



Introduction
Strongyloidiasis affects approximately 370 million people in more than 70 countries, mostly in tropical and sub-tropical regions (Olsen et al., Reference Olsen, van Lieshout, Marti, Polderman, Polman, Steinmann, Stothard, Thybo, Verweij and Magnussen2009; Bisoffi et al., Reference Bisoffi, Buonfrate, Montresor, Requena-Méndez, Muñoz, Krolewiecki, Gotuzzo, Mena, Chiodini, Anselmi, Moreira and Albonico2013). Strongyloides infections in humans are typically caused by two species; predominantly Strongyloides stercoralis Stiles and Hassall 1902, with a smaller proportion caused by Strongyloides fuelleborni von Linstow, Reference Von Linstow1905. Most Strongyloides species exhibit high host specificity (Speare, Reference Speare and Grove1989) but S. stercoralis infections have been reported in humans, dogs, cats and non-human primates. In particular, the role of dogs as reservoirs of zoonotic S. stercoralis has been a matter of contention historically and remains unclear today. Originally thought to infect humans exclusively, a nematode indistinguishable from S. stercoralis was identified in a dog in China in the early 1900s (Fuelleborn, Reference Fuelleborn1914), raising questions as to whether S. stercoralis was responsible for both human and canine strongyloidiasis. The taxonomic separation of Strongyloides infecting humans and dogs was suggested on the basis of experimental and epidemiological evidence (Brumpt, Reference Brumpt1922; Augustine, Reference Augustine1940; Jaleta et al., Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017), but this distinction has not been adopted. Even today, the epidemiology of canine strongyloidiasis remains poorly understood, and strongyloidiasis is generally considered a rare disease of dogs outside of East and Southeast Asia and the USA (Kreis, Reference Kreis1932; Galliard, Reference Galliard1951a). Reports of S. stercoralis or S. stercoralis-like species in domestic cats further complicates the question surrounding possible zoonotic reservoirs and Strongyloides species diversity (Chandler, Reference Chandler1925; Thamsborg et al., Reference Thamsborg, Ketzis, Horii and Matthews2017; Wulcan et al., Reference Wulcan, Dennis, Ketzis, Bevelock and Verocai2019).
Strongyloides stercoralis is currently thought to possess a population structure consisting of lineages A and B (Jaleta et al., Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017; Nagayasu et al., Reference Nagayasu, Htwe, Hortiwakul, Hino, Tanaka, Higashiarakawa, Olia, Taniguchi, Win and Ohashi2017; Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b). Humans and dogs are considered permissive hosts of S. stercoralis lineage A which is genetically characterized by possession of hyper-variable region (HVR)-IV haplotype A in most isolates, with the exception of some recently described Chinese S. stercoralis (Zhou et al., Reference Zhou, Fu, Pei, Kucka, Liu, Tang, Zhan, He, Chan, Rödelsperger, Liu and Streit2019). On the other hand, lineage B seemingly infects only dogs and invariably possesses HVR-IV haplotype B. However, fragmentary experimental evidence from early investigators suggests a more complicated population structure than the recently proposed ‘two lineage’ model implies (Nagayasu et al., Reference Nagayasu, Htwe, Hortiwakul, Hino, Tanaka, Higashiarakawa, Olia, Taniguchi, Win and Ohashi2017).
The less-common zoonotic species, S. fuelleborni (subsp. fuelleborni), is a non-human primate specialist (Pampiglione and Ricciardi, Reference Pampiglione and Ricciardi1971, Reference Pampiglione and Ricciardi1972; Hira and Patel, Reference Hira and Patel1980; Nutman, Reference Nutman2017; Thanchomnang et al., Reference Thanchomnang, Intapan, Sanpool, Rodpai, Tourtip, Yahom, Kullawat, Radomyos, Thammasiri and Maleewong2017). Its global population structure has not been extensively studied and it is not known whether the ability to infect humans varies among populations. This variation might contribute to the relative geographic restriction of S. fuelleborni (subsp. fuelleborni) infections in humans, which are nearly exclusive to sub-Saharan Africa despite the broader occurrence of S. fuelleborni (subsp. fuelleborni) in primates in other parts of the Old World. The enigmatic S. fuelleborni subsp. kellyi Viney, Ashford, and Barnish 1991 has only been reported from humans in Papua New Guinea, and its relation to primate S. fuelleborni is ambiguous, possibly representing another species altogether (Dorris et al., Reference Dorris, Viney and Blaxter2002).
The broad geographic range, complicated taxonomic history, and possible differences in host permissibility raise questions about whether S. fuelleborni represents a complex of species with varying degrees of transmissibility to humans (Hung and Höppli, Reference Hung and Höppli1923; Sandground, Reference Sandground1925; Augustine, Reference Augustine1940; Premvati, Reference Premvati1959; Little, Reference Little1966a). Current taxonomic nomenclature does not distinguish between S. fuelleborni from Asian and African primates (Ashford and Barnish, Reference Ashford, Barnish and Grove1989; Hasegawa et al., Reference Hasegawa, Sato, Fujita, Nguema, Nobusue, Miyagi, Kooriyama, Takenoshita, Noda, Sato, Morimoto, Ikeda and Nishida2010). Like for S. stercoralis, modern genetic approaches could greatly aid in reevaluating the diversity, host and geographic associations of this species.
Hasegawa et al. (Reference Hasegawa, Hayashida, Ikeda and Sato2009) proposed HVRs I to IV of the 18S rDNA as useful markers for Strongyloides species diagnosis, especially HVR-IV, based on the observation that ‘its nucleotide arrangements are mostly species specific’. HVR-I and HVR-IV are now routinely used in Strongyloides genotyping surveys (Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b; Zhou et al., Reference Zhou, Fu, Pei, Kucka, Liu, Tang, Zhan, He, Chan, Rödelsperger, Liu and Streit2019). The mitochondrial cytochrome c oxidase subunit 1 (cox1) locus of Strongyloides spp. is hypervariable and has been used to investigate genetic variation within and among Strongyloides populations by phylogenetic analysis and/or sequence clustering (Jaleta et al., Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017; Frias et al., Reference Frias, Stark, Lynn, Nathan, Goossens, Okamoto and MacIntosh2018; Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b). When used in various combinations, these three multi-locus sequence typing (MLST) markers (cox1, HVR-I and HVR-IV) have demonstrable genotyping utility. However, Strongyloides genotyping surveys undertaken to date have used various combinations of these three loci, with some examining only one (Frias et al., Reference Frias, Stark, Lynn, Nathan, Goossens, Okamoto and MacIntosh2018), or two loci (Sato et al., Reference Sato, Torii, Une and Ooi2007; Hasegawa et al., Reference Hasegawa, Hayashida, Ikeda and Sato2009; Thanchomnang et al., Reference Thanchomnang, Intapan, Sanpool, Rodpai, Tourtip, Yahom, Kullawat, Radomyos, Thammasiri and Maleewong2017). In two recent surveys all three loci were examined (Jaleta et al., Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017; Zhou et al., Reference Zhou, Fu, Pei, Kucka, Liu, Tang, Zhan, He, Chan, Rödelsperger, Liu and Streit2019), and subsequently these three loci were used in a next-generation sequencing approach to attempt to identify all Strongyloides genotypes present in a single sample, directly from faecal DNA extracts (Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b; Beknazarova et al., Reference Beknazarova, Barratt, Bradbury, Lane, Whiley and Ross2019). These studies, which aimed to elucidate relationships between Strongyloides genotypes, hosts and geographic distributions, have led to an abundance of MLST data in public databases, representing hundreds of individual worms.
Individual published MLST studies usually analysed relatively small numbers of worms (often less than 100) from small numbers of hosts and locations, making it difficult to identify clear patterns within these populations. In addition, integrating MLST data from these various studies into a single large dataset is also impractical because the studies sometimes examine overlapping, but different combinations of loci. This means that phylogenetic and other analytic approaches cannot be utilized effectively to explore such datasets in an integrative manner.
A recently described unsupervised machine learning (ML) procedure that calculates a distance statistic from MLST data for downstream clustering, even for datasets composed of different loci and combinations thereof, produced at different timepoints, may offer a solution to this problem. This approach constitutes a novel population genetics tool comprising an ensemble of two ML algorithms (Barratt et al., Reference Barratt, Park, Nascimento, Hofstetter, Plucinski, Casillas, Bradbury, Arrowood, Qvarnstrom and Talundzic2019a). As input, this method requires a set of user-defined haplotypes from large population datasets. An advantage of this method over traditional sequence analysis approaches (e.g. phylogeny) is its ability to calculate distances from MLST data, even when the genotype of some specimens in the dataset is not completely defined (e.g. in the absence of data for a particular marker). Such an approach could facilitate the integration of MLST data for Strongyloides from multiple studies into a single analysis, even though the marker combinations vary among studies. Additionally, this method has an advantage that it can address the challenge of heterozygosity that might be encountered at nuclear loci in sexually reproducing eukaryotes (Barratt et al., Reference Barratt, Park, Nascimento, Hofstetter, Plucinski, Casillas, Bradbury, Arrowood, Qvarnstrom and Talundzic2019a). Thus, data for heterozygous individuals could also be retained for analysis.
In the present study, we applied this ML method to all publicly available MLST datasets for S. stercoralis and S. fuelleborni. We integrated these data to increase our ability to detect novel population-level associations. We hypothesized that the use of this method would enable the identification of relationships among Strongyloides genotypes, hosts and geographic distributions, that were not evident using smaller datasets individually. We propose that this approach could aid in resolving taxonomic questions and controversies surrounding Strongyloides species.
Materials and methods
Data selection
All available sequences of S. stercoralis and S. fuelleborni 18S HVR-I, 18S HVR-IV (n = 218) and mitochondrial cox1 (n = 789) were obtained from GenBank (accessed August of 2019). Strongyloides sp. cox1 sequences obtained from Bornean slow lorises (Nycticebus borneanus, n = 18) were also included for analysis, as they share a relatively close phylogenetic relationship with S. stercoralis and S. fuelleborni despite representing a distinct group (Frias et al., Reference Frias, Stark, Lynn, Nathan, Goossens, Okamoto and MacIntosh2018). In cases where the HVR-I, HVR-IV and/or cox1 sequences were available from the same individual worm, the complete or partial genotype of these worms was recorded (Supplementary File S1, Tabs B and E, column F) after the genotyping approach by Barratt et al. (Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b) and Jaleta et al. (Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017). In instances where specimens were genotyped using polymerase chain reaction (PCR) products amplified directly from stool (Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b; Beknazarova et al., Reference Beknazarova, Barratt, Bradbury, Lane, Whiley and Ross2019), only specimens obtaining a single haplotype for each locus were included in this analysis. The rationale for this criterion was that for amplicons sequenced directly from stool where multiple haplotypes were detected, the underlying genotype of individual worms cannot be elucidated. Sequences in GenBank possessing International Union of Pure and Applied Chemistry (IUPAC) ambiguity codes were unphased into their underlying haplotypes only if a sequence supporting the existence of both possible haplotypes was available in GenBank. If only one of two possible haplotypes were supported by other haplotypes in GenBank for such a sequence, the IUPAC code was changed in favour of the supported haplotype. If there was no support for either of the underlying haplotypes for a sequence in GenBank possessing IUPAC codes, this part of the sequence was trimmed off from the ambiguous base onwards, in the direction that maximized the final length of the sequence. This analysis was inclusive of all S. stercoralis and S. fuelleborni sequences available in GenBank that met these criteria, irrespective of their host origin, and included specimens from humans, non-human primates, dogs and cats (Supplementary File S1, Tabs B and E, column D). Due to the limited sequence data available for S. fuelleborni kellyi, it was excluded from this analysis.
Genotypes were also constructed for five S. fuelleborni specimens from Japanese macaques, using all three MLST markers (i.e. HVR-I, HVR-IV and cox1). These genotypes were constructed from five cox1 sequences generated in one study (Hasegawa et al., Reference Hasegawa, Sato, Fujita, Nguema, Nobusue, Miyagi, Kooriyama, Takenoshita, Noda, Sato, Morimoto, Ikeda and Nishida2010), and 18S rDNA sequences published in different studies presumably from different worms (Sato et al., Reference Sato, Torii, Une and Ooi2007; Hasegawa et al., Reference Hasegawa, Hayashida, Ikeda and Sato2009). Therefore, ‘synthetic’ genotypes were constructed from these sequences to represent S. fuelleborni from the five Japanese macaques (Macaca fuscata). The synthetic genotypes were constructed in this way based on the observation that each of these 18S rDNA sequences was generated from worms collected from different Japanese macaque populations, yet all sequences are identical (GB: AB272235.1, AB453317.1, AB453318.1, AB453319.1). This supports that they represent the haplotypes found in all Japanese macaques (Sato et al., Reference Sato, Torii, Une and Ooi2007; Hasegawa et al., Reference Hasegawa, Hayashida, Ikeda and Sato2009). However, because these genotypes were based on human inference, classification was performed twice on the S. fuelleborni dataset; once including and once excluding these five ‘synthetic’ Japanese S. fuelleborni specimens.
Extracting cox1 and 18s haplotypes from published Strongyloides genomes
Genome sequences of S. stercoralis (Kikuchi et al., Reference Kikuchi, Hino, Tanaka, Aung, Afrin, Nagayasu, Tanaka, Higashiarakawa, Win, Hirata, Htike, Fujita and Maruyama2016) (36 genomes in total) were downloaded from the GenBank SRA database (Supplementary File S1, Tab B, column E) and the raw Illumina reads were subjected to a workflow designed in Geneious Prime (version 11: www.geneious.com). This workflow performed removal of Illumina adapter sequences (using BBDuk – default parameters) and filtering for quality (minimum PHRED score: 20 and minimum read length: 50 bases). Trimmed reads were mapped to references of HVR-I (GenBank: AF279916.2), HVR-IV (GenBank: AF279916.2), and cox1 (GenBank: MK463927.1) using Geneious mapper (default parameters) and the consensus of each mapped assembly was extracted for inclusion in this analysis.
Haplotype definitions
Haplotypes were identified in each specimen using a recently described genotyping system (Jaleta et al., Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017; Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b) with some modifications (Fig. 1). Briefly, for the purposes of this study, sequences of the ~434 bp fragment of HVR-I were divided into four segments, sequences of the ~260 bp fragment of HVR-IV were divided into three segments, and a 217 bp region of cox1 defined elsewhere (Beknazarova et al., Reference Beknazarova, Barratt, Bradbury, Lane, Whiley and Ross2019; Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b) was divided into nine segments (A1 to A3, B1 to B3 and C1 to C3), so that each of these sub-segments was considered a distinct locus. The rationale for dividing the sequences into these segments is based on extensive testing of the ML approach for performance optimization. These tests indicated that performance is best when the number of haplotypes at a given locus is between 10 and 20, and preferably less than 30 (https://github.com/Joel-Barratt/Eukaryotyping). The reasons for this are explained in detail in Supplementary File S2, pages 4–7. The sequence of each haplotype after the division of the three MLST markers into segments (resulting in a final panel of 16 markers) is provided in Supplementary File S2, Appendix (part A).
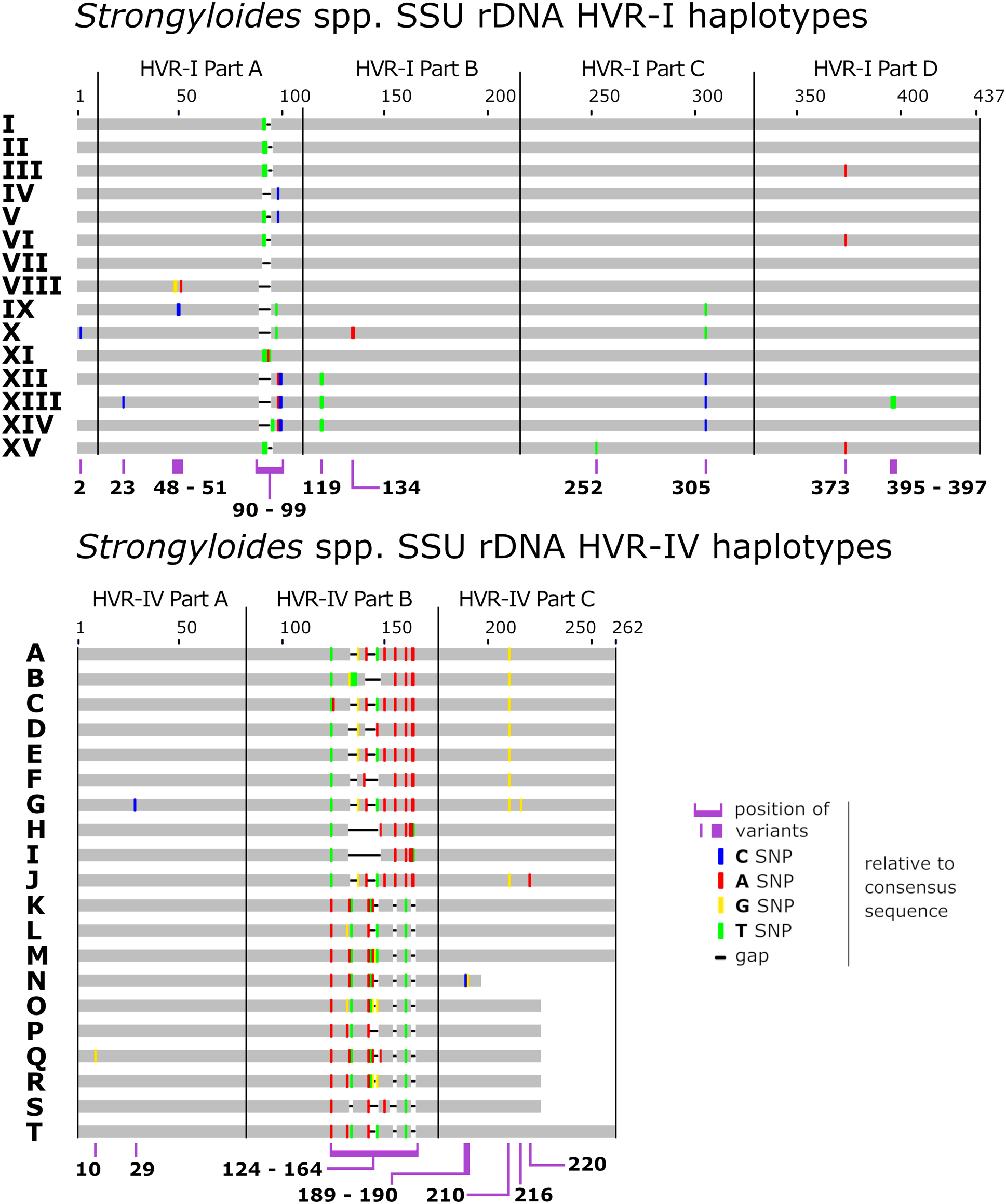
Fig. 1. Schematic of the Strongyloides genotyping scheme referenced here. Graphical representation a Strongyloides sp. genotyping scheme after the description of Barratt et al. (Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b) and Jaleta et al. (Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017). This scheme was expanded here to include haplotype XV of 18S HVR-I (GenBank Accession: MT436714), identified in genomes sequenced from Japanese humans by Kikuchi et al. (Reference Kikuchi, Hino, Tanaka, Aung, Afrin, Nagayasu, Tanaka, Higashiarakawa, Win, Hirata, Htike, Fujita and Maruyama2016). For the purposes of this study, note that the haplotypes originally defined after the description of Barratt and colleagues were split into smaller segments as shown here.
Assessment of population structure
Genotypes were assigned to each specimen using a Geneious workflow developed by the study team. Firstly, the Strongyloides sequences from individual worms were merged into a sequence list and then compared to a BLASTN database containing the FASTA sequences provided in Supplementary File S2, Appendix part A. The results were exported from Geneious in text format (one file for each specimen) and this result was converted to the format shown in Supplementary file S1 (Tabs A and D). The ML procedure (Barratt et al., Reference Barratt, Park, Nascimento, Hofstetter, Plucinski, Casillas, Bradbury, Arrowood, Qvarnstrom and Talundzic2019a) was applied to the resultant Strongyloides dataset using the scripts and instructions available here: https://github.com/Joel-Barratt/Eukaryotyping. Briefly, the haplotype data sheets provided in Supplementary file S1 (Tabs A and D) were exported as .txt files; these files were used directly as the input for the R scripts. This input included 138 specimens from S. fuelleborni and Strongyloides sp. ‘loris’, and 764 from S. stercoralis.
The ML procedure performs an unsupervised similarity-based classification task; it assesses whether any two specimens are related or unrelated on the basis of their genotype. The algorithms underpinning this method do not require that the genotype of every specimen be defined in the same manner; specimens are not required to be sequenced at the same markers (Barratt et al., Reference Barratt, Park, Nascimento, Hofstetter, Plucinski, Casillas, Bradbury, Arrowood, Qvarnstrom and Talundzic2019a), although, realistic minimum data requirements must be set by the user prior to analysis (https://github.com/Joel-Barratt/Eukaryotyping). For example, it would be unrealistic to expect that a specimen would be accurately classified in the event that only 20 bases of a single locus were available for this specimen. In this study, specimens were only retained for classification if they met at least one of two minimum data availability criteria. Firstly (1), when cox1 was the only sequence available for a specimen, or when data was not obtained for Part B of HVR-IV, the availability of nine out of the nine cox1 segments was required for a specimen to be included in the analysis. Secondly (2), if the only cox1 sequence available for a specimen was truncated (i.e. partially overlapping with the 217 bp region of cox1 being analysed here), the specimen could still be analysed provided its cox1 sequence overlapped with this 217 bp region by a minimum of seven out of nine segments. However, specimens with a truncated cox1 sequence were only retained in this analysis if part B of HVR-IV was also available for that specimen. This requirement considers the pertinent observations of Hasegawa et al. (Reference Hasegawa, Hayashida, Ikeda and Sato2009), regarding HVR-IV, indicating that ‘its nucleotide arrangements are mostly species specific’. It also considers that Part B of HVR-I can differentiate lineages A and B of S. stercoralis (Nagayasu et al., Reference Nagayasu, Htwe, Hortiwakul, Hino, Tanaka, Higashiarakawa, Olia, Taniguchi, Win and Ohashi2017; Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b). Therefore, part B of HVR-IV was set as a supplementary data requirement to compensate for information lost in the event of a truncated cox1 sequence. Data for HVR-I and/or additional parts of HVR-IV were considered in the analysis if available for a specimen, but data for these loci were not an absolute requirement for retaining specimens in this analysis.
The classification was performed for S. stercoralis and S. fuelleborni separately using an epsilon value of 0.05 for Plucinski's naïve Bayes classifier (refer to: https://github.com/Joel-Barratt/Eukaryotyping), and the resultant pairwise distance matrices (a pairwise matrix is the standard output of our ML method – Supplementary File S1, Tabs C and F) were clustered. Clustering was performed using the agglomerative nested approach in the ‘agnes’ R package, utilizing Ward's clustering method as described here (Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b). The ‘ggtree’ R package was then used to generate cluster dendrograms. Images of relevant hosts were obtained from PhyloPic (http://phylopic.org) or prepared in-house at the Centers for Disease Control and Prevention (CDC) for annotation of dendrograms. Images were rendered using the freely available GNU Image manipulation program (https://www.gimp.org).
Rationale for study design
Strongyloides cox1 sequences are considered hypervariable; this variability relates to single nucleotide polymorphisms (SNPs). As cox1 is also encoded on the mitochondrion, it is not subject to heterozygosity. These features make cox1 conducive to an analysis by phylogenetic methods or clustering based on sequence similarity (Jaleta et al., Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017; Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b; Beknazarova et al., Reference Beknazarova, Barratt, Bradbury, Lane, Whiley and Ross2019; Zhou et al., Reference Zhou, Fu, Pei, Kucka, Liu, Tang, Zhan, He, Chan, Rödelsperger, Liu and Streit2019). Alternatively, the 18S rDNA locus may be heterozygous in some individuals of S. stercoralis (see Zhou et al., Reference Zhou, Fu, Pei, Kucka, Liu, Tang, Zhan, He, Chan, Rödelsperger, Liu and Streit2019), which is an important confounding factor for phylogenetic methods. Within the Strongyloides genus, the 18S rDNA locus is less diverse than cox1 but it contains nucleotide insertions and deletions (indels) that represent important variants for differentiating Strongyloides species and genotypes. Most phylogenetic algorithms typically treat indels as missing data (Truszkowski and Goldman, Reference Truszkowski and Goldman2016; Donath and Stadler, Reference Donath and Stadler2018), and variability in indel handling between various multiple sequence aligners prior to phylogeny can produce markedly different results (Golubchik et al., Reference Golubchik, Wise, Easteal and Jermiin2007; Ashkenazy et al., Reference Ashkenazy, Cohen, Pupko and Huchon2014). For this reason, algorithms such as Gblocks are often used to identify and remove regions of the alignment containing gaps, making this data more amenable to phylogenetic applications (Castresana, Reference Castresana2000). Unfortunately, this practice would result in a loss of meaningful information when analyzing Strongyloides 18S rDNA HVRs. Furthermore, Strongyloides genotyping surveys published in the last decade display only some consistency in the combinations of the three widely used genotyping loci examined (Hasegawa et al., Reference Hasegawa, Hayashida, Ikeda and Sato2009, Reference Hasegawa, Sato, Fujita, Nguema, Nobusue, Miyagi, Kooriyama, Takenoshita, Noda, Sato, Morimoto, Ikeda and Nishida2010; Schär et al., Reference Schär, Guo, Streit, Khieu, Muth, Marti and Odermatt2014; Laymanivong et al., Reference Laymanivong, Hangvanthong, Insisiengmay, Vanisaveth, Laxachack, Jongthawin, Sanpool, Thanchomnang, Sadaow, Phosuk, Rodpai, Maleewong and Intapan2016; Jaleta et al., Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017; Thanchomnang et al., Reference Thanchomnang, Intapan, Sanpool, Rodpai, Tourtip, Yahom, Kullawat, Radomyos, Thammasiri and Maleewong2017, Reference Thanchomnang, Intapan, Sanpool, Rodpai, Sadaow, Phosuk, Somboonpatarakun, Laymanivong, Tourtip and Maleewong2019; Frias et al., Reference Frias, Stark, Lynn, Nathan, Goossens, Okamoto and MacIntosh2018; Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b; Beknazarova et al., Reference Beknazarova, Barratt, Bradbury, Lane, Whiley and Ross2019). This makes comparative analyses of distinct datasets difficult when using phylogenetic and/or sequence clustering methods. These limitations represent the main impetus for the design of this study, specifically; the ML procedure that was designed to overcome these challenges (Barratt et al., Reference Barratt, Park, Nascimento, Hofstetter, Plucinski, Casillas, Bradbury, Arrowood, Qvarnstrom and Talundzic2019a) (Supplementary File S2).
Results
Data filtering and detection of novel 18s rDNA haplotypes
After filtering the combined MLST dataset according to the minimum data availability requirements, data from 704 of the original 764 genotyped S. stercoralis specimens were retained. For S. fuelleborni, data from 133 of the 138 genotyped specimens were retained including all sequences from Strongyloides sp. ‘Loris’. These specimens had varying combinations of HVR-I, HVR-IV and cox1 data available; the markers available for each individual specimen that was retained for analysis are provided in Supplementary File S1, Tabs B and E, column F.
A novel S. stercoralis 18S rDNA HVR-I haplotype was identified in previously published genome datasets (Kikuchi et al., Reference Kikuchi, Hino, Tanaka, Aung, Afrin, Nagayasu, Tanaka, Higashiarakawa, Win, Hirata, Htike, Fujita and Maruyama2016). This 18S rDNA haplotype was detected in specimens from Okinawa, Japan and was assigned to haplotype XV, which contains a unique SNP at position 252 relative to the alignment shown in Fig. 1. The sequence of haplotype XV is identical to that of HVR-I haplotype III at all other base positions. The Strongyloides sp. genotyping scheme previously described (Jaleta et al., Reference Jaleta, Zhou, Bemm, Schär, Khieu, Muth, Odermatt, Lok and Streit2017; Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b; Beknazarova et al., Reference Beknazarova, Barratt, Bradbury, Lane, Whiley and Ross2019), was thus expanded to include this novel type (GenBank Accession: MT436714). The typing scheme used here (Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b; Beknazarova et al., Reference Beknazarova, Barratt, Bradbury, Lane, Whiley and Ross2019), differs from that described by Zhou et al. (Reference Zhou, Fu, Pei, Kucka, Liu, Tang, Zhan, He, Chan, Rödelsperger, Liu and Streit2019), who described HVR-IV haplotype C, which is identical to haplotype E reported by Beknazarova et al. (Reference Beknazarova, Barratt, Bradbury, Lane, Whiley and Ross2019) (Fig. 1).
The population structure of Strongyloides stercoralis
The final filtered S. stercoralis dataset includes sequences from specimens representing all inhabited continents. However, S. stercoralis sequences from specimens collected in Japan and some Southeast Asian countries were more highly represented (Supplementary File S2, page 13). MLST data from S. stercoralis collected from humans were more highly represented than data collected from dogs (Table 1, Fig. 2). Following classification using ML, the dataset was divided into 7 distinct genetic clusters, each representing a proposed sub-population of S. stercoralis (Fig. 2). The vast majority of S. stercoralis from dogs were assigned to one of two populations based on the MLST data available, represented by clusters 4 and 5 (129 of 146 S. stercoralis from dogs were assigned to these clusters; 88.4%). Most S. stercoralis specimens of human origin (550/554, 99.3%) were assigned to clusters 1, 2, 3, 6 or 7 based on their genotype (Table 1). All specimens assigned to cluster 5 (92/92) were derived from dogs as were 86% of specimens (37/43) assigned to cluster 4. Two specimens assigned to cluster 4 were obtained from cats and only 4 specimens assigned to this cluster were from humans (90.7% of cluster 4 included S. stercoralis collected from dogs or cats). Overall, 95.5% of S. stercoralis assigned to clusters 4 and 5 based on their MLST genotype had been isolated from dogs. Given the geographical sampling bias towards specimens collected in Southeast Asia and Japan (Supplementary File S2, pages 13 and 14), no attempt was made to statistically validate possible associations between genetic cluster assignment and the geographic origin of S. stercoralis specimens. An exception to this is genetic cluster 5, where 90 of 92 specimens assigned to this cluster (98%) were collected from dogs in Cambodia or Myanmar.
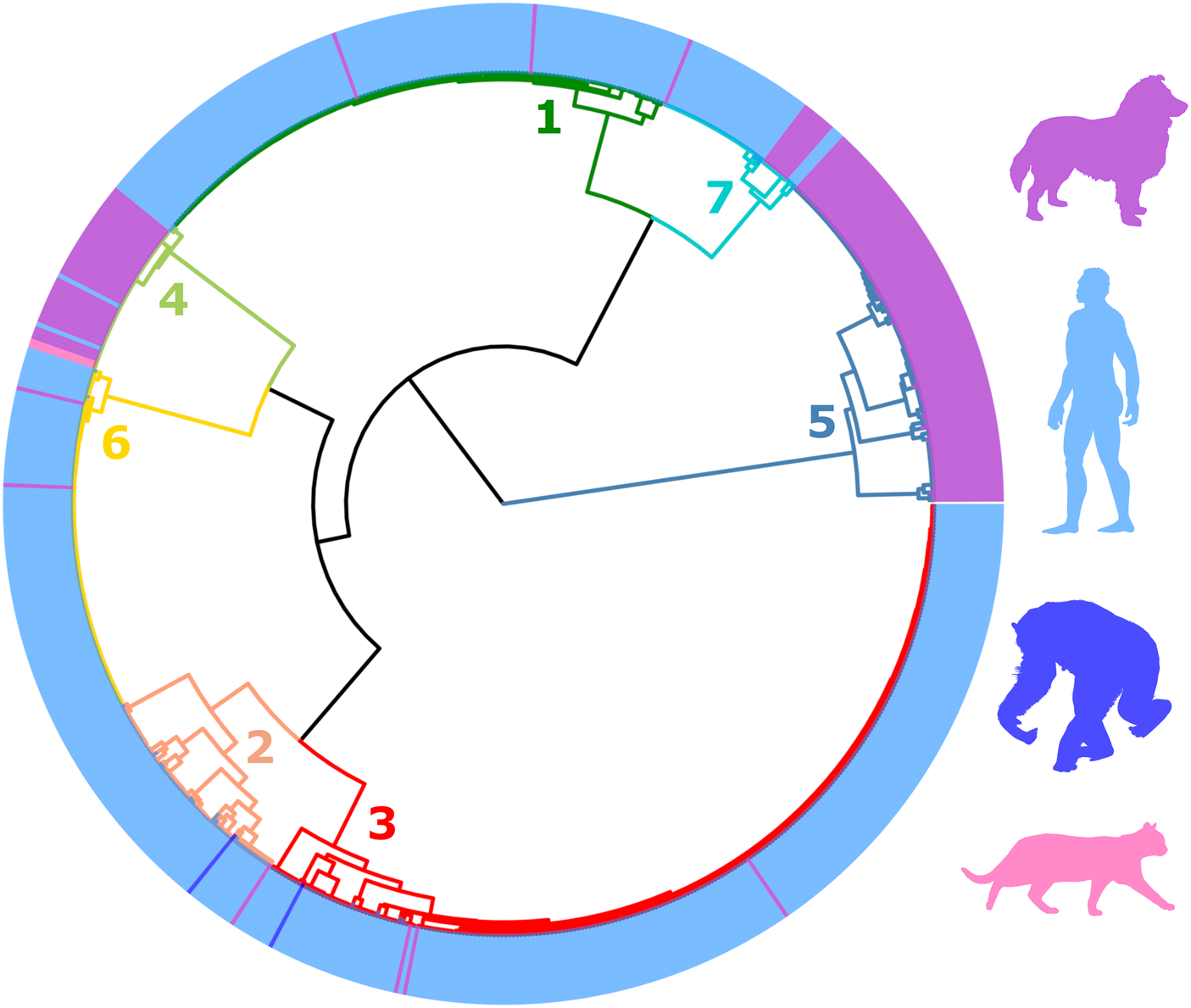
Fig. 2. Dendrogram of clustered distances generated from genotyped Strongyloides stercoralis using ML. This dendrogram was divided into seven distinct clusters. Branches are coloured according to their cluster number and are simply used to differentiate branches. Coloured peripheral bars indicate the host species from which the genotyped S. stercoralis were collected from; either a dog, human, chimpanzee or cat. The colour of these respective silhouettes matches the peripheral bars. For the identity of each specimen in this dendrogram refer to Supplementary File S2, Appendix part B. Cluster 5 is exclusively occupied by S. stercoralis infecting dogs. Specimens assigned to Cluster 4 were more frequently collected from dogs, though were also found in humans and cats. S. stercoralis from chimpanzees were assigned to clusters 2 and 3, though these were less frequently observed in these samples. S. stercoralis from clusters 1, 2, 3, 6 and 7 were more frequently collected from human samples than other hosts. The geographic origin of sequenced specimens included in this dendrogram is shown in Supplementary File S1, Tab B.
Table 1. Frequency of S. stercoralis from dogs and humans assigned to each genetic cluster and their respective χ2 P values
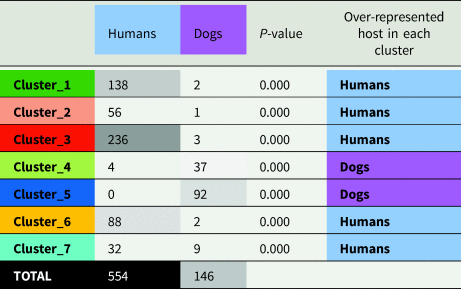
Note: Strongyloides stercoralis from humans were rarely assigned to cluster 4 and never to cluster 5. Strongyloides stercoralis from dogs, were vastly more common in clusters 4 and 5 (88.4% of cases), though 6.2% S. stercoralis from dogs were assigned to cluster 7. Boxes containing numeric values are shaded according to their frequency, with the highest frequencies shown in black and the lowest frequency (zero) in white.
The population structure of Strongyloides fuelleborni
The S. fuelleborni dataset retained for classification includes specimens collected mostly from parts of Southeast Asia and Africa, one specimen from India, and five specimens from Japan. Overall, the study population included 9 specimens from humans and 124 specimens from non-human primates. Long-tailed macaques (Macaca fascicularis) were the most highly sampled host among the specimens included in this analysis. The S. fuelleborni dataset also included 18 specimens obtained from Bornean slow lorises (Nycticebus borneanus). For precise numbers of specimens retained for analysis from each host and country refer to Supplementary File S2, Tables S4 to S7. The genotypes constructed for the five S. fuelleborni specimens from Japanese macaques possess all markers (i.e. HVR-I, HVR-IV and cox1) but these genotypes were generated from five cox1 sequences from one study (Hasegawa et al., Reference Hasegawa, Sato, Fujita, Nguema, Nobusue, Miyagi, Kooriyama, Takenoshita, Noda, Sato, Morimoto, Ikeda and Nishida2010), and 18S rDNA sequences obtained in different studies from different worms. Regarding these ‘synthetic’ genotypes from S. fuelleborni infecting Japanese macaques, classification was performed twice on the S. fuelleborni dataset; once including and once excluding these five ‘synthetic’ specimens. Irrespectively, the presence or absence of these specimens during classification had very little impact on the S. fuelleborni population structure (Supplementary File S2, Appendix part D).
Following classification, the S. fuelleborni specimens (plus Strongyloides sp. from slow lorises) were divided amongst 7 clusters, each representing a distinct population (Fig. 3). Clusters 1 and 2 as well as clusters 6 and 7, showed no clear association with a particular primate host species. Cluster 5 is exclusively occupied by the Strongyloides sp. from lorises, though the distinctness of this group was previously reported (Frias et al., Reference Frias, Stark, Lynn, Nathan, Goossens, Okamoto and MacIntosh2018). Cluster 4 is occupied by only a small number of inferred genotypes from one host species (Japanese macaques), and the specimens obtained from long-tailed macaques assigned to cluster 3 come from a single study surveying only this primate species in Thailand and Laos (Thanchomnang et al., Reference Thanchomnang, Intapan, Sanpool, Rodpai, Tourtip, Yahom, Kullawat, Radomyos, Thammasiri and Maleewong2017). Therefore, the possibility that this is a geographic association as opposed to a host association cannot be excluded. Generally, the data support a population structure for S. fuelleborni based on associations among genotype and geography; clusters 1 and 2 including S. fuelleborni from Africa, clusters 3 and 4 including S. fuelleborni from Southeast Asia and Japan (respectively), and clusters 6 and 7 including S. fuelleborni from Malaysian Borneo, which is suggestive of allopatric speciation.

Fig. 3. Dendrogram of clustered distances generated from genotyped specimens of Strongyloides fuelleborni and Strongyloides sp. from lorises using ML. This dendrogram was divided into seven distinct clusters. Branches are coloured according to their cluster number and the coloured peripheral bars indicate the host species from which the S. stercoralis specimens/sequences were derived, either a human (Hu), gorilla (Go), chimpanzee (Ch), baboon (Ba), orangutan (Or), Long-tailed macaque (Lt), proboscis monkey (Pr), Silvered leaf monkey (Sl), Slow loris (Lo), or Japanese macaque (Jm). Black branches visualize the relationship between clusters. For the identity of each specimen in this dendrogram refer to Supplementary File S1, Appendix part C. Clusters 1 & 2 include specimens from Africa. Cluster 3 includes specimens from Thailand and Laos, and a single specimen from an Indian human. Cluster 4 includes specimens collected in Japan. Clusters 5, 6 and 7 include specimens from Malaysian Borneo. This dendrogram supports that clusters 1 and 2 include S. fuelleborni genotypes that infect humans more commonly than the Strongyloides in other clusters.
Discussion
The ability to integrate large sequence datasets from multiple studies composed of specimens with varying combinations of MLST markers amplified, and specimens that are heterozygous, represents an important advantage of the ML procedure employed here over traditional methods utilized in the field of population genetics. Furthermore, the way in which haplotypes are defined using our ML procedure avoids problems associated with the treatment of indels that are inherent in many phylogenetic methods; indels (alignment gaps) are often treated as missing data in phylogenetic analyses and are excluded altogether (Truszkowski and Goldman, Reference Truszkowski and Goldman2016; Donath and Stadler, Reference Donath and Stadler2018). This exclusion of alignment gaps is problematic for the analysis of Strongyloides HVRs because these loci contain informative indels that differentiate haplotypes. Indels do not represent a problem for the ML approach used here which represents another advantage of this approach over phylogenetic methods. This method facilitated the inclusion of hundreds of genotyped S. stercoralis specimens from multiple studies in a single analysis, despite that many published genotyping studies examined different, yet overlapping, combinations of markers. Therefore, this analysis provides a broader view of potential host-associations and geographic patterns that exist in populations of these worms than could be provided in previous studies given the data analysis challenges mentioned above. The large number of specimens retained for analysis here meant that any observable trends are supported with greater statistical power than could be achieved within the smaller genotyping studies. Consequently, this ML procedure represents a powerful alternative to traditional phylogenetic methods. Using this approach, we provide evidence supporting that S. stercoralis represents a species complex and that African and s.e. Asian S. fuelleborni are distinct. These results are supported by a synthesis of scientific literature, focusing on observed patterns relating to host preference and geography reported on the basis of biological and experimental evidence published by pioneering investigators who were predominantly active in the early to middle 20th century. This synthesis highlights the agreement between our current analysis and the observations of these classic parasitologists who lacked the molecular tools required to help resolve these important taxonomic questions.
Since Fuelleborn's initial discovery of a Strongyloides infection in a Chinese dog, the status of dog-derived isolates as a valid species or as a variant of human S. stercoralis, and thus its zoonotic potential, has remained a subject of debate (Fuelleborn, Reference Fuelleborn1914). While many authors regarded canine and human isolates as more or less morphologically identical strains with differing host specificity within the same species S. stercoralis (Hung and Höppli, Reference Hung and Höppli1923; Kreis, Reference Kreis1932), others recognized the potential existence of ‘geographic races’ or even suggested the separation of canine Strongyloides on experimental and epidemiological grounds in spite of negligible morphological differences (Brumpt, Reference Brumpt1922; Augustine and Davey, Reference Augustine and Davey1939). Brumpt (Reference Brumpt1922) argued for the establishment of a new species Strongyloides canis on the basis of geographic disparities in prevalence, barriers in experimental cross-transmission, and reported developmental differences in vitro. As most early 20th century authors were averse to applying for specific status without important morphological differences (Chandler, Reference Chandler1925; Sandground, Reference Sandground1925; Goodey, Reference Goodey1926; Kreis, Reference Kreis1932), S. canis was never recognized as a valid name. However, the modern availability of genetic data – including our results – and the resulting shift in taxonomic dogmas allows this question to be revisited.
The body of experimental evidence demonstrates varying abilities of human-derived S. stercoralis from different geographic regions to establish infections in dogs. This could correspond with the ‘spectrum’ between human- and apparently canine-restricted genetic lineages presented here (Fig. 2). In Fuelleborn's investigations (Fuelleborn, Reference Fuelleborn1914, Reference Fuelleborn1927), dogs developed only short duration infections (2–3 weeks) with human-derived Chinese S. stercoralis and were refractory to infection with East African S. stercoralis. Sandground successfully infected dogs with a USA human-derived strain, though the majority of dogs eventually self-cured after about 3–10 weeks and were refractory towards repeated exposures (Sandground, Reference Sandground1928; Galliard, Reference Galliard1939, Reference Galliard1951b). Galliard reported ease in infecting dogs with human Strongyloides from Vietnam (Galliard, Reference Galliard1939), but that dogs were more or less refractory to isolates from the West Indies and North Africa (Galliard, Reference Galliard1951a). Also, in his experiments, dogs that were not successfully infected with the African strain developed severe infections after exposure to the Vietnamese strain.
Sandosham (Reference Sandosham1952) reported short-term, unstable infection in a dog repeatedly inoculated with many larvae derived from former prisoners of war in Thailand, concluding that dogs could not have been the reservoir host for S. stercoralis in that setting despite their supposed abundant presence in the prison camps (Sandosham, Reference Sandosham1952). Many years later, attempts to infect laboratory dogs with S. stercoralis derived from a human patient infected in Southeast Asia yielded mixed results; in one dog, a chronic infection lasting at least 15 months was established, but in four others, fecal larval counts peaked at 3 weeks and dropped off drastically after that (Grove and Northern, Reference Grove and Northern1982). A similar pattern of unstable, transient patent infections has been noted previously in human volunteers infected with other animal Strongyloides species, such as Strongyloides procyonis (raccoons) and Strongyloides ransomi (swine), further suggesting that host adaptation strongly influences the duration of infection and larval output (Freedman, Reference Freedman1991). Reciprocally, Augustine and Davey (Reference Augustine and Davey1939) failed to infect a human volunteer, guinea pigs and cats with filariform larvae reared from a naturally-infected dog in Massachusetts but readily infected both young and aged dogs, which developed long-lasting patent infections.
Clearly, historical workers had differing levels of success infecting dogs, and one varying factor was the geographic origin of isolates. Though fragmentary, it is interesting to compare the geographic origins of parasites used in dog infection trials to the emerging picture of the S. stercoralis global population structure shown here. For example, dogs were found to be refractory or developing very short-lived infections when exposed to human-derived isolates of S. stercoralis from China, East Africa, North Africa and the West Indies (Fuelleborn, Reference Fuelleborn1914; Galliard, Reference Galliard1950; Galliard and Berdonneau, Reference Galliard and Berdonneau1953). Notably, the lineages of S. stercoralis, represented by clusters 1, 2, 3 and 6 were almost all obtained from human samples and known to occur in these locations (Fig. 2). Intermediate results (e.g. short-lived infections, low larval outputs, and/or resistance to reinfection in most dogs, with occasional chronic infections) were seen with North American and some Southeast Asian S. stercoralis strains (Sandground, Reference Sandground1925; Faust, Reference Faust1933; Grove and Northern, Reference Grove and Northern1982; Genta, Reference Genta1989); these locations are represented by lineages of S. stercoralis assigned to genetic clusters 4 and 7, where we observe genotypes that are seemingly capable of infecting both hosts, yet with the majority of specimens assigned to these clusters collected from dogs and humans, respectively (Fig. 2, Table 1). The sole trial where dogs were readily infected with no immunity to reinfection used a human-derived S. stercoralis from Vietnam (Galliard, Reference Galliard1939), which is in close geographic proximity to the origin of roughly half of all specimens assigned to cluster 7 (n = 20, Myanmar and Thailand). In summary, the observed historical variations show some overlap with both the locations and host frequencies in our observed S. stercoralis clusters.
Our analysis suggests a disparity between where human and canine S. stercoralis infections are occurring; infections caused by S. stercoralis assigned to cluster 5 are almost exclusive to Southeast Asia (98%). Our analysis also indicates that different genotypes of S. stercoralis are more frequently reported in specific hosts (dogs are over-represented hosts in clusters 4 and 5), based on a χ 2 analysis (Table 1). These observations were also extensively discussed by historical authors who commented on the marked disparity in the occurrence of canine and human S. stercoralis infections i.e. in places where dog S. stercoralis infections occur commonly, human infections are rare, and vice versa. Outside of East and Southeast Asia, natural infections in dogs appeared to only occur in North America, and reports of dog infections were exceedingly rare to absent in areas where prevalence was high in humans (Brumpt, Reference Brumpt1922; Faust, Reference Faust1933; Augustine, Reference Augustine1940; Galliard, Reference Galliard1950; Sandosham, Reference Sandosham1952; Genta and Grove, Reference Genta, Grove and Grove1989). For example, not a single infection was detected in 528 dog faecal specimens during a survey in a highly endemic region of Colombia, where at least 14% of humans were S. stercoralis-positive (Faust and Giraldo, Reference Faust and Giraldo1960); even if some infections were missed due to the use of flotation techniques, infection would still be very rare in dogs vs humans. Recent case reports and molecular evidence show that natural canine infections also occur in Europe and Australia (Paradies et al., Reference Paradies, Iarussi, Sasanelli, Capogna, Lia, Zucca, Greco, Cantacessi and Otranto2017; Basso et al., Reference Basso, Grandt, Magnenat, Gottstein and Campos2019; Beknazarova et al., Reference Beknazarova, Barratt, Bradbury, Lane, Whiley and Ross2019; Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b). However, an important caveat is that results based solely on microscopy of feces must be interpreted with caution as free-living nematode larvae, common contaminants of specimens collected from the ground can easily be confused with Strongyloides spp. (Speare, Reference Speare and Grove1989). Direct epidemiologic evidence for cross-transmissibility is limited, in part due to the apparent variability in host adaptation and geographic occurrence of these strains. In one instance, a kennel worker in the USA with no foreign travel acquired Strongyloides from dogs under his care; this strain was successfully used to infect laboratory dogs (Georgi and Sprinkle, Reference Georgi and Sprinkle1974). A small study in an area of Japan considered endemic found no association in S. stercoralis infection status among owners and dogs (Takano et al., Reference Takano, Minakami, Kodama, Matsuo and Satozono2009). Overall, there are still many knowledge gaps in our understanding of canine strongyloidiasis as a zoonosis and its epidemiology, and interpretation will require further characterization of strains across a diverse geographic range.
While establishing the specific identity of canine Strongyloides will require further investigation, based on historical reports and our novel analysis, it appears that different S. stercoralis lineages display differences in infection frequency for canine and human hosts – which may exclude or drastically limit cross-transmission potential in some cases (Table 1). Given the epidemiological implications of this genetic variability and the now large body of evidence across many independent studies, it now seems appropriate to treat S. stercoralis as a species complex. For example, ‘S. stercoralis sensu lato’ could be useful for referring to all isolates, and ‘S. stercoralis sensu stricto’ could be considered for the human-genotypes represented in 6 of the 7 clusters associated with HVR-IV haplotype A (Fig. 2, excluding cluster 5). This would also include specimens assigned to genetic cluster 4 which seems associated with the 18S rDNA genotype VI + A, and likely represents a canine adapted lineage of S. stercoralis sensu stricto. Subspecific ranks (e.g. S. stercoralis canis) were suggested nearly a century ago by Chandler for S. stercoralis of different animal hosts (Chandler, Reference Chandler1925). This seems reasonable, given the exclusive finding in canine hosts (100% of cluster 5), the restricted geographic range of S. stercoralis assigned to cluster 5 (98% were from Southeast Asia), and the fact this lineage (associated with HVR-IV haplotype B) naturally clusters as an outgroup to all other S. stercoralis (Fig. 2).
An important outstanding question is whether the canine infections in certain human host-dominated populations represent transient, spurious, or incidental infections, or if there exists a substructure that is not clearly apparent due to under-sampling of dog-derived genotypes from specific locations. Based on our analysis, dog-derived genotypes assigned to clusters 1–3 and 6 likely represent transient infections with strains originating from humans; this is supported by the experiments of Fuelleborn, Sanground and Galliard who infected dogs with human-derived S. stercoralis which often resulted in transient infections (Fuelleborn, Reference Fuelleborn1914, Reference Fuelleborn1927; Sandground, Reference Sandground1928; Galliard, Reference Galliard1939, Reference Galliard1951a, Reference Galliard1951b). However, for cluster 7 it seems possible that some additional population substructure exists. Cluster 7 contains a small ‘sub-cluster’ composed of eight cox1 sequences derived exclusively from S. stercoralis from Japanese dogs. Sequencing of HVR-I and/or HVR-IV from these worms, and the sampling of worms from additional dogs in the same geographic area that these specimens were collected, would provide greater clarity as to what this smaller ‘sub-cluster’ of Cluster 7 might represent.
The identity of Strongyloides spp. infecting cats is a more complex question, and it appears multiple species are involved, including S. stercoralis. Chandler first reported S. stercoralis in cats in India (Chandler, Reference Chandler1925), and some attempts to infect cats with S. stercoralis from dogs and humans have been successful; though infections were usually short-lived, and it appears that cats are competent but abnormal hosts for S. stercoralis (Sandground, Reference Sandground1928; Desportes, Reference Desportes1944; Wulcan et al., Reference Wulcan, Dennis, Ketzis, Bevelock and Verocai2019). Strongyloides felis has been described from India and Australia, and is somewhat similar morphologically to S. stercoralis although no molecular data are available (Speare and Tinsley, Reference Speare and Tinsley1986). Strongyloides planiceps occasionally infects cats but is dissimilar in its life cycle and morphologic characteristics to S. stercoralis (Thamsborg et al., Reference Thamsborg, Ketzis, Horii and Matthews2017). A fourth species, Strongyloides tumefaciens, is of interest as it has been reported as causing colonic nodules, an unusual clinical presentation for Strongyloides (Thamsborg et al., Reference Thamsborg, Ketzis, Horii and Matthews2017). However, the original description was incomplete, and a recent study cast doubt on the validity of S. tumefaciens. Strongyloides sp. extracted from nodules of necropsied cats were found to have cox1 sequences that matched S. stercoralis. These sequences were assigned to genetic cluster 4 in this study (Fig. 2), suggesting that S. tumefaciens infections could simply be an unusual pathological presentation of S. stercoralis (Wulcan et al., Reference Wulcan, Dennis, Ketzis, Bevelock and Verocai2019). There is clearly a need for further investigation into Strongyloides spp. of domestic cats, especially in regard to their zoonotic potential. The analytic approach presented here, along with morphological and biological characterization, should prove beneficial in answering these questions.
The other topic of interest in our study was the identity and population structure of S. fuelleborni as it relates to human and other primate infections. The validity and number of species infecting primates have been long debated. Genetic analyses, including the approach used here, have proven valuable in reconciling some of these viewpoints, as morphological comparisons have confounded historical investigations of Strongyloides taxonomy. This challenge mainly owes to the variability of characters and their interpretation – importantly, how much relative weight was given to which characteristics for species designations. For example, both Chandler and Little felt that morphologic features of the parasitic female were most reliable (Chandler, Reference Chandler1925; Little, Reference Little1966a), whereas Looss and Goodey placed more importance on the free-living adult stages (Looss, Reference Looss1911; Goodey, Reference Goodey1926). These discrepancies, along with the broad geographic and host range from which Strongyloides specimens were derived in these investigations, lead to differing conclusions and opinions regarding the number of species from primates.
Formerly, two species names were used for Strongyloides derived from Old World primates – S. fuelleborni Von Linstow, Reference Von Linstow1905, described from Pan troglodytes and Papio cynocephalus from Africa, and S. simiae Hung and Höppli, Reference Hung and Höppli1923 from Asian Macaca sp. The latter species was established on the basis of a smaller oesophagus to total body length ratio in comparison to S. fuelleborni, a lack of prominent constriction behind the vulva of the free-living female, and a ‘finely-striated cuticle’ (the authors considered S. fuelleborni to have a smooth cuticle) (Hung and Höppli, Reference Hung and Höppli1923). However, other authors report that S. fuelleborni and many, if not all other Strongyloides spp., indeed has a striated cuticle though sometimes very difficult to observe (Sandground, Reference Sandground1925; Grove, Reference Grove1989). As such, the validity of S. simiae has been scrutinized by several authors, most of whom regarded the morphological evidence for specific status insufficient or too highly variable for unequivocal species discrimination (Sandground, Reference Sandground1925; Premvati, Reference Premvati1959; Little, Reference Little1966a). Staphylococcus simiae became a junior synonym of S. fuelleborni, which is the name currently applied to all egg-passing Strongyloides from Old World apes and monkeys (Grove, Reference Grove1989).
Some biological evidence suggests there may still be valid differences between ‘S. simiae’ from Macaca spp. and S. fuelleborni. Experimental work by Augustine revealed that crosses between strains derived from Macaca and strains derived from Pan failed to produce offspring – suggesting perhaps that S. simiae is indeed a separate species, but nearly impossible to distinguish morphologically from S. fuelleborni (Augustine, Reference Augustine1940). It was also observed that Cebus-derived strains (later designated S. cebus – a species currently considered valid) also failed to cross with the other primate strains, supporting S. cebus as a distinct species despite morphologic overlap with S. fuelleborni in several characters (Augustine, Reference Augustine1940; Premvati, Reference Premvati1959). Another potential point of interest is that Von Linstow's original description of S. fuelleborni was based on parasites from two African host species (Pan troglodytes and Papio cynocephalus); though no major morphological differences were observed, this could have created inherent variability and lead to assumptions on a lack of host specificity (Von Linstow, Reference Von Linstow1905). While it has been established that New World and Old World-primate-derived Strongyloides (i.e. S. cebus and S. fuelleborni) are not capable of cross-infection (Faust, Reference Faust1931), no experiments have been conducted to compare the host specificity within African and Asian S. fuelleborni from different primate hosts.
The distinction between S. fuelleborni lineages on the basis of geography is supported by our analysis, where specimens obtained from Africa (Fig. 3, clusters 1 and 2) are clearly distinct from Asian genotypes. The finer division of Asian S. fuelleborni into multiple lineages is also supported (Fig. 3). Strongyloides fuelleborni from Bornean monkeys (proboscis monkeys, long-tailed macaques, silvered leaf monkeys) and orangutans form an outgroup to other genotypes (Fig. 3, clusters 6 and 7), and are also distinct from an undescribed Strongyloides sp. collected from lorises as part of the same Bornean survey (Frias et al., Reference Frias, Stark, Lynn, Nathan, Goossens, Okamoto and MacIntosh2018). We also note that the host range of S. fuelleborni from Malaysian Borneo (Fig. 3, clusters 6 and 7) and mainland Southeast Asia (Fig. 3, cluster 3) overlap; both genotypes were found in long-tailed macaques, yet the genetic distinction between Bornean and mainland Southeast Asian genotypes is clear. Japanese macaques (Fig. 3, cluster 4) harbour a lineage of S. fuelleborni that shares an affinity with isolates from mainland Southeast Asia and Southern Asia (Fig. 3, cluster 3), but these two clusters may still represent distinct groups. We note, however, that the specimens from Japanese macaques were based on an inferred genotype so the analysis was performed a second time, excluding data from Japanese macaques. The structure of the resulting dendrograms remained virtually unchanged following exclusion of this data, indicating that the distinction between S. fuelleborni from Malaysian Borneo, Africa and mainland Southeast Asia/Southern Asia remains supported (Supplementary File S2, Appendix part D). Further sampling of primate Strongyloides across these areas of interest and from other primate species, will help to resolve the true picture of species diversity.
Similar to how the genetic differences observed in host varieties raised important questions on the most appropriate naming of S. stercoralis across hosts, the geographic differences we uncovered for S. fuelleborni may also prompt a possible nomenclatural revision. Prior to the availability of molecular analyses, Ashford and Barnish stated that ‘In the event of S. fuelleborni is as thus conceived including more than one species, the name S. simiae is available for the parasites of Asian primates’ (Ashford and Barnish, Reference Ashford, Barnish and Grove1989). In the ‘molecular era’, Hasegawa et al. (Reference Hasegawa, Hayashida, Ikeda and Sato2009, Reference Hasegawa, Sato, Fujita, Nguema, Nobusue, Miyagi, Kooriyama, Takenoshita, Noda, Sato, Morimoto, Ikeda and Nishida2010) also remarked on the considerable diversity of S. fuelleborni across Asian and African varieties, suggesting that subspecific designations might be indicated for varieties they investigated. Though not possible to definitively confirm the specific status and assign ‘S. simiae’ or suggest another name for Asian isolates from this work alone, this opens the possibility for such if further characterization continues to support that conclusion. It is also possible that Macaca spp. and other primates may harbour multiple, possibly cryptic, Strongyloides species. Importantly, genetic differences between and among African and Asian primate S. fuelleborni may lead to corresponding differences in zoonotic potential, which could explain the nearly exclusive restriction of human S. fuelleborni infections to sub-Saharan Africa. If this is the case, recognizing separate species would be useful if further investigation supports this. Presently, S. fuelleborni genotyping datasets are smaller and less complete compared to those available for S. stercoralis. Additional sequence data from morphologically characterized African and Asian S. fuelleborni would be required from a range of hosts before any taxonomic changes are formalised.
Continuing genetic analyses will aid in resolving a major outstanding question on whether the occurrence of S. stercoralis in animals and humans represents zoonotic spillover (animal to human transmission) or spillback (human to animal transmission). Apart from occasional S. stercoralis infections in great apes living in proximity to infected humans, bona fide S. stercoralis infections, or infections with similar species are very seldom detected in herbivores or the omnivorous primates (Grove, Reference Grove1989). Therefore, it seems that S. stercoralis would have originally evolved in canids or allied taxa (e.g. within the suborder Caniformia) rather than in hominids.
Finally, historic experimentation reveals that two Strongyloides species described from procyonids, S. procyonis and S. nasua, show interesting patterns analogous to observations of S. stercoralis of dogs and humans. The raccoon-associated species S. procyonis bears close morphological similarities to S. stercoralis (see Little, Reference Little1966a). In one trial, a short-lived, transient patent S. procyonis infection was successfully established in a human volunteer; one dog also developed a moderate-duration patent infection (3.5 months) (Little, Reference Little1966b). Raccoons may also be susceptible to human S. stercoralis – a young raccoon developed a patent infection of moderate duration (92 days) following exposure to filariform larvae collected from an infected human (Johnson, Reference Johnson1962). Strongyloides nasua of coati (Nasua spp.) – is also highly similar to S. stercoralis and some authors regard Strongyloides nasua as a synonym for S. stercoralis (Moraes et al., Reference Moraes, da Silva, Tebaldi and Lux Hoppe2019). Also, a patent infection was established in a white-nosed coati (Nasua narica) using human-derived S. stercoralis (Sandground, Reference Sandground1926). The observation of morphological overlap and cross-transmissibility among these species of S. stercoralis-like parasites (S. procyonis, S. nasua, S. stercoralis of dogs, and S. stercoralis of humans) supports the idea that the common ancestor of the S. stercoralis-like parasites originated in some ancestral Caniformia carnivore, that adapted to infecting humans sometime during the domestication of dogs. This idea was also suggested by Nagayasu et al. (Reference Nagayasu, Htwe, Hortiwakul, Hino, Tanaka, Higashiarakawa, Olia, Taniguchi, Win and Ohashi2017) on the basis of S. procyonis being the closest extant relative to S. stercoralis, which is also supported by phylogenies constructed from 18S rDNA sequences by Hino et al. (Reference Hino, Tanaka, Takaishi and Fuji2014). Further genetic characterization of the procyoid parasites (S. procyonis, S. nasua) and other currently undescribed Strongyloides of other caniforms, including both true canids and arctoid mammals (e.g. bears, mustelids), will be necessary to investigate this evolutionary hypothesis.
It follows from the evidence above that S. fuelleborni, rather than S. stercoralis, is probably best regarded as the true ‘human Strongyloides’ that has coevolved with our species, given the primate origin of the former, and that S. stercoralis probably represents an evolutionary spillover event. The inability to morphologically distinguish distinct genetic lineages of S. stercoralis, the relationship between S. stercoralis and other Strongyloides infecting mammals of the order Caniformia, in conjunction with the clustering of canine S. stercoralis (cluster 5) as an outgroup in this analysis, support the hypothesis of Nagayasu et al. (Reference Nagayasu, Htwe, Hortiwakul, Hino, Tanaka, Higashiarakawa, Olia, Taniguchi, Win and Ohashi2017) who proposed that the origin of human-infecting S. stercoralis was related to the domestication of dogs circa 14,000–6500 years before present (MacHugh et al., Reference MacHugh, Larson and Orlando2017). However, our data indicate that some genotypes have seemingly adapted to the human host to a point where they rarely infect dogs (Fig. 2, excluding clusters 4 & 5). Therefore, these rare cases of dog infection caused by clearly human-adapted genotypes (e.g. Fig. 2, clusters 1–3 & 6) possibly represent modern and ongoing zoonotic ‘spillback’. This highlights that generalizations regarding the role of dogs in human strongyloidiasis are difficult to make without understanding the population structure of this species complex.
Concluding remarks
Our analysis indicates that the ‘two lineage’ population structure supposing one zoonotic lineage (genotype A) and a second dog-specific lineage (genotype B) represents an over-simplification of the S. stercoralis population structure. Our findings show that a gradient of host permissibility is supported, where one genotype (lineage B; associated with cluster 5, Fig. 2), is dog-specific, producing a natural and distinct outgroup to all of lineage A (Fig. 2, lineage A includes all clusters except 5). Strongyloides stercoralis assigned to clusters 1, 2, 3 and 6 support a ‘spillback’ model; otherwise more dog-derived S. stercoralis specimens would be expected in these clusters. While available data for S. fuelleborni was sparser and less complete in comparison to S. stercoralis, the distinction between African and Asian genotypes seems to support allopatric or vicariant speciation. The separation of Asian genotypes into at least two separate lineages (Fig. 3, clusters 6 and 7 from Malaysian Borneo and clusters 3 and 4 from mainland Asia and Japan), may also be warranted. Close examination of experimental infection and reproductive studies performed by historic investigators generally support the aforementioned trends, as evidenced in our detailed discussion and review of published literature.
The associations between host, geography and genotype reported here, tend to corroborate both early and modern observations that support the proposal that S. stercoralis represents a species complex (S. stercoralis sensu lato), and referring to lineage B (Fig. 2, cluster 5) provisionally by a different designation – for example, Strongyloides stercoralis dog genotype, or S. stercoralis canis as proposed by Chandler (Reference Chandler1925). Results from further morphological and molecular studies as well as modern in vitro and in vivo biological comparative studies of defined isolates might provide support for S. stercoralis from dogs (specifically, lineage B/cluster 5) to be renamed as S. canis as originally suggested by Brumpt (Reference Brumpt1922) and later by Augustine (Reference Augustine1940).
It appears that Strongyloides fuelleborni is the most appropriate name for species infecting African primates, as this species was originally described from African chimpanzees and baboons (Von Linstow, Reference Von Linstow1905). However, the genetic distinctions noted here and elsewhere (Fig. 3) (Barratt et al., Reference Barratt, Lane, Talundzic, Richins, Robertson, Formenti, Pritt, Verocai, Nascimento de Souza, Mato Soares, Traub, Buonfrate and and Bradbury2019b) support that future taxonomic revision is needed (Hung and Höppli, Reference Hung and Höppli1923) for Strongyloides currently designated as ‘S. fuelleborni’ infecting Asian primates, although characterization of additional parasite material is needed. It is also possible that African and Asian S. fuelleborni groups represent a species complexes of their own, further emphasizing the need for resolving taxonomic relationships among these lineages.
To address the remaining unanswered questions raised here regarding the statuses of S. stercoralis and S. fuelleborni as species complexes, we first propose that additional sampling and genotyping is required. This sampling should be focused particularly in areas where there is an ongoing dog and human S. stercoralis sensu lato transmission. Given the strong sampling bias towards S. stercoralis from Southeast Asian countries and Japan, sampling of specimens from other endemic areas would be of great value; data from Africa, the Middle East and South America are particularly sparse. A wider sampling of S. fuelleborni from African humans and other primates is also required. This is especially pertinent to Japanese S. fuelleborni for which very few sequences are currently available. In addition to experimental infections to evaluate host specificity, in vitro fertilization/mating experiments such as those performed by Augustine (Reference Augustine1940), using genetically characterized isolates could provide critical insight into reproductive isolation and therefore species statuses of Strongyloides populations of interest. Finally, sequencing of additional Strongyloides loci (e.g. as per Nagayasu et al., Reference Nagayasu, Htwe, Hortiwakul, Hino, Tanaka, Higashiarakawa, Olia, Taniguchi, Win and Ohashi2017) could provide increased resolution of genetic relationships when analyses like the one described here are performed.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182020000979.
Author contributions
The authors contributed equally to this article. SS and JB prepared the manuscript, reviewed the literature and interpreted the results. JB analysed the data.
Financial support
This research was supported by a grant from the Centers for Disease Control and Prevention Office of Advanced Molecular Detection.
Conflict of interest
The authors have no conflicts of interest to disclose.
Ethical standards
The study uses sequence data that were freely available in a public database (NCBI) and is entirely bioinformatics-based.
Disclaimer
The findings and conclusions in this manuscript are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry.







 Open access
Open access