



Introduction
During the last few decades, evidence derived from both clinical and experimental research has revealed that advanced glycation endproducts (AGE) have profound effects on human pathology. AGE are a large group of heterogeneous compounds formed either endogenously or ingested through the diet, which were initially reported to be involved in the pathogenesis of diabetes complications. However, the role of AGE has spread progressively to many non-transmissible diseases including CVD, obesity, the metabolic syndrome, autoimmune diseases, neurodegenerative diseases, cancer, as well as the normal ageing process, all of which have in common elevated levels of inflammation and oxidative stress (OS)(Reference Uribarri, del Castillo and de la Maza1). In the present review we will describe dietary AGE, how they form and how they are absorbed into the body, their main mechanisms of actions, current evidence on their potential role in the development of chronic non-infectious clinical conditions, suggestions on how to restrict them and evidence supporting potential benefits of lowering their consumption.
What are advanced glycation endproducts?
AGE are a very large and heterogeneous group of compounds that originate from the spontaneous reaction of reducing sugars with free amino groups in amino acids in the so-called Maillard or browning reaction(Reference Lin, Wu and Yen2). The Maillard reaction is well established, but AGE can also form through many other reactions, including oxidation of sugars, lipids and amino acids. Specific AGE such as pentosidine, N ε-carboxymethyllysine (CML), N ε-carboxyethyllysine (CEL) and methylglyoxal derivatives (MG-H1; N δ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine) are some of the most commonly measured and well-described AGE in biological studies(Reference Vistoli, De Maddis and Cipak3).
Endogenous AGE form physiologically in the body at a continuous slow rate, which is markedly increased in conditions of hyperglycaemia or elevated OS. AGE, however, can also form exogenously, outside of the body, in any system when the required reagents for the above reactions are available(Reference O’Brien and Morrissey4-Reference Uribarri, Woodruff and Goodman6). AGE form spontaneously in food, but their rate of formation is markedly increased when food is processed and cooked with heat. Higher temperature, longer time of application, lower moisture, presence of trace metals and higher pH of the cooking system enhance AGE formation(Reference O’Brien and Morrissey4-Reference Uribarri, Woodruff and Goodman6). Therefore, cooking methods that use dry heat, such as broiling, grilling, searing and frying have been shown to have the highest content of AGE, while cooking methods that use low heat application in the presence of high water content such as stewing, poaching or steaming produce significantly less AGE(Reference O’Brien and Morrissey4-Reference Uribarri, Woodruff and Goodman6). Only a fraction of the ingested dietary AGE are absorbed into the body AGE pool, where they become indistinguishable from their endogenous counterparts, both in structure and function(Reference Koschinsky, He and Mitsuhashi7).
Dietary AGE intake can be easily decreased by changing the method of cooking from a high dry heat application to a low heat and high humidity, independent of nutrient composition. Databases with the AGE content of common foods have been published and can be used to calculate dietary AGE intake as well as to provide guidance on how to consume meals with less AGE content(Reference O’Brien and Morrissey4-Reference Uribarri, Woodruff and Goodman6). The essential concept in the low-AGE diet is that the same type and amount of foods consumed can provide very different amounts of AGE depending on the cooking method. For example, using published data(Reference Goldberg, Cai and Peppa5,Reference Uribarri, Woodruff and Goodman6) in which the AGE content of food was measured with ELISA and expressed in arbitrary AGE kilounits (kU)/serving, a 90 g beefsteak contains 720 AGE kU when raw, 2199 kU when stewed and 6731 kU when broiled(Reference Goldberg, Cai and Peppa5,Reference Uribarri, Woodruff and Goodman6) . The temperature and method of cooking appear more critical to AGE formation than time of cooking; for instance, meat samples broiled or grilled at 230°C for shorter cooking times have higher AGE content when compared with samples boiled in liquid media at 100°C for longer periods(Reference Goldberg, Cai and Peppa5). Unfortunately, there is no specific threshold temperature above which AGE start to generate and one can only make the general recommendation that the lower the temperature the less the amount of AGE generated(Reference O’Brien and Morrissey4-Reference Uribarri, Woodruff and Goodman6). At present, there are no official recommendations about acceptable dietary AGE intake. It has been previously proposed that half of the current mean AGE intake, or about 7500 kU/d, would be a realistic goal(Reference Goldberg, Cai and Peppa5,Reference Uribarri, Woodruff and Goodman6) since some studies have shown that dietary AGE reduction of this magnitude is feasible and can significantly alter levels of circulating AGE, while at the same time reducing levels of markers of OS and inflammation and enhancing insulin sensitivity in diabetic patients(Reference Uribarri, Cai and Ramdas8). Because the low-AGE diet depends on the culinary technique, and not on the food being cooked, it can be applied for any group of individuals; the same principles will apply whether the patient has diabetes, CVD or not.
AGE can be measured by several methods including fluorescence, ELISA and MS. Most of the early data on food AGE were based on ELISA measurements, which has raised significant criticism since these assays have more problems in assessing food matrices than methods using MS(Reference Assar, Moloney and Lima9). This controversy has prevented a faster development of this field and a general agreement in terms of actual quantification of the AGE content in different foods is still in discussion. At present, however, databases with the food AGE content measured by MS have been published and can be used by anyone(Reference Scheijen, Clevers and Engelen10).
A frequently ignored source of exogenous AGE is tobacco smoking(Reference Cerami, Founds and Nicholl11). Curing of tobacco generates large amounts of AGE that after smoke inhalation are absorbed into the circulation, increasing the body AGE pool similarly to food-derived AGE(Reference Cerami, Founds and Nicholl11). Once in the bloodstream, these AGE may react with serum proteins such as apoB as well as with vascular wall proteins and thereby accelerate the development of atherosclerosis, whose incidence is increased in smokers. These smoking-derived AGE have been shown to have the same actions as endogenous AGE and therefore may cross-link proteins and activate receptors throughout the body(Reference Cerami, Founds and Nicholl11).
Gastrointestinal absorption of dietary advanced glycation endproducts
Gastrointestinal metabolism of dietary AGE depends on two phenomena: hydrolysis of proteins and absorption of the resulting free amino acids or small peptides containing the AGE. The nutritional value of proteins is reduced by the Maillard reaction, mainly due to a reduction of the digestibility of proteins and the bioavailability of some amino acids(Reference Sharma, Kaur and Thind12,Reference Su, Li and Zhao13) . Accessibility of digestive enzymes for hydrolysis is restricted by chemical modifications of the target amino acids and structural changes of proteins that limit enzyme–substrate interactions(Reference Su, Li and Zhao13-Reference Zhao, Le and Larsen15), all of which reduce the rate of protein hydrolysis(Reference Salazar-Villanea, Bruininx and Gruppen16) and increase the molecular weight of the resulting peptides(Reference Zhao, Li and Le14,Reference Hellwig, Matthes and Peto17,Reference Salazar-Villanea, Bruininx and Gruppen18) . The extent of hydrolysis of a protein containing Maillard modifications depends on the type of AGE that are formed(Reference Zhao, Le and Larsen15,Reference Hellwig, Matthes and Peto17) , the extent of modification of the protein(Reference Zhao, Li and Le14) and the protein source(Reference Hellwig, Matthes and Peto17). For example, after hydrolysis of a CML-modified casein under simulated gastric and intestinal conditions, most of the CML-containing peptides resulted in molecular weights between 250 and 1000 Da, which are assumed to be digested, yet not necessarily absorbable(Reference Hellwig, Matthes and Peto17).
Absorption of dietary AGE depends on the molecular weight of the products of protein hydrolysis(Reference Zhao, Li and Le14,Reference Hellwig, Matthes and Peto17) , the type of transporters present in the gastrointestinal epithelium(Reference Stefanie, Michael and Madlen19,Reference Hellwig, Geissler and Matthes20) and the type of AGE being transported(Reference Förster, Kühne and Henle21). Some free-form AGE can be absorbed by simple diffusion(Reference Grunwald, Krause and Bruch22), while peptide-bound AGE require peptide transporters to cross the epithelium(Reference Hellwig, Geissler and Matthes20), for example the di- and tripeptide transporter PEPT1(Reference Stefanie, Michael and Madlen19). Glycated dipeptides can cross a barrier of epithelial cells probably using the peptide transporter PEPT1(Reference Hellwig, Geissler and Matthes20). This was demonstrated for low molecular-weight peptides containing pyrraline that once inside the cell are hydrolysed allowing free pyrraline to enter the basolateral side by simple diffusion(Reference Hellwig, Geissler and Peto23). Furthermore, the transport of peptide-bound pyrraline across Caco-2 cells was shown to be 15-fold higher compared with free pyrraline(Reference Hellwig, Geissler and Peto23).
In one clinical study, approximately 10 % of the dietary intake of AGE reached the systemic circulation(Reference Koschinsky, He and Mitsuhashi7), which is consistent with a recent study in pigs(Reference Salazar-Villanea, Butré and Wierenga24) showing that the apparent ileal digestibility of CML, N ε-carboxyethyllysine (CEL) and fructose-lysine ranged between 0 and 30 %.
The intestinal in situ formation of fructose-derived AGE has been postulated, which once absorbed, become part of the body AGE pool, similar to dietary AGE(Reference DeChristopher, Uribarri and Tucker25). Intraluminal formation of fructose-derived AGE may be facilitated by increased intake of beverages and food containing high-fructose maize syrup in which the ratio of fructose to glucose is higher than 1:1, therefore creating conditions of potential fructose malabsorption favouring intraluminal generation of fructose-derived AGE(Reference DeChristopher, Uribarri and Tucker25). Preliminary in vitro studies have confirmed this postulation(Reference Bains, Gugliucci and Caccavello26,Reference Martinez-Saez, Fernandez-Gomez and Weijing27) . Increased fructose intake and absorption as such may also potentially increase the endogenous formation of AGE and therefore increase the body AGE pool with all its subsequent effects(Reference Aragno and Mastrocola28).
Once exogenous AGE are absorbed into the circulation, they act in the circulation and at different cellular and tissue levels inducing either protein cross-linking or a variety of cellular effects that eventually lead to increased OS and inflammation, precursors of most non-infectious chronic diseases.
Fig. 1 illustrates the metabolism of dietary AGE within the gastrointestinal tract.

Fig. 1. Metabolism of dietary advanced glycation endproducts (AGE). Panel 1A: Food AGE are ingested. Panel 1B: Some AGE may form in situ within the lumen of the intestine in conditions of high local fructose. Panel 2: Both exogenous and in situ-formed AGE may be partially absorbed through the small intestine into the circulation from where they can be excreted in the urine by the kidneys or reach the tissues where they can produce direct protein cross-linking or react with cellular receptors to induce inflammatory cytokines or oxidative stress. Panel 3: AGE not absorbed in the small intestine move to the colon where several things can happen. 3a: AGE may be broken down by bacteria of the microbiota. 3b: AGE may react with bacteria, releasing compounds that may be absorbed across the colon. 3c: AGE may potentially be absorbed through the colon mucosa or locally activate RAGE. 3d: AGE can be eliminated in the stools. For a colour figure, see the online version of the paper.
The role of unabsorbed dietary advanced glycation endproducts in the colon
The major part of the dietary AGE escapes gastrointestinal absorption with a potential effect of unabsorbed dietary AGE in the colon microbiota and even directly in the colon wall. Some of these AGE could bind to the receptor for AGE (RAGE) or Toll-like receptors in the colon cells inducing a local inflammatory response with subsequent release of inflammatory mediators into the circulation eventually producing chronic diseases, or they could alter the microbiome profile in the gut, which in turn leads to release of toxins or inflammatory mediators into the circulation(Reference Snelson and Coughlan29). We know from studies in mice that activation of RAGE-dependent signalling is key in creating mucosal barrier dysfunction after haemorrhagic shock(Reference Raman, Sappington and Yang30). One could postulate that this RAGE-dependent disruption of the intestinal epithelial barrier may contribute to systemic inflammation by facilitating translocation of endotoxin, microbial fragments and other factors produced locally by the gut flora(Reference Vaziri, Zhao and Pahl31).
Degradation of CML and pyrraline by human gut microbiota has been well documented(Reference Hellwig, Auerbach and Müller32). The products of this degradation of dietary AGE in the large intestine could in turn modulate the profile of the bacterial population and the metabolites produced by them, which in turn could facilitate systemic illnesses(Reference Raman, Sappington and Yang30).
A 2‐week randomised two‐period cross-over study, in which two different diets were consumed by male adolescents, showed negative correlation between faecal lactobacilli and enterobacteria numbers and dietary CML content(Reference Seiquer, Rubio and Peinado33). A different study analysed the effects of a low- v. high-AGE diet for 1 month in a small group of peritoneal dialysis patients(Reference Yacoub, Nugent and Cai34). Dietary AGE restriction altered the bacterial gut microbiota with a significant reduction in Prevotella copri and Bifidobacterium animalis relative abundance and increased Alistipes indistinctus, Clostridium citroniae, C. hathewayi and Ruminococcus gauvreauii relative abundance(Reference Yacoub, Nugent and Cai34). The exact significance and systemic effects of these changes in the faecal microbiota, however, remain to be determined.
The role of the advanced glycation endproduct–receptor for advanced glycation endproduct–reactive oxygen species loop
During the last three decades, compelling evidence derived from both clinical and experimental research has revealed that AGE have profound effects on human pathology, far beyond diabetes complications(Reference Uribarri, del Castillo and de la Maza1,Reference Lin, Wu and Yen2,Reference Yan, Ramasamy and Schmidt35) . High dietary AGE intake produces a sustained oxidant overload that may overwhelm host defences and lead to unopposed OS and chronic subclinical inflammation, which over time raises susceptibility to a variety of chronic clinical conditions(Reference Vlassara and Uribarri36).
AGE exert their action through different mechanisms; one is based on the capacity of these compounds to alter the function of many biomolecules, either by reducing its biological activity or by inducing cross-linking with proteins(Reference Rojas, González, Añazco and Uribarri37). Additionally, several AGE-binding proteins have been identified. Some of these proteins play an important role in the removal of AGE, including the AGE receptor complex, AGE-R1/OST-48, AGE-R2/80K-H, AGE-R3/galectin-3, as well as some members of the scavenger receptor family(Reference Yan, Ramasamy and Schmidt35,Reference Rojas, González, Añazco and Uribarri37) . Very interestingly, the activity of AGE-R1 is not only restricted to clearance of AGE, but also it has further activities linked to counteract the pro-oxidant activities of AGE(Reference Cai, He and Zhu38,Reference Cai, He and Zhu39) . Furthermore, AGE also bind to RAGE on different cell types, thus activating key cell signalling pathways with subsequent modulation of inflammation-related gene expression. The RAGE–ligand interaction triggers activation of NF-κB and other signalling pathways through stimulation of p21ras, extracellular signal-regulated kinase (ERK) 1/2, p38 mitogen-activated protein kinase (p38 MAPK), stress-activated protein kinase (SAPK)/c-Jun N-terminal kinase (JNK), Rho GTPases, phosphoinositide 3-kinase (PI3K) and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways(Reference Rojas, Delgado-López and González40-Reference Ott, Jacobs and Haucke44). Subsequently, expression of many inflammatory gene products is increased, leading to the establishment of an inflammatory niche(Reference Hofmann, Drury and Fu45).
RAGE binds ligands other than AGE, particularly high-mobility group protein box-1 (HMGB1), calgranulins, amyloid-β-protein, triggering cellular responses extensively involved in different pathophysiological scenarios including inflammation, proliferation, angiogenesis, migration and fibrosis(Reference Wautier, Guillausseau and Wautier46-Reference Alleyn, Breitzig and Lockey49). Binding of RAGE not only causes an inflammatory gene expression profile, but also a feed-forward loop, in which inflammatory stimuli activate NF-κB, which induces RAGE expression, subsequently followed by sustained NF-κB activation. Thus, RAGE can function as a master switch, converting short-lasting pro-inflammatory responses into long-lasting cellular dysfunction(Reference Bierhaus, Schiekofer and Schwaninger50,Reference Kierdorf and Fritz51) .
The complexity of signalling is further demonstrated by the presence of alternative adaptor molecules such as DAP10, and even by the recruitment of TIRAP (Toll/IL-1 receptor domain-containing adapter protein) and MyD88, well-known adaptor proteins for downstream signal transduction of Toll-like receptor (TLR)-2 and TLR-4, which are critical for enhancing RAGE-mediated signalling(Reference Sakaguchi, Murata and Aoyama52,Reference Sakaguchi, Murata and Yamamoto53) .
RAGE and TLR also share common ligands and signalling pathways, and accumulating evidence points towards their cooperative interaction. This fact is supported by several experimental findings, such as the capacity of AGE to induce PPARγ down-regulation-mediated inflammatory signalling via TLR4 and RAGE(Reference Chen, Sheu and Tsai54), as well as that AGE-modified molecules activate TLR signalling through binding to either TLR receptors on the cell surface or the intracellular adaptor proteins of TLR(Reference Cheng, Dong and Zhu55).
The RAGE gene undergoes extensive alternative splicing to produce a variety of transcripts with diverse functions. In this context, soluble variants include a secreted form RAGE v1 (previously named as sRAGE, secretory C-truncated RAGE, esRAGE, hRAGE sec or sRAGE1/2/3) and an N-terminally truncated isoform RAGE v2 (previously named Nt-RAGE, N-RAGE or N-truncated RAGE)(Reference González, Romero and Rodríguez56). However, endogenous soluble RAGE isoforms may be generated by mechanisms other than alternative splicing, such as membrane-associated proteases, including the sheddase A disintegrin, metalloprotease 10 (ADAM-10) and matrix metalloproteinase-9 (MMP-9)(Reference Raucci, Cugusi and Antonelli57,Reference Zhang, Bukulin and Kojro58) .
Soluble RAGE is thought by many authors to function as a decoy receptor, and thus preventing the interaction with the membrane-anchored full-length RAGE(Reference Salazar-Villanea, Butré and Wierenga24). In this context, there is evidence supporting a role for soluble RAGE contributing to the removal/detoxification of AGE(Reference Santilli, Vazzana and Bucciarelli59).
In the 1990s, it was initially reported that RAGE activation by AGE was associated with increased OS, as demonstrated either by the use of blocking antibodies to RAGE or antioxidants, both in vivo and in vitro (Reference Yan, Schmidt and Anderson60). As a consequence of RAGE engagement, multiple signalling pathways are induced, including the generation of reactive oxygen species (ROS), mainly due to the activation of the NADPH oxidase (NOX) pathway(Reference Guo, Ananthakrishnan and Qu61,Reference Wautier, Wautier and Schmidt62) . Interestingly, AGE–RAGE-induced cytosolic ROS production also facilitates mitochondrial superoxide production in hyperglycaemic environments, which play a central role in inducing diabetic-associated organ damage(Reference Coughlan, Thorburn and Penfold63). Of note, the cytoplasmic domain of RAGE binds to the formin molecule mDia1, through its conserved poly-proline rich formin homology-1 (FH1) domain, and this interaction is strictly required for RAGE ligands to activate cell signalling responses. Formins such as mDia1 are actin-binding molecules that contribute to signal transduction mechanisms, in part via Rho GTPase signals(Reference Young and Copeland64), and particularly Rac1, which is a key component in NADPH oxidase activation(Reference Hordijk65-Reference Acevedo and González-Billault67). There is increasing evidence that ROS induced by NADPH oxidases not only causes cell damage, but also acts as a second messenger implicated in proliferation, differentiation and apoptosis, which finally may lead to disease onset and progression(Reference Elnakish, Hassanain and Janssen68-Reference Hoffmann and Griffiths72).
OS and inflammation are inseparable actors in the pathogenesis of many human diseases. For instance, by activating NF-κB, a redox-sensitive transcription factor of major importance in inflammation, OS promotes the expression of pro-inflammatory cytokines and chemokines, increasing the recruitment and activation of leucocytes and resident cells, thereby fuelling inflammatory processes(Reference Gloire, Legrand-Poels and Piette73-Reference Gloire and Piette77). Additionally, plasma proteins are prone to be oxidised by ROS. These oxidised protein products, termed advanced oxidation protein products (AOPP), have been linked to vascular lesions in diabetes, chronic renal disease, obesity, immune-mediated inflammatory diseases, neurodegenerative diseases, cancer, the metabolic syndrome and atherosclerosis(Reference Cao, Hou and Nie78-Reference Witko-Sarsat, Friedlander and Nguyen Khoa81).
Of note, RAGE has also been described as a receptor of AOPP-modified albumin(Reference Guo, Niu and Hou82,Reference Zhou, Cao and Xie83) . Furthermore, RAGE expression on kidney podocytes is highly increased in response to chronic loading of AOPP. The AOPP–RAGE interaction activates NADPH oxidase, leading to ROS production and to a perpetuation of renal OS(Reference Yamamoto and Yamamoto84-Reference Wu, Zhong and Zhu86).
Finally, oxidants, derived from either the phagocyte NADPH oxidase or the myeloperoxidase–H2O2–chloride system, may lead to AGE production, particularly CML. These CML adducts of proteins act as ligands for RAGE and could potentially generate an important amplifying loop in tissue damage at inflammation sites(Reference Anderson, Requena and Crowley87,Reference Anderson and Heinecke88) (Fig. 2).
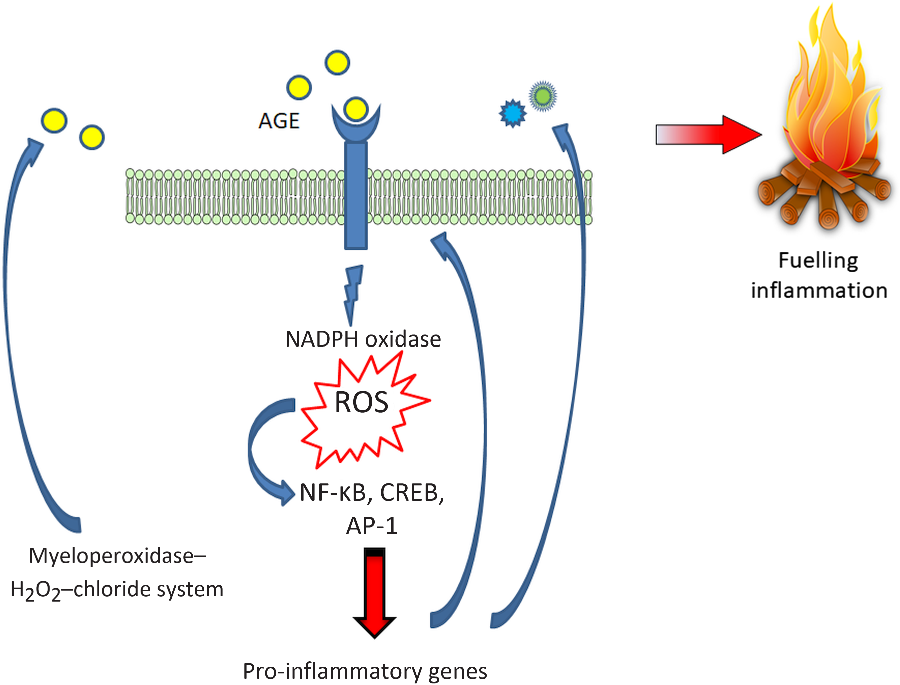
Fig. 2. Advanced glycation endproduct–receptor for advanced glycation endproduct–reactive oxygen species (AGE–RAGE–ROS) loops. RAGE engagement by AGE produces an early increase in NADPH oxidase-derived ROS which in turn, activate the redox-sensitive transcription factor NF-κB, and thus favouring a transcriptional pro-inflammatory profile fuelling inflammation and a positive feed-forward loop for RAGE itself. Additionally, ROS are also involved in another relevant amplifying loop, by promoting the formation of AGE, particularly N ε-carboxymethyllysine. CREB, cAMP response element-binding protein; AP-1, activator protein 1. For a colour figure, see the online version of the paper.
Potential role of dietary advanced glycation endproducts in chronic diseases
The effect of dietary AGE on a variety of clinical conditions in human subjects has been studied by many, using either acute oral loads of AGE(Reference Koschinsky, He and Mitsuhashi7,Reference Schiekofer, Franke and Andrassy89-Reference Davis, Prasad and Vijayagopal93) or dietary interventions(Reference Vlassara, Cai and Goodman94-Reference Yacoub, Nugent and Cai106), ranging from a few weeks up to 1 year consisting essentially of changing the way of cooking food without interfering with the actual energy and macronutrient intake. A summary of these studies with their main findings is listed in Table 1. One of the critiques of these clinical studies is that changing the way of cooking may change not only AGE but also many other factors such as vitamins and phytochemicals contained in food that may confound the interpretation of the effects resulting only as the results of the changes in food AGE content(Reference Kellow and Coughlan107). In order to address this critique, studies were performed in mice by providing a chronic load of a pure synthetic AGE precursor that produced the same effect as those of a high-AGE diet created by cooking with high heat(Reference Cai, Ramdas, Zhu and Chen108). However, no similar studies in human subjects are available.
Table 1. Dietary advanced glycation endproduct (AGE) interventions in human subjects
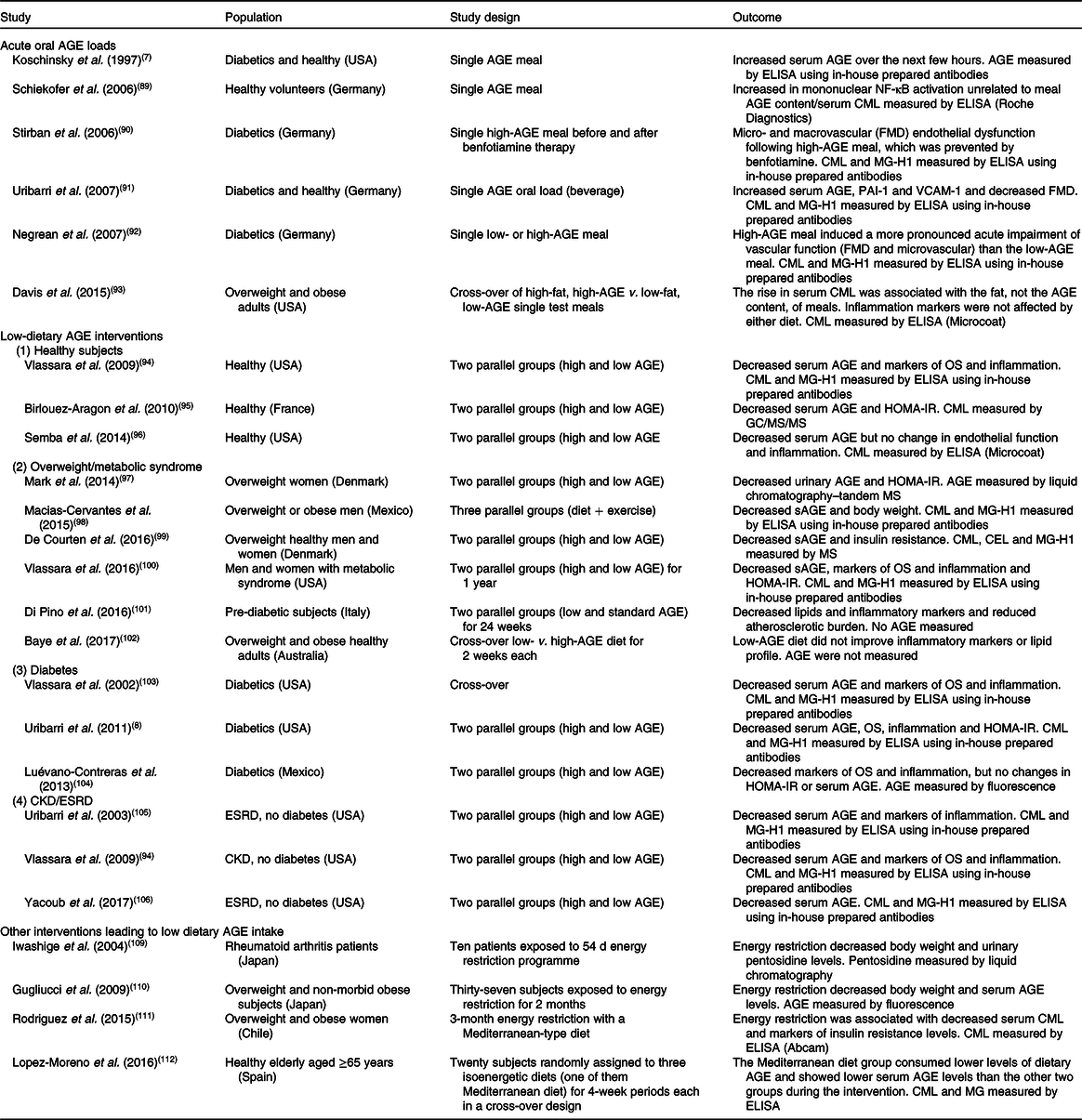
CML, N ε-carboxymethyllysine; FMD, flow-mediated vasodilatation; MG-H1, N δ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine; PAI-1, plasminogen activator-1; VCAM-1, vascular cell adhesion protein-1; OS, oxidative stress; HOMA-IR, homeostatic model assessment of insulin resistance; sAGE, serum AGE; CEL, N ε-carboxyethyllysine; CKD, chronic kidney disease; ESRD, end-stage renal disease.
The variability of method used in estimating dietary AGE content, mentioned above, also extends to measurements of AGE is serum, plasma or urine. Different methodologies including ELISA with commercially prepared kits or in-house developed antibodies as well as more precise MS/HPLC-based assays have been used by different groups. In order to give the readers a more balanced view of the different outcomes we are also including the AGE measurement methodology for each study listed in Table 1.
Four of the six acute oral AGE load studies showed that this load significantly increased circulating AGE levels in parallel with increases in markers of inflammation and endothelial dysfunction(Reference Koschinsky, He and Mitsuhashi7,Reference Stirban, Negrean and Stratmann90-Reference Negrean, Stirban and Stratmann92) , while two studies did not find this association(Reference Schiekofer, Franke and Andrassy89,Reference Davis, Prasad and Vijayagopal93) .
In Table 1 we also briefly mention two studies with energy restriction that demonstrated that this intervention is associated with reduced dietary AGE intake, probably related to the low AGE content of the replacement meals used for the studies(Reference Iwashige, Kouda and Kouda109,Reference Gugliucci, Kotani and Taing110) . Another study looked at the effect of energy restriction with a Mediterranean-type diet and showed a significant fall of serum CML levels; subjects were not given the actual meals but only instructions on how to eat(Reference Rodriguez, Leiva Balich and Concha111). The effect of the Mediterranean diet was tested in healthy elderly subjects in Spain and also showed that this intervention decreased serum AGE levels, although the magnitude of the change was small(Reference Lopez-Moreno, Quintana-Navarro and Delgado-Lista112).
In the next few sections we will review the above information, first in healthy subjects and in individuals with the metabolic syndrome, diabetes and kidney disease where evidence for a potential effect from dietary AGE comes from intervention studies (Table 1), and then in conditions such as CVD, dementia, sarcopenia and cancer in which intervention studies have not been performed, but there is a rationale for the potential involvement of dietary AGE.
Healthy subjects
Three studies have studied the effect of the low-AGE diet in healthy subjects(Reference Vlassara, Cai and Goodman94-Reference Semba, Gebauer and Baer96). In all three studies the restricted dietary AGE intervention led to decreased circulating levels of AGE. In one of the studies(Reference Vlassara, Cai and Goodman94), this occurred in parallel with decreasing levels of markers of OS and inflammation, while no effects on these parameters were observed in another study(Reference Semba, Gebauer and Baer96). Levels of AGE decreased in parallel with levels of homeostatic model assessment of insulin resistance (HOMA-IR), a marker of insulin resistance, in another study(Reference Birlouez-Aragon, Saavedra and Tessier95).
Overweight and/or metabolic syndrome
At least six studies have looked at the effect of a low-AGE diet on overweight and/or metabolic syndrome subjects(Reference Mark, Poulsen and Andersen97-Reference Baye, de Courten and Walker102). In four of these studies the low-AGE diet was associated with lower serum or urinary levels of AGE as well as a parallel decrease in HOMA-IR(Reference Semba, Gebauer and Baer96-Reference De Courten, de Courten and Soldatos99). In the fifth study(Reference Di Pino, Currenti and Urbano101) decreased levels of lipids and inflammatory markers were reported, but serum AGE were not measured. In the last study the low-AGE diet did not improve inflammatory markers or lipid profile, but the trial was rather short (2 weeks) and AGE were not measured(Reference Baye, de Courten and Walker102).
Diabetes
In 2016, an estimated 1·6 million deaths in the world were directly caused by diabetes(113). Therefore, the search for strategies to reduce incidence and complications of this disease is of great public health importance. Endogenous production of AGE is elevated in individuals with diabetes because of elevated blood glucose levels, but several studies in animals and in human subjects suggest a significant contribution of dietary AGE to the body pool of AGE in individuals with diabetes(Reference Uribarri, del Castillo and de la Maza1,Reference Vlassara and Uribarri36) .
A strong argument for the role of dietary AGE in diabetes has been demonstrated in clinical trials testing the effect of AGE restriction(Reference Uribarri, Cai and Ramdas8,Reference Vlassara, Cai and Crandall103,Reference Uribarri, Peppa and Cai105) . Two diets with the same energy and nutrient intake but differing only in their AGE content, one high AGE, and the other with 5-fold lower AGE content, were tested in twenty-four ambulatory individuals with diabetes(Reference Vlassara, Cai and Crandall103). During the high-AGE diet, serum AGE increased in parallel with levels of circulating markers of inflammation and endothelial dysfunction, while the opposite occurred in patients exposed to the low-AGE diet(Reference Vlassara, Cai and Crandall103). Before this study, high serum AGE levels in individuals with diabetes were thought to result exclusively from hyperglycaemia-induced endogenous overproduction. Therefore, the significant fall of serum AGE levels induced by the low-AGE diet, while maintaining the same overall glycaemic control, was a novel finding.
A randomised 6-week prospective study in type 2 diabetes subjects with standard diet (n 13) v. low-AGE diet (n 13) showed a significant decrease of TNF-α and malondialdehyde in the low-AGE diet group without a significant change in HOMA-IR or serum AGE(Reference Luévano-Contreras, Garay-Sevilla and Wrobel104). A 4-month randomised study of an AGE-restricted diet on thirty-six subjects with diabetes showed that these patients had lower levels of serum CML, MGH1, 8-isoprostanes and HOMA-IR than subjects on regular AGE intake(Reference Uribarri, Cai and Ramdas8). Reduction of HOMA-IR with the low-AGE diet in patients with type 2 diabetes raises the hypothesis that AGE have an important role in modifying insulin resistance itself in individuals with diabetes. Consistent with this hypothesis, an AGE-restricted diet, as shown above, has also been demonstrated to decrease HOMA-IR in subjects with the metabolic syndrome(Reference Mark, Poulsen and Andersen97-Reference Vlassara, Cai and Tripp100), broadening the potential beneficial effects of a low-AGE diet into pre-diabetes. If this effect of the low-AGE diet is further confirmed in larger clinical trials, it opens an opportunity for a safe, inexpensive and effective dietary modulation to prevent or improve diabetes and its secondary co-morbidities.
Chronic kidney disease
Circulating AGE levels increase with progressive chronic kidney disease(Reference Lopez-Moreno, Quintana-Navarro and Delgado-Lista112,Reference Vlassara, Torreggiani and Post114) . At least three clinical trials have tested the effects of an AGE-restricted diet in patients with renal disease in the absence of diabetes(Reference Vlassara, Cai and Goodman94,Reference Uribarri, Peppa and Cai105,Reference Yacoub, Nugent and Cai106) . A group of stage 3 chronic kidney disease patients was randomly assigned to either a regular diet or an isoenergetic diet containing 50 % lower AGE for a period of 4 weeks. The low-AGE diet patients had markedly decreased markers of inflammation and OS, including AGE, TNFα, vascular cell adhesion protein-1 (VCAM-1) and RAGE compared with the regular-diet group(Reference Baye, de Courten and Walker102). The number of patients was too small and duration of study too short to allow demonstration of an effect on the diet on proteinuria and/or actual kidney function. In another study, a group of non-diabetic end-stage renal disease patients on maintenance peritoneal dialysis was randomised to follow either a regular- or a low-AGE diet for 4 weeks; the low-AGE diet group had significant falls of levels of circulating AGE and C-reactive protein(Reference Uribarri, Peppa and Cai105). Another similar intervention on peritoneal dialysis patients again demonstrated an effect of the low-AGE diet in decreasing circulating AGE levels, but other markers were not measured in this study(Reference Yacoub, Nugent and Cai106).
CVD
CVD, such as CHD, cerebrovascular disease and chronic heart failure are major causes of death globally(Reference Henning115). The CVD rate in adults with diabetes is two to three times greater than in adults without diabetes(Reference Turner, Millns and Neil116). Several clinical studies have linked AGE to CVD, particularly in patients with diabetes mellitus. Observational studies have found associations of dietary AGE with markers of endothelial dysfunction, inflammation and OS such as TNF-α, VCAM-1, 8-isoprostane and mRNA RAGE, all consistently associated with CVD(Reference Stirban, Negrean and Götting117,Reference Baye, Kiriakova and Uribarri118) . Additionally, a high-AGE meal was shown to induce a more pronounced acute impairment of vascular function than an otherwise identical low-AGE meal in patients with diabetes(Reference Uribarri, Stirban and Sander91). Moreover, diets with 5-fold lower AGE content significantly decreased serum levels of AGE, VCAM-1 and C-reactive protein, compared with equivalent regular-AGE diets in diabetic patients(Reference Stirban, Negrean and Stratmann90-Reference Negrean, Stirban and Stratmann92).
The postprandial response to a high-AGE meal has been measured and shown acute decreases in flow-mediated vasodilatation and increased levels of intercellular adhesion molecule-1 (ICAM-1) and VCAM-1(Reference Uribarri, Stirban and Sander91). In a similar study, levels of ICAM-1 and VCAM-1 increased 4 h after the high-AGE meal, while flow-mediated dilatation and microvascular function decreased by 20·9 and 67·2 %, respectively(Reference Negrean, Stirban and Stratmann92). Furthermore, serum CML levels were associated with increased arterial stiffness in relatively healthy, community-dwelling adults(Reference Stirban, Negrean and Götting117). These findings suggest a pathological role of dietary AGE in vascular and cardiac stiffness in humans.
A recent meta-analysis of randomised controlled trials examining the effects of consumption of diets low in AGE on cardiometabolic parameters showed that low-AGE diets decreased insulin resistance, total cholesterol and LDL; in a subgroup of patients with type 2 diabetes, they also decreased fasting insulin, TNFα, VCAM-1, 8-isoprostane, leptin, circulating AGE and RAGE, while increasing protective factors adiponectin and sirtuin-1(Reference Baye, Kiriakova and Uribarri118). Whether these alterations ultimately affect cardiometabolic health, i.e. decreasing heart disease and death from heart disease, is yet to be determined.
Alzheimer’s disease
Very little investigation has been done on the potential role of serum, diet or brain AGE levels on cognitive decline and incident Alzheimer’s disease (AD). In a small sample of older human subjects, elevated dietary AGE, measured by a validated FFQ, were associated with faster cognitive decline(Reference West, Moshier and Lubitz119). There is cross-sectional evidence for higher levels of methylglyoxal and CML in cerebrospinal fluid of AD patients(Reference Ahmed, Ahmed and Thornalley120,Reference Bär, Franke and Wenda121) . Elevated serum AGE levels have been associated with a lower level of cognitive functioning in older adults(Reference Meli, Perier and Ferron122,Reference Cai, Uribarri and Zhu123) but serum pentosidine levels did not differ between AD patients and normal controls(Reference Thome, Munch and Muller124). Longitudinal studies have shown that elevated serum levels of MG-H1 (N δ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine) and of pentosidine are associated with more rapid cognitive decline in older adults with and without diabetes(Reference Yaffe, Lindquist and Schwartz125).
Several mechanisms may link elevated levels of AGE with impaired cognition and AD. Experimental studies in mice suggest that impaired clearance of β-amyloid may play a role. Mice fed a high-AGE diet had significantly higher hippocampal levels of β-amyloid and AGE compared with controls(Reference Cai, Uribarri and Zhu123). Human studies report that brain AGE are found in neurons, astrocytes and glial cells in AD brain(Reference Luth, Ogunlade and Kuhla126-Reference Girones, Guimera and Cruz-Sanchez128). AGE are suggested to take part in the transformation of soluble into insoluble β-amyloid and the aggregation of microtubule-associated protein τ(Reference Yan, Chen and Schmidt129,Reference Yan, Chen and Fu130) . Histochemical and immunoquantitative studies show that brain levels of AGE are increased in AD and that they constitute components of senile plaques and neurofibrillary tangles(Reference Vitek, Bhattacharya and Glendening131,Reference Sasaki, Fukatsu and Tsuzuki132) . AGE-modified β-amyloid peptide seems to serve as a ‘seed’ to accelerate and stabilise the formation of amyloid plaques in the AD brain(Reference Yan, Chen and Fu130). Some, but not all, studies suggest that brain AGE colocalise with amyloid or τ in AD brains(Reference Takeda, Wakai and Niwa127).
There are animal data pointing to a link between AGE with proteins related to other dementias including α-synuclein(Reference Arvanitakis, Tounsi and Pillon133). AGE may affect cognition via accelerated cerebrovascular disease, as elevated AGE are associated with vascular risk factors, incident stroke and small vessel disease(Reference Cai, Uribarri and Zhu123). Human studies show brain AGE in small vessels of AD brain(Reference Cai, Uribarri and Zhu123,Reference Luth, Ogunlade and Kuhla126,Reference Girones, Guimera and Cruz-Sanchez128) . Moreover, human brain imaging studies suggest that AGE may accelerate regional cortical atrophy in both diabetics and non-diabetics(Reference Moran, Munch and Forbes134). AGE may also impair blood–brain barrier and white matter integrity(Reference Hudson, Moon and Kalea135).
While these data link AGE and cognition in older adults and while there is strong biological plausibility for the contribution of AGE to neuropathological substrates underlying cognitive impairment and AD, there are critical gaps in our knowledge. No study has shown that serum AGE are related to incident AD in older adults. Moreover, while AGE have been documented in human AD brains(Reference Luth, Ogunlade and Kuhla126), no study has documented an association between brain AGE levels and cognitive decline. Finally, the inter-relationship of dietary, serum and brain levels of AGE in older adults with cognition is unknown. Since AD is a major epidemic, disentangling the role of these three pools of AGE on cognition may open new venues for treatment against this disease.
Cancer
Over time the potential contribution of AGE to the onset and progression of other chronic and non-infectious diseases, such as cancer, has been widely documented(Reference Uribarri, del Castillo and de la Maza1). Since the pioneering report showing that AGE taken up through RAGE played a role in promoting the growth of renal carcinoma cells(Reference Miki, Kasayama and Miki136), a large body of both clinical and experimental evidence supports the role of the activation of the AGE–RAGE axis on cancer onset and progression, by promoting protein damage, aberrant cell signalling, increased inflammatory responses, increased oxidative damage, changes in extracellular matrix composition and decreased genetic fidelity(Reference Abe and Yamagishi137-Reference Ahmad, Khan and Rafi141). In addition, there is evidence indicating that dietary AGE are associated with a higher cancer incidence and mortality in modern society(Reference Turner142).
A recent study has shed light into how circulating AGE may promote resistance to tamoxifen in breast cancer patients(Reference Walter, Ford and Gregoski143). Because dietary AGE restriction has been shown to be a successful approach to reduce the levels of circulating AGE(Reference Uribarri, del Castillo and de la Maza1), any intervention focused on lowering AGE levels may become an attractive option to improve the prognosis of breast cancer patients.
It is important to highlight the potential contribution of dietary AGE in explaining the disproportionately high levels of cancer incidence and mortality in modern society(Reference Turner142). While both the socio-economic and environmental factors are now well-recognised contributors to this cancer disparity, the role of dietary AGE deserves special attention as a novel component of this disparity since populations of lower socio-economic status seem to have higher dietary AGE intake as well as higher cancer incidence(Reference Turner142).
Sarcopenia
In 2018, the European Working Group on Sarcopenia in Older People (EWGSOP) revised its consensus on the definition and diagnosis of sarcopenia, which in the past was viewed as a geriatric syndrome of loss of muscle mass and function, and has now been more specifically redefined as a generalised skeletal muscle disease(Reference Cruz-Jentoft, Bahat and Bauer144). Sarcopenia has been associated with multiple adverse health outcomes including falls and fractures, impaired mobility and activities of daily living, cardiac disease, respiratory disease, longer hospitalisation, cognitive impairment and higher mortality(Reference Rosenberg and Roubenoff145).
Evidence for the contribution of AGE to sarcopenia includes the associations of higher plasma CML with lower handgrip strength among elderly, higher skin autofluorescence (a measure of skin AGE) with lower skeletal muscle mass measured by dual-energy X-ray absorptiometry in middle-aged adults, and with reduced muscle strength in type 1 diabetic patients(Reference Dalal, Ferrucci and Sun146-Reference Mori, Kuroda and Araki148). Higher levels of muscle pentosidine were linked to lower muscle strength but not to loss of muscle mass among elderly, suggesting that collagen cross-links can produce rigidity of the muscle fibre, primarily affecting function and, less so, muscle mass(Reference Haus, Carrithers and Trappe149). Serum pentosidine was also associated with osteoporotic and sarcopenic degenerative lumbar scoliosis(Reference Eguchi, Toyoguchi and Inage150). A systematic review on the subject indicated that higher levels of AGE are moderately but independently related to decline in walking abilities, activities of daily living, decreased muscle properties (strength, power and mass) and increased physical frailty among elderly subjects. In fact, AGE can alter the biomechanical properties of muscle tissue, increasing stiffness and reducing elasticity through cross-linking and up-regulated inflammation by RAGE binding and endothelial dysfunction in the intra-muscular microcirculation(Reference Drenth, Zuidema and Bunt151).
A direct role for AGE of dietary origin in sarcopenia remains to be established.
Conclusions
In recent years, numerous small clinical trials have measured the effects of a low-AGE dietary intervention on a variety of clinical conditions. These trials have demonstrated that a simple low-AGE dietary intervention decreases circulating levels of AGE, markers of inflammation and OS in healthy, chronic kidney disease and diabetic patients, and improves insulin resistance in diabetic and pre-diabetes patients. These data have generated a new paradigm of disease widely unrecognised, suggesting that excessive consumption of dietary AGE secondary to a ‘Western lifestyle’ represents an independent risk factor for inappropriate chronic mild OS and inflammation during life, which over time facilitates the emergence of the chronic diseases of the modern world, especially diabetes and CVD (Fig. 3). An emerging role for dietary AGE is being postulated even in seemingly disconnected areas such as cancer. Simultaneously, we are seeing a rise in the processed food sector revealing that an increasing number of individuals are consuming modern processed foods, laden with AGE. This potential increase in health hazard should also be a concern for processed food industries and not only for the health industry(Reference Sharma, Kaur and Thind12). Reducing the AGE content of common foods by simple changes in culinary techniques is a feasible, safe and easily applicable intervention in both health and disease. Large-scale clinical trials must be performed to replicate the small clinical trials that have been performed so far and provide broader evidence for the deleterious role of dietary AGE on chronic disease. If this evidence continues being reiterated, then reduction of AGE content in common foods may convey an enormous public health impact.
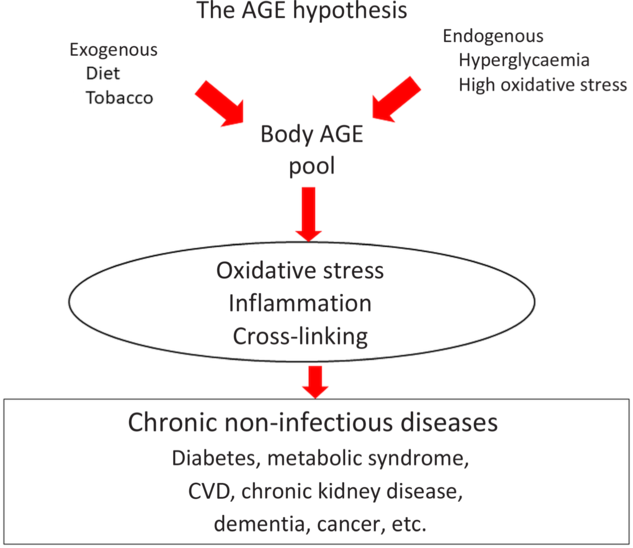
Fig. 3. The advanced glycation endproduct (AGE) hypothesis. Endogenous and exogenous AGE contribute to the body AGE pool that will affect body homeostasis through direct protein cross-linking as well as through several cellular receptors, which will in turn generate inflammatory cytokines and oxidative stress. All these factors eventually contribute to the genesis of chronic non-infectious diseases. For a colour figure, see the online version of the paper.
Acknowledgements
No funding was received for the present review.
Each author contributed equally.
The authors declare that there were no conflicts of interest.




