



No CrossRef data available.
Article contents
Fine mapping and discovery of candidate genes for rind trait in watermelon
Published online by Cambridge University Press: 01 December 2023
Abstract
Fine mapping and discovery of watermelon rind trait candidate genes are of great significance for modern watermelon breeding and development. In this study, we used the high-resolution genetic mapping and genome-wide genetic variation detection technology, combined with genome survey and sequencing technology, to locate and discover the candidate genes for rind traits of star watermelon varieties ‘Su XuanBai’ and ‘SHLX21’. Firstly, we identified a total of eight quantitative trait loci (QTLs) related to watermelon rind traits on chromosome 6. Secondly, a total of 208,240 single nucleotide polymorphisms and 75,345 small Indels (insertions/deletions) were detected in the two parents by high-coverage re-sequencing, respectively. Based on the genetic variation of the two parents and combined with Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis using the planta database, the QTL region was reduced to 0.02 Mb. Finally, we identified the six potential regulatory factors for watermelon rind traits using real-time quantitative PCR. In conclusion, our results revealed the fine localization of candidate genes for watermelon rind traits and the successful discovery of candidate genes for regulating watermelon rind traits, which is of importance for watermelon rind traits and breeding-improved watermelon varieties.
- Type
- Research Article
- Information
- Copyright
- Copyright © The Author(s), 2023. Published by Cambridge University Press on behalf of National Institute of Agricultural Botany
References
Chang, J, Guo, Y, Yan, J, Zhang, Z, Yuan, L, Wei, C, Zhang, Y, Ma, J, Yang, J, Zhang, X and Li, H (2021) The role of watermelon caffeic acid O-methyltransferase (ClCOMT1) in melatonin biosynthesis and abiotic stress tolerance. Horticulture Research 8, 210.Google ScholarPubMed
Cui, H, Ding, Z, Zhu, Q, Wu, Y, Qiu, B and Gao, P (2021) Comparative analysis of nuclear, chloroplast, and mitochondrial genomes of watermelon and melon provides evidence of gene transfer. Scientific Reports 11, 1595.CrossRefGoogle ScholarPubMed
Dia, M, Wehner, TC, Perkins-Veazie, P, Hassell, R, Price, DS, Boyhan, GE, Olson, SM, King, SR, Davis, AR, Tolla, GE, Bernier, J and Juarez, B (2016) Stability of fruit quality traits in diverse watermelon cultivars tested in multiple environments. Horticulture Research 3, 16066.Google ScholarPubMed
Dong, Y, Wang, Y, Jin, F-W, Xing, L-J, Fang, Y, Zhang, Z-Y, Zou, J-J, Wang, L and Xu, M-Y (2020) Differentially expressed miRNAs in anthers may contribute to the fertility of a novel Brassica napus genic male sterile line CN12A. Journal of Integrative Agriculture 19, 1731–1742.Google Scholar
Guo, S, Zhang, J, Sun, H, Salse, J, Lucas, WJ, Zhang, H, Zheng, Y, Mao, L, Ren, Y, Wang, Z, Min, J, Guo, X, Murat, F, Ham, B-K, Zhang, Z, Gao, S, Huang, M, Xu, Y, Zhong, S, Bombarely, A, Mueller, LA, Zhao, H, He, H, Zhang, Y, Zhang, Z, Huang, S, Tan, T, Pang, E, Lin, K, Hu, Q, Kuang, H, Ni, P, Wang, B, Liu, J, Kou, Q, Hou, W, Zou, X, Jiang, J, Gong, G, Klee, K, Schoof, H, Huang, Y, Hu, X, Dong, S, Liang, D, Wang, J, Wu, K, Xia, Y, Zhao, X, Zheng, Z, Xing, M, Liang, X, Huang, B, Lv, T, Wang, J, Yin, Y, Yi, H, Li, R, Wu, M, Levi, A, Zhang, X, Giovannoni, JJ, Wang, J, Li, Y, Fei, Z and Xu, Y (2013) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nature Genetics 45, 51–58.CrossRefGoogle ScholarPubMed
Guo, S, Zhao, S, Sun, H, Wang, X, Wu, S, Lin, T, Ren, Y, Gao, L, Deng, Y, Zhang, J, Lu, X, Zhang, H, Shang, J, Gong, G, Wen, C, He, N, Tian, S, Li, M, Liu, J, Wang, Y, Zhu, Y, Jarret, R, Levi, A, Zhang, X, Huang, S, Fei, Z, Liu, W and Xu, Y (2019) Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nature Genetics 51, 1616–1623.CrossRefGoogle ScholarPubMed
Kim, M, Nguyen, TTP, Ahn, JH, Kim, GJ and Sim, SC (2021) Genome-wide association study identifies QTL for eight fruit traits in cultivated tomato (Solanum lycopersicum L.). Horticulture Research 8, 203.CrossRefGoogle ScholarPubMed
Li, X, An, M, Xia, Z, Bai, X and Wu, Y (2017) Transcriptome analysis of watermelon (Citrullus lanatus) fruits in response to Cucumber green mottle mosaic virus (CGMMV) infection. Scientific Reports 7, 16747.CrossRefGoogle ScholarPubMed
Li, N, Shang, J, Wang, J, Zhou, D, Li, N and Ma, S (2018) Fine mapping and discovery of candidate genes for seed size in watermelon by genome survey sequencing. Scientific Reports 8, 17843.CrossRefGoogle ScholarPubMed
Li, H, Guo, Y, Lan, Z, Xu, K, Chang, J, Ahammed, GJ, Ma, J, Wei, C and Zhang, X (2021) Methyl jasmonate mediates melatonin-induced cold tolerance of grafted watermelon plants. Horticulture Research 8, 57.CrossRefGoogle ScholarPubMed
Liu, M, Philp, J, Wang, Y, Hu, J, Wei, Y, Li, J, Ryder, M, Toh, R, Zhou, Y, Denton, MD, Wu, Y and Yang, H (2022) Plant growth-promoting rhizobacteria Burkholderia vietnamiensis B418 inhibits root-knot nematode on watermelon by modifying the rhizosphere microbial community. Scientific Reports 12, 8381.CrossRefGoogle ScholarPubMed
Mandal, MK, Suren, H and Kousik, C (2020) Elucidation of resistance signaling and identification of powdery mildew resistant mapping loci (ClaPMR2) during watermelon-Podosphaera xanthii interaction using RNA-Seq and whole-genome resequencing approach. Scientific Reports 10, 14038.Google ScholarPubMed
Park, S-W, Kim, K-T, Kang, S-C and Yang, H-B (2016) Rapid and practical molecular marker development for rind traits in watermelon. Horticulture, Environment and Biotechnology 57, 385–391.CrossRefGoogle Scholar
Qin, J-X, Jiang, Y-J, Lu, Y-Z, Zhao, P, Wu, B-J, Li, H-X, Wang, Y, Xu, S-B, Sun, Q-X and Liu, Z-S (2020) Genome-wide identification and transcriptome profiling reveal great expansion of SWEET gene family and their wide-spread responses to abiotic stress in wheat (Triticum aestivum L.). Journal of Integrative Agriculture 19, 1704–1720.CrossRefGoogle Scholar
Ramos, A, Fu, Y, Michael, V and Meru, G (2020) QTL-seq for identification of loci associated with resistance to Phytophthora crown rot in squash. Scientific Reports 10, 5326.Google ScholarPubMed
Santos-Garcia, D, Mestre-Rincon, N, Zchori-Fein, E and Morin, S (2020) Inside out: microbiota dynamics during host-plant adaptation of whiteflies. ISME Journal 14, 847–856.CrossRefGoogle ScholarPubMed
Sun, Y, Zhang, H, Fan, M, He, Y and Guo, P (2020) A mutation in the intron splice acceptor site of a GA3ox gene confers dwarf architecture in watermelon (Citrullus lanatus L.). Scientific Reports 10, 14915.CrossRefGoogle ScholarPubMed
Tian, S, Ge, J, Ai, G, Jiang, J, Liu, Q, Chen, X, Liu, M, Yang, J, Zhang, X and Yuan, L (2021b) A 2.09 Mb fragment translocation on chromosome 6 causes abnormalities during meiosis and leads to less seed watermelon. Horticulture Research 8, 256.CrossRefGoogle ScholarPubMed
Umer, MJ, Bin Safdar, L, Gebremeskel, H, Zhao, S, Yuan, P, Zhu, H, Kaseb, MO, Anees, M, Lu, X, He, N, Gong, C and Liu, W (2020) Identification of key gene networks controlling organic acid and sugar metabolism during watermelon fruit development by integrating metabolic phenotypes and gene expression profiles. Horticulture Research 7, 193.CrossRefGoogle ScholarPubMed
Volino-Souza, M, de Oliveira, GV, Conte-Junior, CA and Alvares, TS (2023) Effect of microencapsulated watermelon (Citrullus lanatus) rind on flow-mediated dilation and tissue oxygen saturation of young adults. European Journal of Clinical Nutrition 77, 71–74.CrossRefGoogle ScholarPubMed
Wang, Y, Bo, K, Gu, X, Pan, J, Li, Y, Chen, J, Wen, C, Ren, Z, Ren, H, Chen, X, Grumet, R and Weng, Y (2020) Molecularly tagged genes and quantitative trait loci in cucumber with recommendations for QTL nomenclature. Horticulture Research 7, 3.CrossRefGoogle ScholarPubMed
Wang, J, Wang, Y, Zhang, J, Ren, Y, Li, M, Tian, S, Yu, Y, Zuo, Y, Gong, G, Zhang, H, Guo, S and Xu, Y (2021a) The NAC transcription factor ClNAC68 positively regulates sugar content and seed development in watermelon by repressing ClINV and ClGH3.6. Horticulture Research 8, 214.CrossRefGoogle ScholarPubMed
Wang, D, Zhang, M, Xu, N, Yang, S, Dou, J, Liu, D, Zhu, L, Zhu, H, Hu, J, Ma, C, Yang, L and Sun, S (2022) Fine mapping a ClGS gene controlling dark-green stripe rind in watermelon. Scientia Horticulturae 291, 36–48.CrossRefGoogle Scholar
Xiao, J, Chen, SY, Sun, Y, Yang, SD and He, Y (2022) Differences of rhizospheric and endophytic bacteria are recruited by different watermelon phenotypes relating to rind colors formation. Scientific Reports 12, 6360.CrossRefGoogle ScholarPubMed
Xie, D, Xu, Y, Wang, J, Liu, W, Zhou, Q, Luo, S, Huang, W, He, X, Li, Q, Peng, Q, Yang, X, Yuan, J, Yu, J, Wang, X, Lucas, WJ, Huang, S, Jiang, B and Zhang, Z (2019) The wax gourd genomes offer insights into the genetic diversity and ancestral cucurbit karyotype. Nature Communications 10, 5158.CrossRefGoogle ScholarPubMed
Yan, J, Xu, P, Zhang, F, Huang, X, Cao, Y and Zhang, S (2022) The effects of aqueous extract from watermelon (Citrullus lanatus) peel on the growth and physiological characteristics of Dolichospermum flos-aquae. Scientific Reports 12, 8086.CrossRefGoogle ScholarPubMed
Zhou, C-Y, Xiong, H-C, Li, Y-T, Guo, H-J, Xie, Y-D, Zhao, L-S, Gu, J-Y, Zhao, S-R, Ding, Y-P, Song, X-Y and Liu, L-X (2020) Genetic analysis and QTL mapping of a novel reduced height gene in common wheat (Triticum aestivum L.). Journal of Integrative Agriculture 19, 1721–1730.CrossRefGoogle Scholar

Wang et al. supplementary material 1
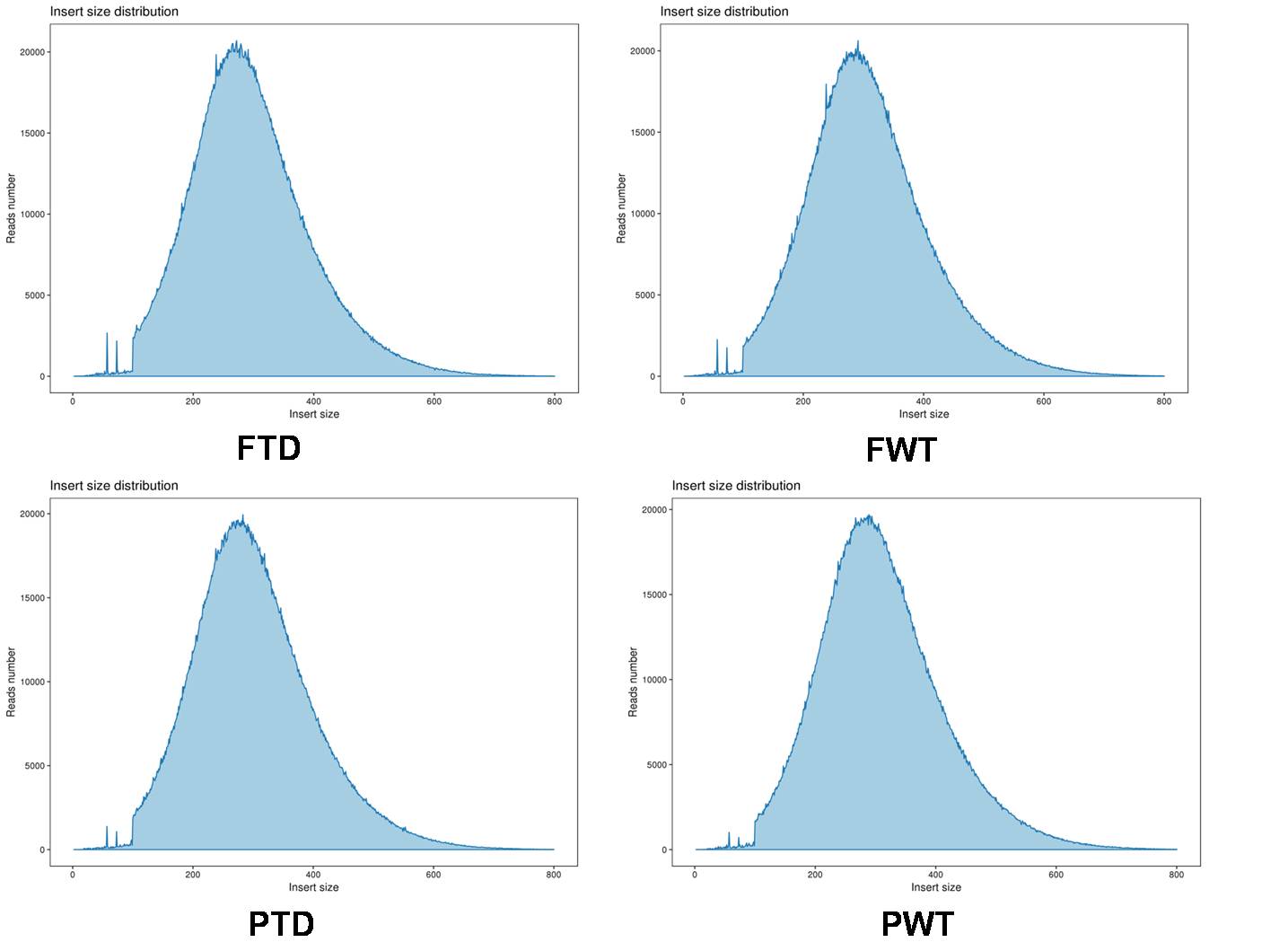
File
77.2 KB
Wang et al. supplementary material 2
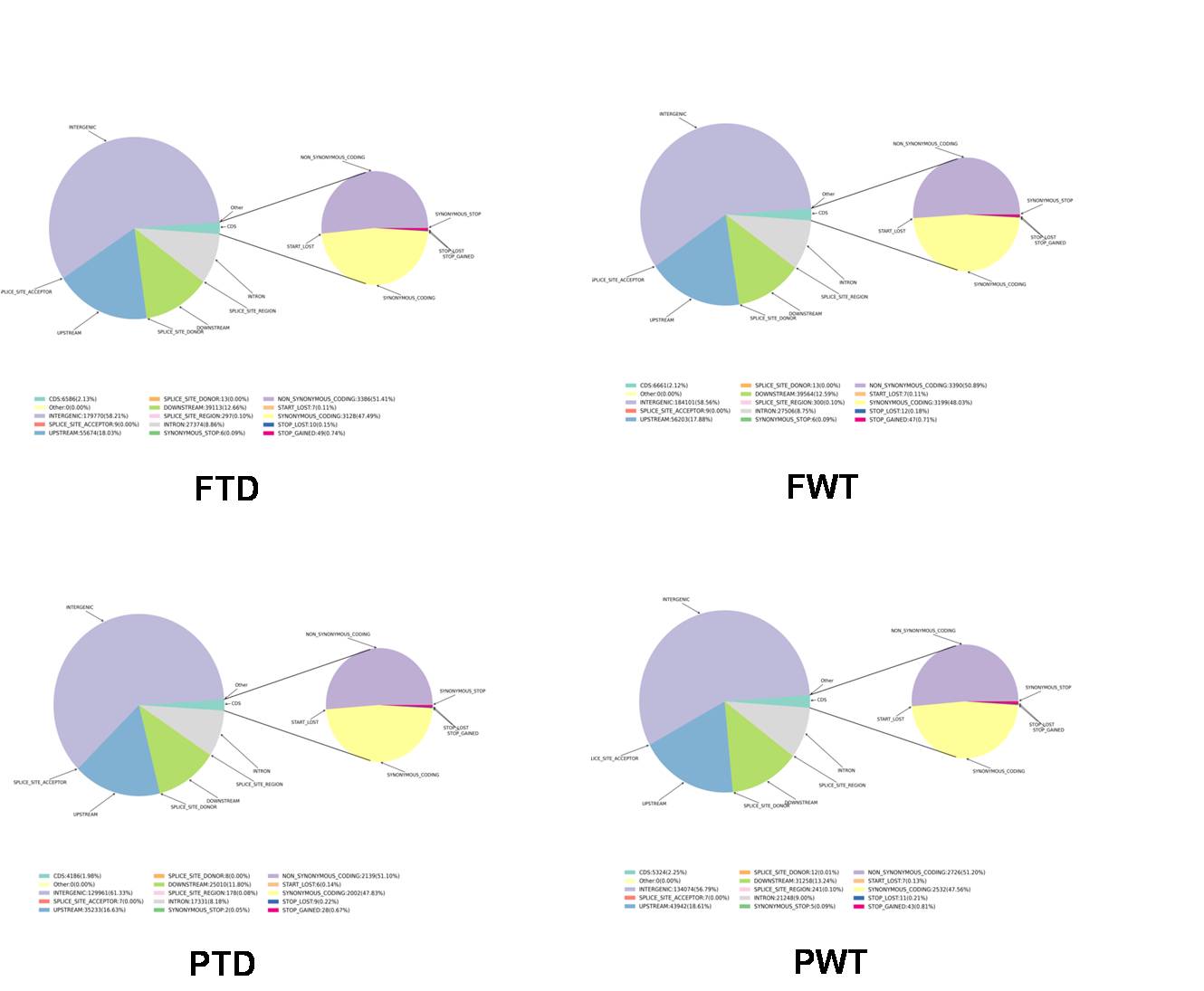
File
75.7 KB
Wang et al. supplementary material 3
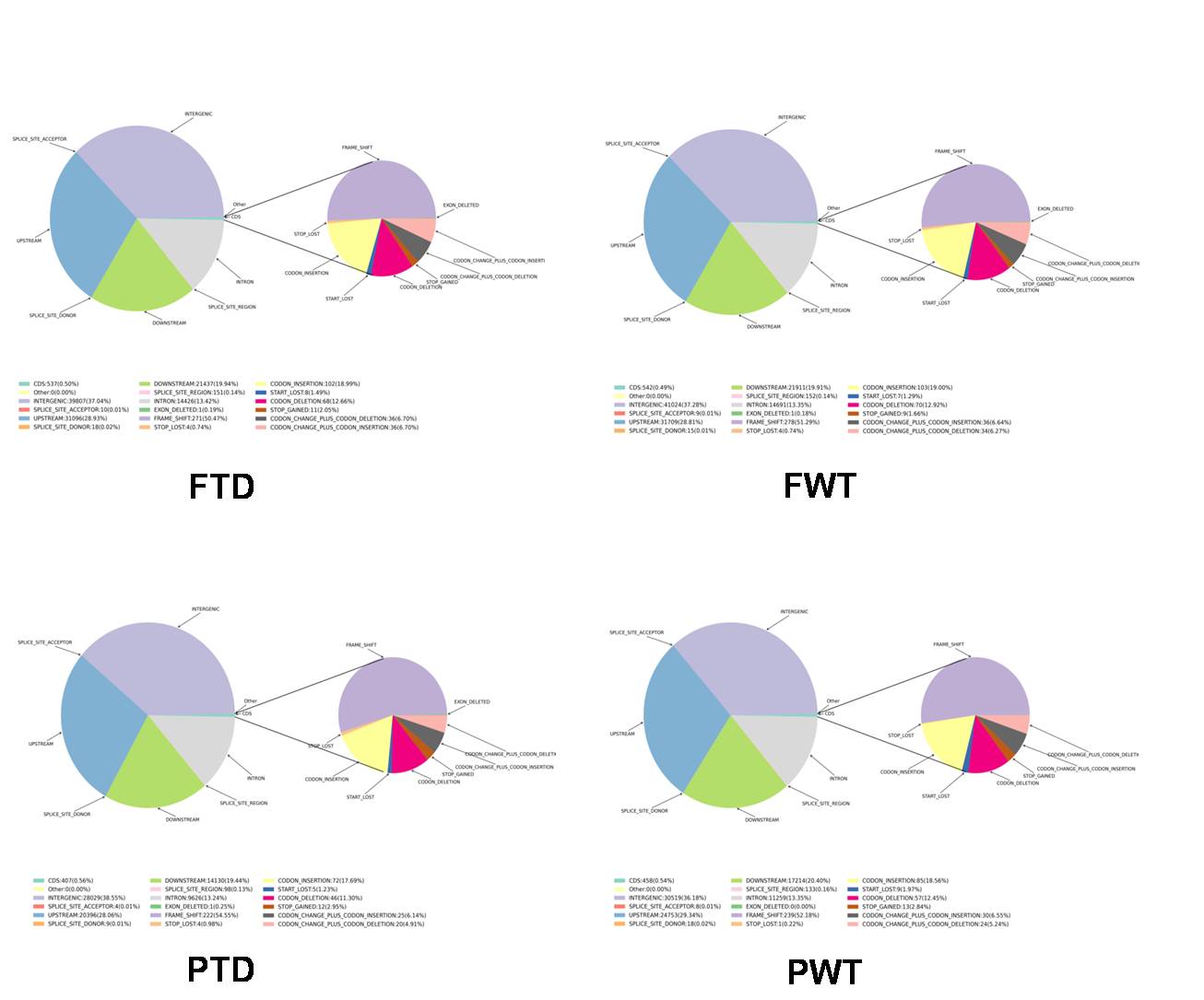
File
85.4 KB
Wang et al. supplementary material 4

File
253.3 KB