


INTRODUCTION
Lymphatic filariasis (LF), onchocerciasis, soil-transmitted helminthiases (STHs) and schistosomiasis are four major helminth diseases in humans targeted for control with preventive chemotherapy (PC) by mass drug administration (MDA). Following the London Declaration on Neglected Tropical Diseases (NTDs) in 2012, the World Health Organization (WHO) developed a roadmap to prioritize and guide the scaling-up of interventions for control and elimination of NTDs by 2020 (World Health Organization, 2015). Current goals include elimination of LF and control of onchocerciasis, soil-transmitted helminths and schistosomiasis by sustaining, expanding and extending MDA alongside condition-specific interventions including vector control and water, sanitation and hygiene measures to abate transmission.
Access to and deployment of appropriate diagnostic tools is crucial within disease control and elimination programmes throughout their inherent four key time points: (i) initial mapping to establish disease prevalence, (ii) monitoring the impact of interventions after they are started, (iii) deciding when to stop interventions and (iv) post-elimination surveillance (Solomon et al. Reference Solomon, Engels, Bailey, Blake, Brooker, Chen, Chen, Churcher, Drakeley, Edwards, Fenwick, French, Gabrielli, Grassly, Harding-Esch, Holland, Koukounari, Lammie, Leslie, Mabey, Rhajaoui, Secor, Stothard, Wei, Willingham, Zhou and Peeling2012). Diagnostic tools which are both highly sensitive and cost-effective, particularly when scaling-up MDA and control interventions and within ‘endgame’ final stages of elimination scenarios and post-MDA surveillance, are key to the impact and success of control and elimination programmes (McCarthy et al. Reference McCarthy, Lustigman, Yang, Barakat, García, Sripa, Willingham, Prichard and Basáñez2012). The expected reduction in both infection prevalence and intensity in communities will result in the need to detect very low levels of infection and screen larger numbers of people or vectors, often returning a much greater number of negative tests to provide confidence in excluding presence of infections. The reliability of diagnostics under such scenarios is vital when the decision to stop MDA in an area has to be made, as well as in the post-MDA monitoring stages: residual low levels of infection, if overlooked, can cause resurgence of transmission and subsequent endemic infection. More sensitive diagnostics can also aid in the early detection of drug resistance potentially emerging in parasite populations as a result of increased exposure to drugs due to scaling-up of MDA. Additionally, given the nature of integrated MDA, further unique challenges exist in devising strategies, protocols and tests for each disease in parallel when appropriate.
Currently, for MDA-targeted helminths the pragmatic ‘gold standards’ for human diagnosis as endorsed by the WHO for implementation in resource-poor settings include the microscopic identification of parasite larvae in blood (LF) or skin snips (onchocerciasis), eggs or larvae in stools or urine (STHs and schistosomiasis) or the detection of antibodies or worm antigens circulating in blood (LF, onchocerciasis and schistosomiasis) or urine (schistosomiasis) (McCarthy et al. Reference McCarthy, Lustigman, Yang, Barakat, García, Sripa, Willingham, Prichard and Basáñez2012). All these ‘gold standard’ tools, although appropriate for mapping disease in moderate-to-high intensity and prevalence areas, show their limitations for impact monitoring and post-elimination stages: in particular for the latter stage, it is proposed that the ideal test format would be a high-throughput laboratory assay (Solomon et al. Reference Solomon, Engels, Bailey, Blake, Brooker, Chen, Chen, Churcher, Drakeley, Edwards, Fenwick, French, Gabrielli, Grassly, Harding-Esch, Holland, Koukounari, Lammie, Leslie, Mabey, Rhajaoui, Secor, Stothard, Wei, Willingham, Zhou and Peeling2012).
Research requirements to advance current diagnostic tools for human helminthiases in the context of control and elimination programmes have been extensively reviewed (McCarthy et al. Reference McCarthy, Lustigman, Yang, Barakat, García, Sripa, Willingham, Prichard and Basáñez2012; World Health Organization, 2012). However, there remains lack of funding commitment despite recognition of the fundamental role of diagnostics in meeting the 2020 goals for NTDs (Hotez et al. Reference Hotez, Pecoul, Rijal, Boehme, Aksoy, Malecela, Tapia-Conyer and Reeder2016). With more countries scaling up MDA and associated interventions and approaching elimination of at least some of these infections, an important question is whether efforts should be focused on developing novel sensitive diagnostics for resource-poor and/or field settings, or validating throughput, cost-effectiveness and building required capacity in implementing existing tools.
Future solutions to the need for more sensitive, high-throughput diagnostics could include development of antibody, antigen and nucleic acid detection-based methods as all these approaches could lend themselves to levels of automation, handling of larger sample numbers, and sensitivity that helminth elimination programmes will need. These approaches, however, would require investment in capacity building with equipment, training and general resources as well as the necessary initial research to develop, validate, standardize and quality assure assays. Whilst antibody and antigen detection assays are of great potential value and most certainly warrant further investment, it is the nucleic-acid-based methods that arguably may currently offer the most rapid route to multiplexing and implementation within helminth control and elimination programmes. Furthermore, nucleic-acid-based methods may also offer ancillary benefit through their potential to monitor population genetic variations in helminths through possible drug pressure selection changes and thus inform strategies for judicious use of the few efficacious anthelmintics upon which we rely heavily.
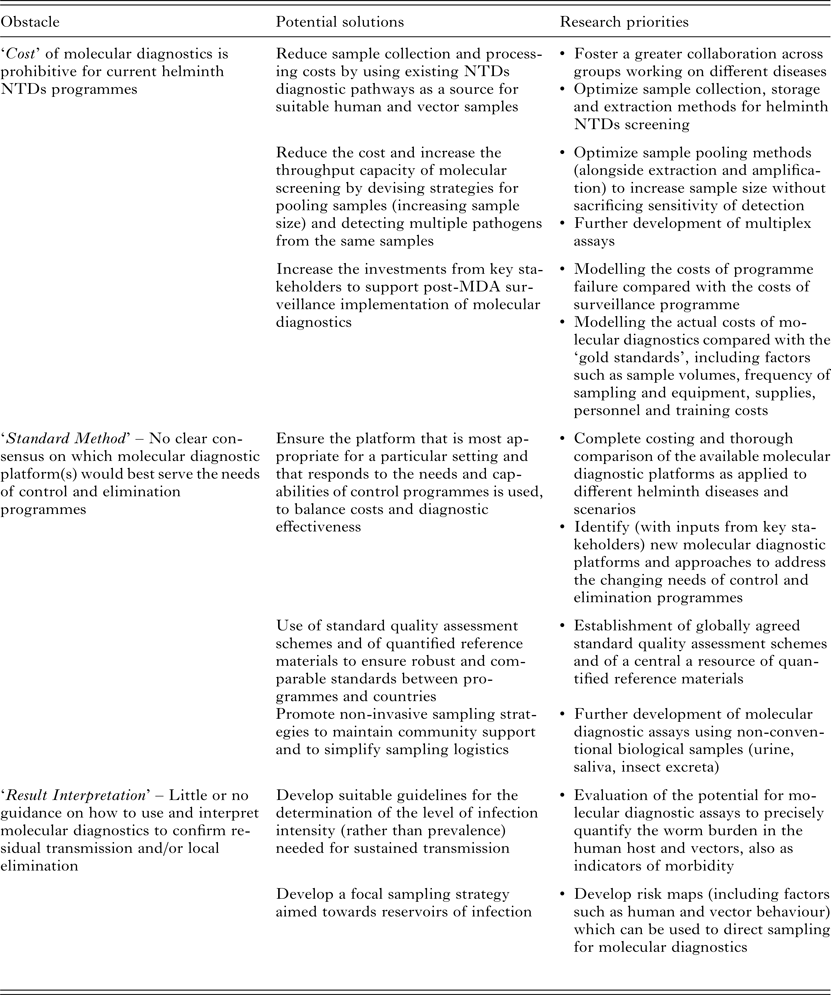
Proposed nucleic-acid-based molecular approaches as candidates for sensitive, high-throughput diagnostics, include (multiplex) real-time quantitative polymerase chain reaction (qPCR) assays or isothermal amplification methods, such as loop-mediated isothermal amplification (LAMP), nucleic acid sequence-based amplification (NASBA) and recombinase polymerase amplification (RPA). However, although these molecular methods do offer the required sensitivity and high-throughput, their acceptance and uptake is hampered by three main ‘obstacles’, that may be categorized as; ‘Cost’, through the expense of equipment, reagents and training; ‘Standard Method’, in terms of there being as yet no agreement on a specific ‘best approach/method and quality standards’; and ‘Result Interpretation’ with regard to how control/elimination programmes might best use molecular data to inform decision making (summarized in Table 1).
Table 1. Current obstacles, potential solutions and research priorities for the large-scale implementation of molecular diagnostics in MDA-targeted helminths control and elimination programmes
PCR is one of the most promising approaches to detect parasite DNA or RNA in human or vector samples due to its higher diagnostic sensitivity and specificity compared to microscopy or immunodiagnostics through antibody or antigen detection. In particular real-time qPCR has become the mainstay of molecular diagnostics in many clinical microbiology laboratories across the world for detecting bacteria, viruses, fungi and parasites in clinical samples (Espy et al. Reference Espy, Uhl, Sloan, Buckwalter, Jones, Vetter, Yao, Wengenack, Rosenblatt, Cockerill and Smith2006; Verweij and Stensvold, Reference Verweij and Stensvold2014), with a comprehensive number of published qPCR assays for pathogen detection.
However, operational issues such as the high cost of the instrumentation required for thermal cycling and signal detection and the need for well-equipped laboratories, have so far hindered use of qPCR in resource-poor settings. In the past few years, isothermal methods allowing the amplification of nucleic acids at constant temperature and with minimal equipment have been described and presented as more ‘field-friendly’ and cost-effective alternatives to qPCR, particularly for their potential use as point-of-care tests (Craw and Balachandran, Reference Craw and Balachandran2012). Amongst these methods, the LAMP (Notomi et al. Reference Notomi, Okayama, Masubuchi, Yonekawa, Watanabe, Amino and Hase2000) is the most researched and characterized for parasite diagnostics. For example, a variety of LAMP assays have been published for the detection of malaria (Han, Reference Han2013) and trypanosomes (Deborggraeve and Büscher, Reference Deborggraeve and Büscher2012). Another isothermal amplification technique, known as NASBA is a method, which amplifies RNA templates, mimicking the retroviral replication mechanism, and is particularly suited for the diagnosis of RNA viruses and in detecting viable targets (Craw and Balachandran, Reference Craw and Balachandran2012). Although NASBA has been successfully applied to the diagnosis of parasitic infections such as leishmaniasis (de Ruiter et al. Reference de Ruiter, Van Der Veer, Leeflang, Deborggraeve, Lucas and Adams2014) and trypanosomiasis (Mugasa et al. Reference Mugasa, Katiti, Boobo, Lubega, Schallig and Matovu2014), it has yet to be applied to human helminths. More recently a further isothermal method, the RPA (Piepenburg et al. Reference Piepenburg, Williams, Stemple and Armes2006) has come to the attention of the scientific community. In RPA, target amplification is rapidly accomplished (20–40 min) using a combination of target-specific primers and a recombinase enzyme (which form a recombinase filament inserting the primers when the specific target is encountered via a strand exchange mechanism) and single-strand DNA-binding proteins, which stabilizes and prevents re-annealing of the DNA double-strand (Craw and Balachandran, Reference Craw and Balachandran2012). The RPA approach has been applied for the detection of a variety of viral pathogens, including Ebola (Faye et al. Reference Faye, Faye, Soropogui, Patel, El Wahed, Loucoubar, Fall, Kiory, Magassouba, Keita, Kondé, Diallo, Koivogui, Karlberg, Mirazimi, Nentwich, Piepenburg, Niedrig, Weidmann and Sall2015) and also of urinary schistosomiasis (Rosser et al. Reference Rosser, Rollinson, Forrest and Webster2015). With increasing focus towards in-country laboratory capacity building and the necessity for integrating interventions and monitoring activities for different NTDs, molecular diagnostic methods are now suitable to explore in terms of value and effectiveness in helminth control and elimination programmes.
AIMS OF THE REVIEW
The aims of this review are threefold. First, to compare the features of qPCR and the two isothermal tools for DNA amplification (LAMP and RPA) for which examples in human helminths exist, and so are relevant for their potential use in control and elimination programmes of the four major human helminthiases. Second, to review the qPCR, LAMP and RPA assays currently available for patient and/or community diagnosis or xenomonitoring of LF, onchocerciasis, STHs and schistosomiasis. Third, to identify the research needs and implementation issues which should be considered for the large-scale adoption of such molecular diagnostic tools to assist the control and elimination programmes, particularly during the post-MDA surveillance stages.
WHAT FEATURES REALLY MATTER: qPCR VS LAMP VS RPA
When comparing the features of different molecular detection formats or assays, it is important to consider the characteristics most relevant to the purpose for which the tools are meant to be used. Analytical or diagnostic sensitivity and specificity are influenced by how the assay is designed and optimized and how samples are processed before testing (Verweij and Stensvold, Reference Verweij and Stensvold2014). For example, choosing a molecular target with multi-copy genes and using an appropriate method to extract nucleic acids efficiently (in terms of both yield and purity) from samples will allow higher sensitivity of detection. Following careful design and extensive testing of extraction methods and reaction set up, similar and very low analytical sensitivities can be accomplished using either qPCR or isothermal assays (Han, Reference Han2013). However, the lack of standardization in protocols between laboratories and published papers makes a direct comparison of assays based on the above characteristics difficult: variability between laboratories is commonly observed in terms of sample volumes, storage conditions and extraction methods (de Ruiter et al. Reference de Ruiter, Van Der Veer, Leeflang, Deborggraeve, Lucas and Adams2014). Also, when comparing the sensitivity of detection between a new molecular assay and one that is already available, the choice of the comparison assay is crucial. For example, the sensitivity of LAMP assays is commonly reported against a PCR performed using the same two primers employed in the LAMP reaction. In general and whenever possible, comparison should be made against the most sensitive assay available.
Cost-effectiveness and scalability of molecular diagnostics will depend on equipment and reaction costs, throughput capacity and the ability for tests to be multiplexed to amplify multiple targets from the same sample. It will be crucial to optimize these features for use in control and elimination campaigns of MDA-targeted helminths. A general comparison of qPCR, LAMP and RPA based on some of these features is shown in Table 2.
Table 2. Qualitative comparison of qPCR, LAMP and RPA based on features relevant to implementation of molecular diagnostics for MDA-targeted helminths control and elimination
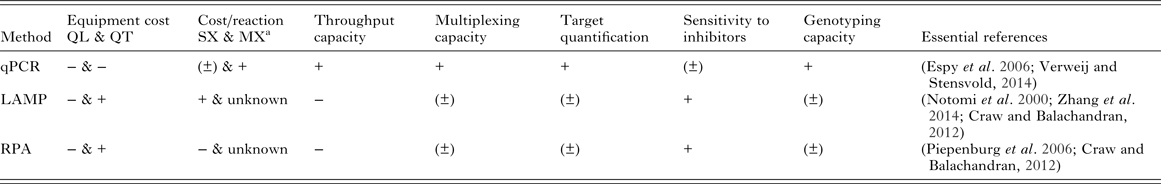
The ‘positive’/‘negative’ symbols (‘+’, ‘±’, ‘−’) indicate qualitatively assigned comparative suitability, performance, benefit and/or advantage of methods for the indicated features as follows: ‘+’ indicates relative comparable superior suitability performance, increased benefit and/or advantage; ‘(±)’ indicates relative comparable/no difference in suitability, performance, similar benefit and/or equitable vantage; ‘−’ indicates relative comparable inferior suitability, performance, reduced benefit and/or disadvantage.
a Excluding sample collection, extraction, consumables and primers costs: qPCR SX $0.55 (intercalating dye detection) – $0.81 (probe detection), MX (two targets, probe detection) $0.87 (Source: http://blog.biosearchtech.com/how-much-is-your-qpcr-assay-really-costing-you); LAMP SX $0.26 (estimated using the Bst 2·0 DNA polymerase kit from New England Biolabs: $264 for 1 mL of 8000 units mL−1 of enzyme for 1000 reactions at 1 µL of enzyme in 25 µL total reaction volume per manufacturer's standard protocol); RPA SX $4.27 (estimated using the TwistAmp® Basic kit from TwistDX: $410 for 96 reactions). qPCR, quantitative (real-time) polymerase chain reaction; LAMP, loop-mediated isothermal amplification; RPA, recombinase polymerase amplification; QL, qualitative detection; QT, quantitative detection; SX, singleplex reaction (one target); MX, multiplex reaction (more than one target); mL, millilitre; μL, microlitre; $, US dollar
Cost of reagents and equipment and the issue of target quantification
Real-time qPCR has a high cost associated with reagents and equipment needed for thermal cycling and signal detection, although the cost can vary between models and according to the machine multiplex capacity. During qPCR the amplified target detection happens ‘real-time’ using fluorescent intercalating dyes binding to the nucleic acids (such as SYBR-Green) or fluorescence-labelled probes (such as TaqMan probes) (Verweij and Stensvold, Reference Verweij and Stensvold2014): this feature makes the detection of amplification quantitative, allowing the determination of the pathogen load in samples. Quantifying parasite load is a key aspect in the diagnostics of helminths and assessment of morbidity and control, where determining infection intensity is more important than calculating prevalence (McCarthy et al. Reference McCarthy, Lustigman, Yang, Barakat, García, Sripa, Willingham, Prichard and Basáñez2012).
Isothermal methods can amplify nucleic acids at a constant (and relatively lower, if compared with qPCR) temperature using a simple water bath or heat block: in the case of RPA, successful amplification using simply the body heat of the operator has been proven possible (Crannell et al. Reference Crannell, Rohrman and Richards-Kortum2014). A wide range of approaches are available for the qualitative detection of LAMP and RPA products, including gel electrophoresis, in-tube fluorescence, lateral flow detection with immunochromatographic strips (Craw and Balachandran, Reference Craw and Balachandran2012) and also by naked eye using pH-sensitive dyes (Tanner et al. Reference Tanner, Zhang and Evans2015). The ability to perform nucleic acid amplification in the absence of expensive instrumentation, under field conditions in resource-poor setting, has been the major selling point for LAMP and similar methods as a ‘new generation’ of molecular diagnostic tools. However, if quantification of the target is needed, the advantage of isothermal methods over qPCR in terms of equipment requirements vanishes since detection must be accomplished by turbidimetric or fluorometric approaches requiring similar or identical equipment to qPCR (Zhang et al. Reference Zhang, Lowe and Gooding2014).
One or more than one pathogen(s)? The issue of multiplexing
Another important consideration to be made when estimating the cost per reaction for each platform is whether the aim is to detect one (singleplex) or more than one (multiplex) targets (parasite species in this case) simultaneously. Currently, RPA has the highest cost per sample and LAMP the lowest for a single target reaction (Table 2). The cost-effectiveness of qPCR at less than a dollar for a singleplex reaction increases as more targets are amplified and detected in the same tube: recent estimates showed that the cost per reaction tube increases only slightly from a single target ($ 0·82) up to, for example, four targets amplified ($ 1·01). As a consequence, the multiplexing capacity of qPCR implies a significant decrease in the hands-on time and increase in throughput capacity. The upper limit of targets that can be amplified in a multiplex qPCR assay using, for example, TaqMan probes depends on the number of different dyes the machine is able to detect, but using microfluidic technologies such as TaqMan Array Cards (TACs) can allow the detection of up to 19 different targets (Liu et al. Reference Liu, Gratz, Amour, Kibiki, Becker, Janaki, Verweij, Taniuchi, Sobuz, Haque, Haverstick and Houpt2013). Whether the same multiplexing dynamics can in time be applied to the isothermal tools remain to be seen. Although the possibility of amplifying multiple targets has been shown in both LAMP (Aonuma et al. Reference Aonuma, Yoshimura, Kobayashi, Okado, Badolo, Nelson, Kanuka and Fukumoto2010; Zhang et al. Reference Zhang, Lowe and Gooding2014) and RPA (Crannell et al. Reference Crannell, Castellanos-Gonzalez, Nair, Mejia, White and Richards-Kortum2016), both the multiplexing and sample throughput capacity of these approaches are still reduced if compared with qPCR and are not well established at present.
Parasite genotyping: an added value
The possibility of obtaining additional information on genetic makeup of parasites alongside detection should be considered an added value too. For example, molecular diagnostics can be applied to the detection of single nucleotide polymorphisms (SNPs) in the beta-tubulin gene sequence, which has been shown to be associated with resistance to benzimidazole drugs in a variety of animal nematode parasites (Kotze et al. Reference Kotze, Hunt, Skuce, von Samson-Himmelstjerna, Martin, Sager, Krücken, Hodgkinson, Lespine, Jex, Gilleard, Beech, Wolstenholme, Demeler, Robertson, Charvet, Neveu, Kaminsky, Rufener, Alberich, Menez and Prichard2014), and potentially STHs (Diawara et al. Reference Diawara, Halpenny, Churcher, Mwandawiro, Kihara, Kaplan, Streit, Idaghdour, Scott, Basáñez and Prichard2013) and Wuchereria bancrofti (Schwab et al. Reference Schwab, Boakye, Kyelem and Prichard2005). The detection of SNPs by qPCR using TaqMan probes is well established (Espy et al. Reference Espy, Uhl, Sloan, Buckwalter, Jones, Vetter, Yao, Wengenack, Rosenblatt, Cockerill and Smith2006), whereas it is still in its infancy for isothermal tools such as LAMP and RPA (Craw and Balachandran, Reference Craw and Balachandran2012). Where and when drug resistance genes and alleles are known, inclusion of such molecular tests within standard diagnostic monitoring surveys could ensure early detection of emergence of potential drug resistance and thus inform appropriate responsible strategies in judicious use of existing precious drugs on which MDA programmes rely significantly.
Are we really considering all costs?
It is important to stress that the real cost-effectiveness of molecular diagnostics must also include costs, which are usually overlooked, such as those associated with sample collection, storage and extraction. Particularly in relation to assay sensitivity and throughput capacity, the extraction of DNA and/or RNA from the samples is a crucial step (Verweij and Stensvold, Reference Verweij and Stensvold2014). Depending on the nature and number of samples processed, extractions can be expensive and time-consuming. The possibility of running molecular assays using unprocessed or minimally processed samples is highly attractive in terms of reducing the cost and time of extractions. It is a common belief that isothermal methods are more robust than PCR-based methods are to amplification inhibition due to sample contaminants, as shown in publications reporting pathogen detection by RPA directly from urine (Krõlov et al. Reference Krõlov, Frolova, Tudoran, Suhorutsenko, Lehto, Sibul, Mäger, Laanpere, Tulp and Langel2014) or by LAMP from crude or boiled insects (Alhassan et al. Reference Alhassan, Makepeace, LaCourse, Osei-Atweneboana and Carlow2014; Nzelu et al. Reference Nzelu, Gomez, Cáceres, Sakurai, Martini-Robles, Uezato, Mimori, Katakura, Hashiguchi and Kato2014) or freshwater samples (Koloren et al. Reference Koloren, Sotiriadou and Karanis2011). Although these results are promising, the effect of amplification inhibition on qPCR, LAMP and RPA must be thoroughly tested in different samples to avoid compromising the sensitivity of detection in seeking to reduce extraction costs.
OVERVIEW OF AVAILABLE qPCR, LAMP AND RPA ASSAYS FOR EACH HELMINTHIASIS
The major qPCR, LAMP and RPA assays described for the human diagnosis and xenomonitoring of LF, onchocerciasis, STHs and schistosomiasis are summarized in Table 3.
Table 3. Overview of qPCR, LAMP and RPA assays published for diagnosis and xenomonitoring of the MDA-targeted helminths
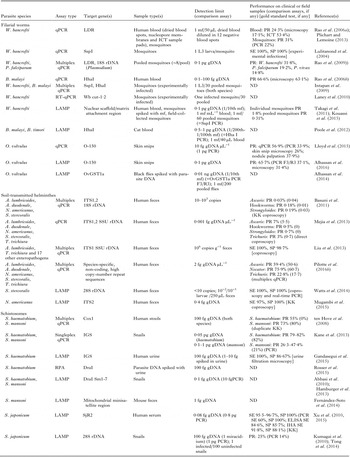
ND, not determined; gDNA, parasite genomic DNA; SE, sensitivity; SP, specificity; PR, prevalence; PCR, polymerase chain reaction; PCR F3/R3, PCR using the LAMP outer primers F3 and R3; qPCR, real-time quantitative polymerase chain reaction; RT–qPCR, reverse transcriptase quantitative PCR; LAMP, loop-mediated isothermal amplification; RPA, recombinase polymerase amplification; ELISA, enzyme-linked immunosorbent assay; IHA, indirect haemagglutination assay; KK, Kato-Katz; LDR, SspI, W. bancrofti repeat sequences; Hha I, Brugia repeat sequence; Wb cut-1·2, W. bancrofti cuticular collagen gene; O-150, O. volvulus tandem repeat; OvGST1a, O. volvulus glutathione S-transferase 1a; SSU rDNA, small subunit ribosomal DNA (nuclear); 12S rDNA, 12S ribosomal DNA (mithocondrial); 16S rDNA, 16S ribosomal DNA (mithocondrial); 28S rDNA, 28S ribosomal DNA (nuclear); IGS, intergenic spacer region of the ribosomal DNA; ITS1,2, internal transcribed spacer region 1 or 2 of the ribosomal DNA; Cox1, cytochrome c oxidase subunit 1; SjR2, S. japonicum repetitive retrotransposon; DraI, S. haematobium tandem repeat; Sm1,7, S. mansoni tandem repeat; g, gram; ng, nanogram; pg, picogram; fg, femtogram; mL, millilitre; mf, microfilariae; L3, L3 larva.
Lymphatic filariasis
PCR detection of the causative agents of LF W. bancrofti and Brugia malayi in blood samples or mosquitoes has been widely explored through a variety of formats and gene targets (Alhassan et al. Reference Alhassan, Li, Poole and Carlow2015). Real-time PCR assays have been developed using highly repeated sequences in parasites’ genomes, namely SspI (Lulitanond et al. Reference Lulitanond, Intapan, Pipitgool, Choochote and Maleewong2004) and LDR (Rao et al. Reference Rao, Atkinson, Ramzy, Helmy, Farid, Bockarie, Susapu, Laney, Williams and Weil2006a ) in W. bancrofti and HhaI (Rao et al. Reference Rao, Weil, Fischer, Supali and Fischer2006b ) in Brugia. The W. bancrofti LDR qPCR has been successfully used to amplify W. bancrofti DNA from dried nucleopore membranes stored from 6 months up to 4 years, as well as from dried blood on ICT sample application pads and individual mosquitoes (Rao et al. Reference Rao, Atkinson, Ramzy, Helmy, Farid, Bockarie, Susapu, Laney, Williams and Weil2006a ). The same assay later allowed a quantification of microfilarial load from dried blood spots, and proved to be suitable for screening pooled blood spots with a detection limit of one positive spot detected in a pool of 12 negative aliquots (Plichart and Lemoine, Reference Plichart and Lemoine2013). The detection of B. malayi in blood by qPCR has also been successfully achieved (Rao et al. Reference Rao, Weil, Fischer, Supali and Fischer2006b ). In this study, 17 out of 31 (54·8%) amicrofilaremic blood samples collected during the day tested positive by PCR. A PCR-positivity rate in day-collected blood samples similar to night-collected samples has been also observed in areas with nocturnally periodic W. bancrofti (Lucena et al. Reference Lucena, Dhalia, Abath, Nicolas, Regis and Furtado1998).
Xenomonitoring will play a major role in the ‘endgame’ final stages of elimination programmes and post-MDA surveillance of LF, since it gives proxy estimates of disease burden and existing potential for transmission while also being less invasive for the community (Okorie and de Souza, Reference Okorie and de Souza2016). Furthermore, a reduction in infection rates in vectors offers a valuable demonstration of interruption of transmission (Owusu et al. Reference Owusu, de Souza, Anto, Wilson, Boakye, Bockarie and Gyapong2015). The detection of filarial DNA or RNA in mosquitoes has several advantages over the traditional method of dissections. For example, it is relatively faster since it can be used on pooled mosquitoes and is less reliant on labour intensive and highly skilled dissection methods; it is more sensitive and allows the species-specific identification of filarial worms (Okorie and de Souza, Reference Okorie and de Souza2016). During an entomological survey in American Samoa, the estimated infection rate in pooled Aedes polynesiensis was 0·68% compared with the overall 0·11% rate observed by dissection of individual mosquitoes (Chambers et al. Reference Chambers, McClintock, Avery, King, Bradley, Schmaedick, Lammie and Burkot2009). Real-time qPCR has recently been used for detection of W. bancrofti in mosquito populations across the world, including Sri Lanka (Rao et al. Reference Rao, Nagodavithana, Samarasekera, Wijegunawardana, Premakumara, Perera, Settinayake, Miller and Weil2014), Samoa (Hapairai et al. Reference Hapairai, Plichart, Naseri, Silva, Tesimale, Pemita, Bossin, Burkot, Ritchie, Graves, Melrose and Joseph2015), Ghana (Owusu et al. Reference Owusu, de Souza, Anto, Wilson, Boakye, Bockarie and Gyapong2015) and Tanzania (Irish et al. Reference Irish, Stevens, Derua, Walker and Cameron2015). Under the perspective of a programmatic use in detection of filarial DNA from both blood and mosquitoes as part of post-MDA surveillance and verification of LF elimination, the possibility of applying the same molecular test to both sample types could potentially constitute a cost and operational advantage since laboratories in developing countries will be required to be proficient in only one assay.
Multiplex assays are also available, including the simultaneous detection of both W. bancrofti and B. malayi with a sensitivity of a single L3 larva of either species in up to 30 pooled mosquitoes (Intapan et al. Reference Intapan, Thanchomnang, Lulitanond and Maleewong2009). Of note is the duplex qPCR assay co-amplifying DNA from W. bancrofti and two species of Plasmodium (Plasmodium falciparum or P. vivax) in mosquito pools (Rao et al. Reference Rao, Huang, Bockarie, Susapu, Laney and Weil2009), which also showed a better sensitivity compared to the respective singleplex assays. This method has yet to be tested for its applicability in diagnosing filariasis and malaria coinfection in human blood, but it may prove useful in determining infection rates in mosquitoes from areas of filariasis and malaria co-endemicity. The possibility of diagnosing coinfections has been explored before using a ligation detection reaction-fluorescent microsphere assay (LDR-FMA) multiplex PCR on blood samples of 517 people from Papua New Guinea (Mehlotra et al. Reference Mehlotra, Gray, Blood-Zikursh, Kloos, Henry-Halldin, Tisch, Thomsen, Reimer, Kastens, Baea, Baea, Baisor, Tarongka, Kazura and Zimmerman2010). The assay, capable of detecting simultaneously W. bancrofti and four species of Plasmodium, revealed a frequent co-occurrence of bancroftian filariasis and any of the four malaria parasites in 29% of samples.
The ability to discriminate infective mosquitoes (harbouring the L3 infective stage of the parasite and thus contributing to transmission) from mosquitoes which, although having taken an infected blood meal do not support the development of the parasite and thus remain unable to infect the human host, is essential to obtain more precise estimates of actual disease transmission potential from xenomonitoring data (Okorie and de Souza, Reference Okorie and de Souza2016). For this purpose, reverse transcriptase (RT) PCR and qPCR assays have been developed for detection of L3 larvae-specific RNA transcripts in mosquitoes (Laney et al. Reference Laney, Buttaro, Visconti, Pilotte, Ramzy, Weil and Williams2008, Reference Laney, Ramzy, Helmy, Farid, Ashour, Weil and Williams2010; Vasuki et al. Reference Vasuki, Hoti and Patra2008). The assays showed a detection limit of a single L3 larva in pools of up to 25 mosquitoes (Laney et al. Reference Laney, Buttaro, Visconti, Pilotte, Ramzy, Weil and Williams2008; Vasuki et al. Reference Vasuki, Hoti and Patra2008) or one infected mosquito in a group of 30 uninfected insects (Laney et al. Reference Laney, Ramzy, Helmy, Farid, Ashour, Weil and Williams2010). The qPCR assay for W. bancrofti has also been multiplexed to detect both one L3 larvae-specific transcript and one further transcript present in all parasite stages (Laney et al. Reference Laney, Ramzy, Helmy, Farid, Ashour, Weil and Williams2010) thus allowing simultaneous detection of infected and infective insects in one reaction. Neither of the RT–PCR assays has been tested to date on field-collected mosquitoes from entomological surveys.
In terms of isothermal diagnostic tools, only one LAMP assay has been developed for bancroftian filariasis (Takagi et al. Reference Takagi, Itoh, Kasai, Yahathugoda, Weerasooriya and Kimura2011). Targeting a portion of the parasite nuclear scaffold/matrix attachment region, the method successfully amplified the DNA extracted from a single W. bancrofti microfilaria spiked in one millilitre of human blood or in a pool of 60 Culex mosquitoes. This LAMP assay has recently been used in Ghana screening for W. bancrofti infection in 112 individual Anopheles and 3635 Culex mosquitoes (arranged in 184 pools, with a range of 7–20 insects per pool) (Kouassi et al. Reference Kouassi, de Souza, Goepogui, Narh, King, Mamadou, Diakité, Dadzie, Boakye, Utzinger, Bockarie and Koudou2015). However, the very low number of positive mosquitoes (1·8%) and pools (0·31%) and the absence of a reference test prevented sufficient evaluation of the assay performance. Nevertheless, the two single mosquitoes and seven out of 11 (64%) of pools testing positive by LAMP, subsequently tested negative by PCR.
A LAMP assay has also been developed for the detection of B. malayi and B. timori, based on the tandem repeat HhaI (Poole et al. Reference Poole, Tanner, Zhang, Evans and Carlow2012). The assay showed the same limit of detection (corresponding to approximately 1/100th of a microfilaria) as the original HhaI PCR assay (Lizotte et al. Reference Lizotte, Supali, Partono and Williams1994) when pure parasite genomic DNA was used, but a lower limit (1/200th of a microfilaria) was observed when diluted cat blood was used as template due perhaps to dead parasite DNA circulating in blood (Poole et al. Reference Poole, Tanner, Zhang, Evans and Carlow2012).
Despite the potential applications and the possibility of developing a less invasive testing, the amplification of filarial DNA from samples such as sputum or urine has been attempted only in a few instances. Using PCR assays targeting the W. bancrofti repeated elements SspI and AccI, DNA of the parasite was amplified from the sputum of 32 (94%) out of 34 patients with confirmed bancroftian filariasis with a detection limit of one infected sputum sample mixed with 14 uninfected samples (Abbasi et al. Reference Abbasi, Githure, Ochola, Agure, Koech, Ramzy, Williams and Hamburger1999). In a latter study involving 304 people from Kenya, the sputum SspI PCR detected the presence of the parasite in more patients (42·8%) compared with ICT (39·1%) and blood microscopy (22%) (Kagai et al. Reference Kagai, Mpoke, Muli, Hamburger and Kenya2008). Thus, taking ICT as ‘gold standard’, the calculated sensitivity and specificity for sputum PCR was 97·5 and 92·4%, respectively. The DNA of W. bancrofti has also been successfully detected in urine from 39 people from Brazil using the same PCR assay (Lucena et al. Reference Lucena, Dhalia, Abath, Nicolas, Regis and Furtado1998) for an overall detection rate of 48·7–59% (in the first and second urine samples collected in the morning, respectively).
The possibility of detecting filarial DNA shed in the insect excreta and/or feces as a form of passive xenomonitoring tool (allowing then to bypass the need for pooling and extracting large numbers of mosquitoes) has been recently explored (Pilotte et al. Reference Pilotte, Zaky, Abrams, Chadee and Williams2016a ). In this study, the authors showed that using the HhaI repeat qPCR assay the DNA of B. malayi can be detected in the excreta/feces of a single mosquito (fed with microfilaraemic blood) in pooled excreta from up to 500 uninfected insects. The same approach was also used to detect the presence of DNA from the malaria parasite Plasmodium vivax (Pilotte et al. Reference Pilotte, Zaky, Abrams, Chadee and Williams2016a ). The high throughput and level of adaptability of excreta/feces collection and DNA testing to a variety of mosquito trapping systems makes this novel approach to xenomonitoring of LF very attractive and worth investigating in the near future, bearing also in mind its potential applicability for the surveillance of other blood-borne and mosquito-borne pathogens such as malaria or arboviruses.
Onchocerciasis
Molecular detection tools for Onchocerca volvulus have largely been based on the PCR amplification of the parasite-specific O-150 tandem repeat from skin snips, particularly in the assessment of skin microfilarial load reduction following PC (Alhassan et al. Reference Alhassan, Li, Poole and Carlow2015), and also from individual and pooled black flies in the Americas (Rodríguez-Pérez et al. Reference Rodríguez-Pérez, Danis-Lozano, Rodríguez, Unnasch and Bradley1999). The O-150 PCR has been adapted into a qPCR format, showing increased sensitivity of detection of infection in skin snips compared with either microscopy (Fink et al. Reference Fink, Fahle, Fischer, Fedorko and Nutman2011; Lloyd et al. Reference Lloyd, Gilbert, Taha, Weil, Meite, Kouakou and Fischer2015) and also nodule palpation (Lloyd et al. Reference Lloyd, Gilbert, Taha, Weil, Meite, Kouakou and Fischer2015). The TaqMan assay developed allowed a semi-quantitative estimation of the microfilarial load from skin snips in only 30 min (Lloyd et al. Reference Lloyd, Gilbert, Taha, Weil, Meite, Kouakou and Fischer2015).
The applicability of the O-150 assay for onchocerciasis xenomonitoring in Africa is partially hindered by the need for a DNA probe hybridization step to distinguish O. volvulus from the often co-endemic cattle parasite Onchocerca ochengi, which is transmitted by the same vectors (Alhassan et al. Reference Alhassan, Li, Poole and Carlow2015). However, the recently developed TaqMan assay did not cross-amplify DNA from O. ochengi (Lloyd et al. Reference Lloyd, Gilbert, Taha, Weil, Meite, Kouakou and Fischer2015) and so its future evaluation for the detection of O. volvulus in African black flies appears promising. The detection of Onchocerca DNA in the urine of 38 people from an endemic area in Cameroon has also been reported, but with a significantly lower sensitivity (14%) compared with either skin snip PCR (90%) or serum antigen detection (100%) (Vincent et al. Reference Vincent, Lustigman, Zhang and Weil2000).
To date, only one LAMP assay has been developed for specific detection of O. volvulus DNA in black flies (Alhassan et al. Reference Alhassan, Makepeace, LaCourse, Osei-Atweneboana and Carlow2014). The assay was capable of amplifying an amount of DNA equivalent to a single microfilaria spiked in a pool of up to 200 flies, irrespective of whether purified DNA or simply boiled flies were used as template. In the latter case using boiled flies, a reduction in analytical sensitivity was observed for PCR (one mf in 150 flies) (Alhassan et al. Reference Alhassan, Makepeace, LaCourse, Osei-Atweneboana and Carlow2014).
The applicability of LAMP to detect Onchocerca microfilariae in skin snips has also been tested using the O-150 tandem repeat as a target (Alhassan et al. Reference Alhassan, Osei-Atweneboana, Kyeremeh, Poole, Li, Tettevi, Tanner and Carlow2016). When applied to skin snips from 70 individuals living in endemic areas of Ghana, the LAMP assay showed a significantly higher sensitivity (67·5% prevalence) compared with both PCR using the external LAMP primers (37·1%) and microscopy (31·4%). Additionally, the detection of amplified parasite DNA was accomplished visually using pH-sensitive dyes (Alhassan et al. Reference Alhassan, Osei-Atweneboana, Kyeremeh, Poole, Li, Tettevi, Tanner and Carlow2016). To date, the amplification of L3 larvae-specific transcripts by RT–PCR for the detection of infective black flies (as it has been for LF in mosquitoes) has yet to be reported.
Soil-transmitted helminths (STHs)
Infection with the soil-transmitted helminths Ascaris lumbricoides, the hookworms (Ancylostoma duodenale, A. ceylanicum, Necator americanus), Trichuris trichiura and Strongyloides stercoralis is usually determined by finding the parasite eggs or larvae in stools by microscopy or culture (for hookworms and Strongyloides). However, coproscopy (either by concentrated or direct wet smear, egg flotation or Kato-Katz method) is relatively insensitive and time consuming, being most suitable for only moderate-to-heavy intensity egg loads, and thus light infections may typically be missed.
Molecular and biochemical assays that may confirm the presence of fecally-diagnosed helminths (STHs and schistosomes under our consideration in this review) through detection of antigens or DNA could therefore offer greater sensitivity. It is of course important to acknowledge detection limitations, as is the case in microscopic detection of eggs, when considering parasite antigens and DNA in low parasite burdens. As host parasite numbers fall, eggs become more difficult to detect and so DNA levels are reduced also. In the case of STHs, antigens and DNA would be present in feces from both eggs and the bodies of parasites themselves (through excretion/secretion, regurgitation, sloughing off from cuticle turnover, etc.), whilst schistosome antigens and DNA in feces would largely derive from eggs. Coproantigen detection methods are currently not commercially available or adopted for STHs or S. stercoralis diagnosis. Serological assays to detect antibodies against exposure to parasite antigens are also not available for STHs although several have been demonstrated in use for Strongyloides infections where the clinical importance of this infection may require greater attention [for recent reviews see (Buonfrate et al. Reference Buonfrate, Formenti, Perandin and Bisoffi2015) and (Krolewiecki et al. Reference Krolewiecki, Lammie, Jacobson, Gabrielli, Levecke, Socias, Arias, Sosa, Abraham, Cimino, Echazú, Crudo, Vercruysse and Albonico2013)]. Since co-infection with multiple STH species is common in endemic areas and the intensity of infection (worm burden) is the most important parameter to assess the efficacy of STH control programmes, the development, testing and refinement of multiplex and quantitative molecular assays should be considered a priority for these parasites.
In recent years a variety of PCR assays, both conventional and real-time, have been developed for the molecular detection of STHs in human stools (Verweij and Stensvold, Reference Verweij and Stensvold2014). Singleplex qPCR assays have been described and tested on the stools of 400 asymptomatic children from Ecuador (Mejia et al. Reference Mejia, Vicuña, Broncano, Sandoval, Vaca, Chico, Cooper and Nutman2013) and 99 people from Argentina (Cimino et al. Reference Cimino, Jeun, Juarez, Cajal, Vargas, Echazú, Bryan, Nasser, Krolewiecki and Mejia2015). In both studies, qPCR detected additional infections for all STHs, which were missed by microscopy, and there was significant correlation between the egg counts of Ascaris, Trichuris and hookworms in samples and concentration of parasite DNA determined by qPCR. In the Ecuador study, qPCR was also used to confirm complete clearance of Ascaris infection in a cohort of 125 children treated with albendazole and ivermectin (Mejia et al. Reference Mejia, Vicuña, Broncano, Sandoval, Vaca, Chico, Cooper and Nutman2013).
Multiplex PCR and qPCR assays have also been described and applied successfully in epidemiological studies globally (Basuni et al. Reference Basuni, Muhi, Othman, Verweij, Ahmad, Miswan, Rahumatullah, Aziz, Zainudin and Noordin2011; Taniuchi et al. Reference Taniuchi, Verweij, Noor, Sobuz, Van Lieshout, Petri, Haque and Houpt2011; Liu et al. Reference Liu, Gratz, Amour, Kibiki, Becker, Janaki, Verweij, Taniuchi, Sobuz, Haque, Haverstick and Houpt2013; Gordon et al. Reference Gordon, McManus, Acosta, Olveda, Williams, Ross, Gray and Gobert2015; Llewellyn et al. Reference Llewellyn, Inpankaew, Nery, Gray, Verweij, Clements, Gomes, Traub and McCarthy2016). One particular assay (targeting Ascaris, Ancylostoma, Necator and Strongyloides) showed an overall sensitivity of detection eight times higher than using a combination of three wet smears and one Kato-Katz slide on 77 stool samples collected in Malaysia (Basuni et al. Reference Basuni, Muhi, Othman, Verweij, Ahmad, Miswan, Rahumatullah, Aziz, Zainudin and Noordin2011). In another study on 680 stool samples from Timor-Leste and Cambodia, the prevalence of Ascaris and hookworm infections determined by multiplex PCR was respectively 1·2 and 2·9 higher than by microscopy (Llewellyn et al. Reference Llewellyn, Inpankaew, Nery, Gray, Verweij, Clements, Gomes, Traub and McCarthy2016). A multiplex PCR assay with Luminex beads was developed for detection of Ascaris, Ancylostoma, Necator, Strongyloides and three intestinal protozoa, showing high sensitivity (90–100%) and specificity (95–100%) on 319 stool samples previously tested by multiplex qPCR (Taniuchi et al. Reference Taniuchi, Verweij, Noor, Sobuz, Van Lieshout, Petri, Haque and Houpt2011). Two qPCR assays amplifying Ascaris and Trichuris DNA have also been incorporated into a TAC allowing the simultaneous detection of other 17 other pathogens (including amongst others rotavirus, Salmonella, Vibrio cholerae, Entamoeba histolytica, Cryptosporidium and Giardia) (Liu et al. Reference Liu, Gratz, Amour, Kibiki, Becker, Janaki, Verweij, Taniuchi, Sobuz, Haque, Haverstick and Houpt2013). In the Philippines, the simultaneous detection in stool samples of the two cestode parasites Taenia saginata and T. solium alongside Ascaris and hookworms has also been successful using a multiplex approach (Gordon et al. Reference Gordon, McManus, Acosta, Olveda, Williams, Ross, Gray and Gobert2015). In all the above studies, qPCR showed an increased capacity in detecting polyparasitism compared with coproscopy.
In a study on 796 stool samples from Kenya, qPCR was significantly more sensitive than two Kato-Katz slides from a single stool sample in detecting Ascaris and hookworm infections (Easton et al. Reference Easton, Oliveira, O'Connell, Kepha, Mwandawiro, Njenga, Kihara, Mwatele, Odiere, Brooker, Webster, Anderson and Nutman2016). Furthermore significant correlation was also shown between these egg counts by Kato-Katz, the DNA concentration determined by qPCR and the number of expelled Ascaris worms, showing that qPCR can be a good predictor for worm burden. The possibility of using multiplex qPCR to monitor changes in STH infection prevalence and intensity over time and in particular following MDA should be considered a priority (Easton et al. Reference Easton, Oliveira, O'Connell, Kepha, Mwandawiro, Njenga, Kihara, Mwatele, Odiere, Brooker, Webster, Anderson and Nutman2016). A recent study clearly showed the importance of using novel technologies to refine and improve the diagnostic potential of multiplex qPCR as applied to STHs (Pilotte et al. Reference Pilotte, Papaiakovou, Grant, Bierwert, Llewellyn, McCarthy and Williams2016b ), where next generation sequencing data were used to identify non-coding, high copy-number repeated DNA sequences in Ascaris, Ancylostoma, Necator, Strongyloides and Trichuris, which were then included in a novel multiplex qPCR assay. When compared with a previous pentaplex assay (Llewellyn et al. Reference Llewellyn, Inpankaew, Nery, Gray, Verweij, Clements, Gomes, Traub and McCarthy2016) using field collected stool samples, the new assay showed improved sensitivity detecting consistently down to 2 femtograms (fg) of parasite DNA (corresponding to a small fraction of the estimated DNA content of a fertilized egg or single L1 larva of the species considered) (Pilotte et al. Reference Pilotte, Papaiakovou, Grant, Bierwert, Llewellyn, McCarthy and Williams2016b ).
In contrast to qPCR the use of isothermal amplification methods for STH stool diagnosis has been overlooked so far, with the exception of two LAMP assays targeting S. stercoralis (Watts et al. Reference Watts, James, Sultana, Ginn, Outhred, Kong, Verweij, Iredell, Chen and Lee2014) and N. americanus (Mugambi et al. Reference Mugambi, Agola, Mwangi, Kinyua, Shiraho and Mkoji2015), respectively. The former assay, amplifying a region of the S. stercoralis 28S ribosomal DNA sequence, showed excellent performance (100% sensitivity and specificity) when tested on 28 stool specimens from patients with S. stercoralis infection confirmed by a combination of microscopy and real-time PCR (Watts et al. Reference Watts, James, Sultana, Ginn, Outhred, Kong, Verweij, Iredell, Chen and Lee2014). The latter assay, targeting the internal transcribed spacer region 2 of the N. americanus ribosomal DNA region, was used on 86 positive and 20 negative specimens (confirmed by Kato-Katz microscopy) detecting the parasite DNA with a 97% sensitivity and 100% specificity (Mugambi et al. Reference Mugambi, Agola, Mwangi, Kinyua, Shiraho and Mkoji2015). The N. americanus ITS2 LAMP was not tested on DNA from the other two hookworms infecting humans (A. duodenale and A. ceylanicum), so its applicability as a general hookworm diagnostic tool from feces has yet to be verified.
Schistosomiasis
Conventional and real-time PCR assays have been developed for the detection of Schistosoma spp. DNA in numerous types of clinical sample (feces, urine, plasma, serum, vaginal lavages) over the years, showing overall increased sensitivity of detection compared with either stool/urine microscopy or serology and antigen detection (Verweij and Stensvold, Reference Verweij and Stensvold2014). In only one case the amplification of different schistosomes, specifically S. haematobium and S. mansoni, has been attempted with a multiplex qPCR approach (ten Hove et al. Reference ten Hove, Verweij, Vereecken, Polman, Dieye and van Lieshout2008), using 176 stool samples collected over 2 weeks from 88 people in Senegal. Although the method failed to detect a small number of S. mansoni infections (confirmed by duplicate Kato-Katz), the DNA of S. haematobium was frequently detected in stools in the absence of eggs from this species (including people in which eggs were found in the urine and also a few in which the urine was egg-negative) (ten Hove et al. Reference ten Hove, Verweij, Vereecken, Polman, Dieye and van Lieshout2008).
Few LAMP assays have been published for schistosomal DNA detection in clinical samples. The first described assay was developed to target S. japonicum DNA in human serum (Xu et al. Reference Xu, Rong, Zhang, Shi, Zhu and Xia2010). The assay was used on sera from 20 infected and 30 uninfected patients diagnosed by Kato-Katz microscopy with only one false negative detected by LAMP, which otherwise displayed high sensitivity and specificity (96·7 and 100%, respectively) compared to conventional PCR. In a successive study using sera from 110 infected and 42 uninfected patients (Xu et al. Reference Xu, Guan, Zhao, Wang, Cao, Zhang, Zhu, He and Xia2015), the same LAMP assay had a diagnostic performance (95·5% sensitivity and 100% specificity) which surpassed both enzyme-linked immunosorbent assay (ELISA) (84·6% sensitivity and 85·7% specificity) and the indirect haemagglutination assay (91·8% sensitivity and 88·1% specificity). In the same study, the LAMP assay also detected S. japonicum DNA in 10 out of 60 (16·7%) serum samples from people that tested negative by triplicate Kato-Katz, ELISA and IHA. The minimum time required to detect infection with the three methods was also assessed using experimentally infected rabbits (Xu et al. Reference Xu, Guan, Zhao, Wang, Cao, Zhang, Zhu, He and Xia2015). While ELISA and IHA were positive only at 5 and 4 weeks post-infection respectively, LAMP was positive already at three days. The isothermal detection by LAMP of S. haematobium DNA in human urine has also been described (Gandasegui et al. Reference Gandasegui, Fernández-Soto, Carranza-Rodríguez, Pérez-Arellano, Vicente, López-Abán and Muro2015). With the ability to detect as little as 1 fg of parasite DNA using just the heated urine supernatant as template, the LAMP amplified all 18 S. haematobium-confirmed patient samples tested and eight out of 53 (15%) specimens negative for eggs by microscopy (Gandasegui et al. Reference Gandasegui, Fernández-Soto, Carranza-Rodríguez, Pérez-Arellano, Vicente, López-Abán and Muro2015).
The RPA method has also been tested for its potential to diagnose urinary schistosomiasis using oligochromatographic lateral flow strips (Rosser et al. Reference Rosser, Rollinson, Forrest and Webster2015). Although the sensitivity and specificity of the method was not evaluated on patients’ samples, the amplification efficiency for parasite DNA was tested adding variable volume percentages of crude urine (from 1·25 to 10% of total reaction volume) at two parasite DNA concentrations [100 fgs and 10 picograms (pg) L−1]. Significant amplification was observed even in the presence of 10% of added volume of crude urine where 10 pg of DNA was used, but not at lower DNA concentrations (Rosser et al. Reference Rosser, Rollinson, Forrest and Webster2015). These preliminary results suggest further optimization of this RPA assay could provide a rapid diagnostic test requiring only minimal processing of urine samples. To date, only one LAMP assay is described for detection of S. mansoni in stool samples, and is described being tested using mice feces (Fernández-Soto et al. Reference Fernández-Soto, Gandasegui Arahuetes, Sánchez Hernández, López Abán, Vicente Santiago and Muro2014). Providing a detection limit of 1 fg of parasite DNA the LAMP diagnostic gave positive results in mice at 1-week post-infection in comparison with the 4 weeks required for ELISA and 6 weeks for Kato-Katz microscopy (Fernández-Soto et al. Reference Fernández-Soto, Gandasegui Arahuetes, Sánchez Hernández, López Abán, Vicente Santiago and Muro2014).
Xenomonitoring of schistosomes in the snail intermediate host has been advocated as a key component of monitoring and surveillance when emphasis shifts from disease control to elimination (Rollinson et al. Reference Rollinson, Knopp, Levitz, Stothard, Tchuem Tchuenté, Garba, Mohammed, Schur, Person, Colley and Utzinger2013). Molecular tools also allow detection of prepatent infections in snails that do not shed parasite cercariae (Stothard et al. Reference Stothard, Stanton, Bustinduy, Sousa-Figueiredo, Van Dam, Betson, Waterhouse, Ward, Allan, Hassan, Al-Helal, Memish and Rollinson2014). Furthermore, they also allow the precise identification of the infecting schistosome species which is crucial in areas where the snails act also as intermediate hosts for other animal schistosomes (Abbasi et al. Reference Abbasi, Hamburger, Kariuki, Mungai, Muchiri and King2012). PCR and multiplex PCR methods are published for schistosome DNA detection in snails, with some allowing concomitant snail species identification (Stothard et al. Reference Stothard, Stanton, Bustinduy, Sousa-Figueiredo, Van Dam, Betson, Waterhouse, Ward, Allan, Hassan, Al-Helal, Memish and Rollinson2014). Particularly interesting are two qPCR assays allowing detection of S. haematobium and S. mansoni DNA from Bulinus spp. and Biomphalaria spp. snails by either fluorescence or oligochromatographic lateral flow strips (Kane et al. Reference Kane, Stothard, Rollinson, Leclipteux, Evraerts, Standley, Allan, Betson, Kaba, Mertens and Laurent2013). Both assays showed a comparable or even higher sensitivity of detection of parasite DNA than traditional PCR approaches.
Schistosome DNA detection in snail vectors by LAMP has been attempted too. One assay was capable of detecting DNA from either a single S. japonicum miracidium or one infected snail added to a pool of up to 100 uninfected snails (Kumagai et al. Reference Kumagai, Furushima-Shimogawara, Ohmae, Wang, Lu, Chen, Wen and Ohta2010). The assay has also been tested alongside a nested PCR assay using field-collected snails from different sites to determine the prevalence of S. japonicum (Tong et al. Reference Tong, Chen, Zhang, Yang, Kumagai, Furushima-Shimogawara, Lou, Yang, Wen, Lu, Ohta and Zhou2014). Overall in pools of 50 snails from each of the 28 sampled sites, parasite DNA was detected in seven out of 28 sites (25% prevalence) compared with only 4 (14·3% prevalence) detected by PCR. Two LAMP assays targeting S. haematobium and S. mansoni are also published and were compared for detection in snails (Abbasi et al. Reference Abbasi, King, Muchiri and Hamburger2010). Both methods showed a detection limit 10 times lower (at 1 fg) than the standard PCR assays (at 10 fg). These two methods have also been used to test individual field-collected snails in Kenya (Hamburger et al. Reference Hamburger, Abbasi, Kariuki, Wanjala, Mzungu, Mungai, Muchiri and King2013) where the LAMP assay returned infection prevalence estimates comparable with those obtained through PCR and qPCR. Interestingly, authors reported the possibility of pre-mixing all reaction components and stabilizing them with the addition of 17·5% sucrose to attain prolonged storage at room temperature (Hamburger et al. Reference Hamburger, Abbasi, Kariuki, Wanjala, Mzungu, Mungai, Muchiri and King2013).
Concluding remarks and future directions
It is clear that a series of obstacles in terms of costs, agreement on methods/standards, interpretation and strategic application of result data must be overcome to pave the way for nucleic acid-based molecular diagnostics to become valuable tools for the control and elimination programmes of MDA-targeted helminths in general (as summarized in Table 1, with individual parasite-specific research priorities summarized in Box 1). In general, besides technical feasibility and laboratory capacity, the major issue related to the large-scale and routine implementation of molecular diagnostics is its cost-effectiveness. Robust assessment of cost-effectiveness will require careful consideration of several factors and may need to be viewed on a case-by-case basis depending upon the situation, stage and needs of the programmes, and stakeholder requirements. Aside from costs associated with equipment and reagents for sample extraction and amplification, other factors to consider include the time involved for the predicted number of samples to be screened and the sampling frequency, whether the samples are used to screen for multiple pathogens and all costs associated with planning and executing the sampling (supplies, transport, personnel and training). Since these aspects could be overlooked, applying both mathematical modelling and full cost-analysis should be considered pivotal in generating evidence about actual cost-effectiveness of molecular diagnostics and set against a numerical sampling framework to judge affordability when used in discrete applications, for example, to certify elimination. We propose that with a greater focus on laboratory capacity building, cost-effectiveness analysis, agreements on standards in methods (including in particular the formation of quality assessment schemes and the use of quantified reference material) and their strategic application, the full potential of current and future nucleic acid-based molecular diagnostics for MDA-targeted helminths could now realize the more rapid situational assessment required in achieving press timeframes stipulated in the WHO 2020 Roadmap.
Box 1. Proposed research priorities for the molecular diagnostics of specific MDA-targeted helminths as applied to control and elimination programmes
-
• LF: optimization of mosquito pooling strategies and pool extractions, thorough field evaluation of the L3 larvae-specific RT–qPCR to assess mosquito infectivity as compared with the simple detection of DNA in vectors, explore the possibility of molecular xenomonitoring via indirect sampling strategies (insect excreta), exploration of less invasive sampling methods to determine infection prevalence in the communities (use of urine and saliva), using blood samples for the simultaneous screening of LF, malaria and schistosomiasis.
-
• Onchocerciasis: testing the existing O-150 qPCR assay for the xenomonitoring of onchocerciasis in African blackflies, developing an O. volvulus L3 larvae-specific RT–qPCR assay to detect infective black flies.
-
• STHs: Optimization of extant multiplex assays and development of new platforms for the simultaneous detection of all STHs (and schistosomes) from stools.
-
• Schistosomiasis: development of highly specific molecular assays for the xenomonitoring of schistosomes in snail populations, and assessment of their value for interruption of transmission evaluation, test snail pooling strategies for molecular detection using a similar approach as for LF.
-
• All helminths: further evaluation of the use of molecular detection as a quantitative indicator of worm burden and potentially morbidity.
ACKNOWLEDGEMENTS
C. M., L. R. and J. R. S. received funding from DFID, UK as part of the COUNTDOWN implementation research consortium on neglected tropical diseases (NTDs).
FINANCIAL SUPPORT
This work was supported by the Liverpool School of Tropical Medicine Research Development Fund (grant number RDF150303Jla02 to J. L., R. S. and C. M.)
CONFLICT OF INTEREST
None.
ETHICAL STANDARDS
Not applicable.




 Open access
Open access