Keating71). Future studies employing wild-type mice should consider how sex and age differences might affect the study outcome.
‘Westernised’ diet
Combination of a HFD with added fructose or sucrose is considered to be more reflective of a ‘Westernised’ diet and has been characterised(Reference Tetri, Basaranoglu and Brunt72, Reference Yang, Miyahara and Takeo73). After 16 weeks of feeding the American lifestyle-induced obesity syndrome (ALIOS) diet in male C57BL/6 mice, increased weight gain, elevated serum glucose levels and higher ALT levels compared with chow-fed mice were observed(Reference Tetri, Basaranoglu and Brunt72). The ALIOS diet is composed of 45 % of energy from fat as well as high-fructose maize syrup added in the drinking water. Mice fed the ALIOS diet develop macrovesicular steatosis in zones one and two and microvesicular steatosis predominately in zone three(Reference Tetri, Basaranoglu and Brunt72). The development of NASH was limited to few inflammatory foci but there was increased expression of TNF-α within hepatocytes(Reference Tetri, Basaranoglu and Brunt72). It has been observed that even after 24 weeks of HFD feeding plus addition of fructose in the drinking water (5 %, w/v), approximately 30 % of mice in the cohort resisted excessive weight gain and remained insulin sensitive(Reference Tan, McLennan and Williams74). This evidence suggests that ‘Westernised’ diet models should be fed for minimally 4 months and span upwards of 9 months(Reference Tetri, Basaranoglu and Brunt72). In addition each cohort of mice should be stratified based on weight gain to identify mice that are prone to obesity compared with mice that remain lean following high-energy-diet feeding(Reference Tan, McLennan and Williams74). We have found that feeding male C57BL/6 mice a high-fat high-sucrose diet for 10 weeks results in mild steatosis and slightly elevated serum ALT levels (Fig. 3(a)–(c)).
Fig. 3 Formalin-fixed, paraffin-embedded liver sections stained with Gomori’s Trichrome from C57BL/6 mice fed either a standard chow diet or a high-fat, high-sucrose (HF-HS) diet. All images are at 20X magnification. (a) C57BL/6 mice fed a standard chow diet. (b) C57BL/6 fed a HF-HS diet for 10 weeks, demonstrating mild steatosis. (c) Serum alanine aminotransferase (ALT) levels of mice fed a standard chow diet or a HF-HS diet. Values are means, with standard errors represented by vertical bars. No statistical difference was detected (P=0·14). CV, central vein; PT, portal tract.
Carbohydrate-enriched diet
Diets enriched with refined carbohydrates such as sucrose and fructose have been found to increase the susceptibility of mice to develop steatosis and insulin resistance similar to that of the human condition(Reference Spruss, Henkel and Kanuri75–Reference Oliveira, Menezes-Garcia and Henriques79). Mice fed a diet high in fat and refined carbohydrates developed hyperglycaemia and impaired glucose tolerance compared with HFD alone in C57BL/6 mice(Reference Hodgson, Govan and Ketheesan76). Supplementing both male and female C57BL/6 mice with 30 % fructose drinking water for 16 weeks led to greater weight gain in male mice but when normalised to liver weight, female mice were found to have a significantly greater liver:body-weight ratio(Reference Spruss, Henkel and Kanuri75). Histological assessment of liver tissue demonstrated significant development of steatosis in both male and female mice; however, serum ALT and inflammation scores did not differ significantly between mice on the fructose solution compared with normal drinking water(Reference Spruss, Henkel and Kanuri75). In addition, chronic fructose feeding led to increased portal endotoxin levels in both male and female mice, TNFα concentration; also Toll-like receptor adaptor molecule MyD88 expression was elevated in these groups(Reference Spruss, Henkel and Kanuri75). Heightened changes in female mice over male mice was linked to decreased adiponectin levels in adipose tissue as well as increased levels of acute-phase protein-1 in the liver and lower phosphorylation of 5' AMP-activated protein kinase (AMPK) in the liver of female mice(Reference Spruss, Henkel and Kanuri75). A HFD along with the addition of fructose leads to substantial hepatic steatosis and represents a good model to study the initiation of NAFLD(Reference Spruss, Henkel and Kanuri75).
Murine models best for diet-induced non-alcoholic steatohepatitis
Methionine- and choline-deficient diet
Methionine and choline deficiency is a well-characterised dietary rodent model of NASH(Reference Heinrichs, Berres and Coeuru80). Choline is considered an essential nutrient that is required for cell membrane integrity, the neurotransmitter acetylcholine and synthesis of VLDL(Reference Buchman15). As well, choline deficiency has been linked to cellular apoptosis as evident by hepatocyte cell death when placed in a choline-deficient medium. When choline deficiency is coupled with the lack of the essential amino acid methionine, mice develop steatosis and inflammation within 3 weeks and can progress to fibrosis with longer duration of feeding (6–8 weeks on average)(Reference Weltman, Farrell and Liddle81, Reference Bellafante, Murzilli and Salvatore82) (Fig. 4(a)–(c)). A similar trend has been observed in patients receiving TPN(Reference Buchman, Ament and Sohel83). Choline supplementation in TPN feeding was shown to improve liver biochemistry and should be added to long-term TPN to help prevent liver disease(Reference Buchman, Ament and Sohel83). The pathogenesis of NASH in the methionine- and choline-deficient (MCD) diet model is thought to be due to impaired β-oxidation and heightened production of reactive oxygen species leading to oxidative stress. Development of NASH in this model has also been associated with the secretion of proinflammatory cytokines and chemokines and the subsequent recruitment of leucocytes into the liver(Reference Farrell, van Rooyen and Gan38).
Fig. 4 Formalin-fixed, paraffin-embedded liver sections stained with Picrosirius Red from C57BL/6 mice fed either a standard chow diet or a methionine- and choline-deficient (MCD) diet. All images are at 20X magnification. (a) C57BL/6 fed a standard chow diet. (b) C57BL/6 mice fed a MCD diet for 3 weeks. Mild fibrosis is evident forming around lipid-laden hepatocytes. (c) C57BL/6 mice fed a MCD diet for 6 weeks. Extended diet treatment results in the development of bridging fibrosis within the liver reflective of chronic inflammation and hepatocellular damage.
Recently MCD diet feeding was associated with disrupted vascular architecture within the liver, suggesting that angiogenesis may be connected to the progression of liver fibrosis, cirrhosis and HCC. After 4 weeks of MCD diet feeding C57BL6/J mice had increased vascular endothelial growth factor (VEGF) in the liver and treatment with αVEGFR2 was able to reduce the degree of inflammation and hepatocellular ballooning after 8 weeks of MCD diet feeding(Reference Coulon, Legry and Heindryckx84). The availability and ease of use makes the MCD diet model a valuable tool to investigate the role of inflammation during NASH. However, animals become cachectic and may lose up to 50 % of their body weight, ameliorating any obese or insulin-resistant phenotype. We have found that following 3 weeks of MCD diet treatment, diet-induced obese mice develop steatohepatitis; however, their blood glucose levels normalise within the MCD diet feeding period (DT Reid, unpublished results). Recently a new diet combining choline deficiency with HFD was demonstrated to lead to hepatic steatosis, inflammation and fibrosis while the mice retained a stable body weight(Reference Matsumoto, Hada and Sakamaki85). Further experimentation is required to characterise the metabolic status of this dietary treatment; however, this diet represents a novel approach to studying NASH in the context of the metabolic syndrome(Reference Matsumoto, Hada and Sakamaki85).
Use of the db/db mouse in combination with the MCD diet treatment has also been used as an attractive model of obesity, insulin resistance and NASH(Reference Sahai, Malladi and Pan86, Reference Rinella, Elias and Smolak87). In this case insulin resistance is manifested by the hyperphagic behaviour of the mice while at the same time the MCD diet feeding induces inflammation within the liver. Following 4 weeks of diet, db/db mice are susceptible to NASH and are found to have steatosis, inflammatory granulomas present in liver tissue and elevated ALT levels(Reference Salamone, Galvano and Marino Gammazza88). However, we have found that after only 3 weeks of MCD diet feeding, db/db mice lose approximately 15 % of their original body weight and have significantly lower glucose AUC compared with HFD-fed db/db mice following a 90 min oral glucose tolerance test. This suggests that although db/db mice develop NASH on the MCD diet, their insulin sensitivity has improved and may no longer represent a model of the metabolic syndrome.
Extended duration of high-fat diets
Long-term HFD studies have become a popular model to study the development of NASH. Use of a ‘fast food’ diet rich in saturated fat and fructose in C57BL/6 mice for 25 weeks resulted in greater body weight, elevated aminotransferase levels and homeostatic model assessment of insulin resistance (HOMA-IR) scores compared with chow-fed mice(Reference Charlton, Krishnan and Viker89). This diet led to steatosis and evidence of ballooning hepatocytes. As well, bridging fibrosis was evident following 25 weeks of HFD feeding. The combination of high fat plus fructose(Reference Fujii, Enomoto and Fukushima90) in this diet was central to the development of NASH within these mice. Compared with the fast food diet, HF diet-fed mice did not develop fibrosis and had substantially less inflammation and hepatocyte ballooning even after 25 weeks of HF diet treatment. Histological comparison between MCD diet and HF diet feeding demonstrates similar disease progression; however, HF diet treatment requires longer treatment duration and on average leads to less severe NASH. Vitamin D deficiency has been found to accelerate NASH in a HFD model(Reference Kong, Zhu and Bai22). This phenotype was attributed to the loss of bile acid uptake through the ileal apical Na-dependent bile acid cotransporter (iABST) and increased de novo lipogenesis leading to heightened inflammation and exacerbation to steatohepatitis(Reference Kong, Zhu and Bai22).
High-cholesterol and -cholic acid diets
Similar to the HFD model, diets high in cholesterol and cholic acid are considered a more atherogenic diet and have been developed as a model of NASH(Reference Matsuzawa, Takamura and Kurita91). A period of 30 weeks of high-fat/high-cholesterol feeding (15 %, 1 % (w/w), respectively) resulted in weight gain and steatohepatitis including elevated ALT levels, steatosis, lobular inflammation and perisinusoidal fibrosis(Reference Savard, Tartaglione and Kuver92). Critically, the development of NASH was dependent on the combined high-fat and high-cholesterol feeding as neither a HFD or high-cholesterol diet alone led to steatohepatitis(Reference Savard, Tartaglione and Kuver92). A combination of high-fat, high-cholesterol and high-fructose diet resulted in hepatic steatosis, exacerbated TNF-related apoptosis-inducing ligand (TRAIL)-mediated liver injury, increased expression of proinflammatory genes and elevated Mac-2 staining in the liver after 12 weeks of treatment(Reference Hirsova, Ibrahim and Bronk93). Further study of the altered metabolism in mice fed this diet is required to fully elucidate the disease model.
Development of hepatocellular carcinoma during non-alcoholic fatty liver disease
Mirroring the increasing rates of obesity, type 2 diabetes and NAFLD is the increased incidence of HCC(Reference Streba, Vere and Rogoveanu94). NAFLD is now considered a risk factor for the development of HCC as well as both cirrhotic and non-cirrhotic NASH(Reference Streba, Vere and Rogoveanu94–Reference Alexander, Torbenson and Wu96). Animal models have been used to study HCC in the context of NAFLD(Reference Streba, Vere and Rogoveanu94). In one case male neonatal mice exposed to streptozotocin followed HFD feeding develop insulin resistance and hepatic steatosis that can progress to fibrosis and HCC (STAM model)(Reference Fujii and Kawada97). NASH-related HCC in the absence of cirrhosis has also been described in patient populations; however, animal models to investigate this relationship have not been reported(Reference Kawada, Imanaka and Kawaguchi98).
Conclusion
Multiple rodent models can be used to study the pathogenesis of NAFLD and NASH in the context of obesity. In particular, long-term feeding of certain dietary components such as saturated fat, cholesterol and fructose can lead to steatosis as well as mild inflammation and hepatocyte damage. Diets deficient in methionine and choline provide a robust model to study leucocyte infiltration during NASH. However, particular care should be taken when selecting an animal model to study NAFLD and NASH as current available models do not represent the full disease spectrum. Continued evolution of animal models to investigate NAFLD and NASH is required.
Acknowledgements
The present review was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) fellowship (to D. T. R.) and a Canadian Institutes of Health Research (CIHR) Signature Initiative Team grant (to B. E.).
D. T. R. and B. E. have no financial disclosures relating to this body of work.