



No CrossRef data available.
Article contents
Genetic diversity and population structure analysis in early generations maize inbreds derived from local germplasm of Eastern Himalayan regions using microsatellite markers
Published online by Cambridge University Press: 29 November 2023
Abstract
The North-Eastern region (NER) of India falls under the Eastern Himalayan region and it is a bio-diversity hub. Diverse maize landraces with wide adaptability to extreme climatic and soil scenario like heavy rainfall, drought and acidic soil conditions have been grown in NER since time immemorial. However, maize diversity in NER region has drastically reduced due to introduction of high yielding varieties and hybrids. Modern maize breeding programmes are focused on high yield but other unique traits like stay green trait, prolificacy (more than one fertile ear per plant), self-fertilizing ability are also important and the local germplasm of the NER region can contribute with these unique traits. Prior to the selection of any lines in several breeding programmes, assessment of genetic diversity and population structure are basic requirements. Hence, in the present study assessment of genetic diversity and population structure study in 30 maize inbreds developed from different germplasm of NER was undertaken using SSR markers, selected for their broad distribution throughout the genome, in order to assess the extent of allelic diversity among the lines and whether any population structure could be established. In addition to assessing molecular diversity, the study aims to evaluate the potential for yield and other beneficial and unique alleles that have high potential to contribute in the genetic enhancement programme of maize.
- Type
- Research Article
- Information
- Copyright
- Copyright © The Author(s), 2023. Published by Cambridge University Press on behalf of National Institute of Agricultural Botany
Footnotes
*
E. Lamalakshmi Devi and Ngangkham Umakanta would like to share the first authorship together.
References
Adu, GB, Awuku, FJ, Amegbor, IK, Haruna, A, Manigben, KA and Aboyadana, PA (2019) Genetic characterization and population structure of maize populations using SSR markers. Annals of Agricultural Sciences 64, 47–54.CrossRefGoogle Scholar
Badu-Apraku, B and Fakorede, MAB (2017) Advances in Genetic Enhancement of Early and Extra-Early Maize for sub-Saharan Africa. Cham: Springer, 1–604 p.CrossRefGoogle Scholar
Badu-Apraku, B, Garcia-Oliveira, AL, Petroli, CD, Hearne, S, Adewale, SA and Gedil, M (2021) Genetic diversity and population structure of early and extra-early maturing maize germplasm adapted to sub-Saharan Africa. BMC Plant Biology 21, 96. https://doi.org/10.1186/s12870-021-02829-6CrossRefGoogle ScholarPubMed
Belalia, N, Lupini, A, Djemel, A, Morsli, A, Mauceri, A, Lotti, C, Khelifi-Slaoui, M, Khelifi, L and Sunseri, F (2019) Analysis of genetic diversity and population structure in Saharan maize (Zea mays L.) populations using phenotypic traits and SSR markers. Genetic Resources and Crop Evolution 66, 243–257.CrossRefGoogle Scholar
Bharadwaj, DN (2018) Advanced Molecular Plant Breeding: Meeting the Challenge of Food Security, 1st Edition. Florida, USA: Apple Academic Press, pp. 726.CrossRefGoogle Scholar
Boakyewaa Adu, G, B-Apraku, B, Akromah, R, Garcia-Oliveira, AL, Awuku, FJ and Gedil, M (2019) Genetic diversity and population structure of early-maturing tropical maize inbred lines using SNP markers. PLoS ONE 14, e0214810. https://doi.org/10.1371/journal.pone.0214810CrossRefGoogle ScholarPubMed
Borah, R, Bhattacharjee, A, Rao, SR, Kumar, V, Sharma, P, Upadhaya, K and Choudhury, H (2021) Genetic diversity and population structure assessment using molecular markers and SPAR approach in Illicium griffithii, a medicinally important endangered species of Northeast India. Journal of Genetic Engineering and Biotechnology 19, 118. doi: 10.1186/s43141-021-00211-5CrossRefGoogle ScholarPubMed
Boukteb, A, Sakaguchi, S, Ichihashi, Y, Kharrat, M, Nagano, AJ, Shirasu, K and Bouhadida, M (2021) Analysis of genetic diversity and population structure of Orobanche foetida populations from Tunisia Using RADseq. Frontiers in Plant Science 12. https://www.frontiersin.org/articles/10.3389/fpls.2021.618245CrossRefGoogle ScholarPubMed
Devi, EL, Devi, CP, Kumar, S, Sharma, SK, Beemrote, A, Chongtham, SK, Singh, CH, Ch, T, Singh, TB, Ningombam, A, Akoijam, R, Singh, IM, Singh, YR, Monteshori, S, Omita, Y, Prakash, N and Ngachan, SV (2017b) Marker assisted selection (MAS) towards generating stress tolerant crop plants. Plant Gene 11, 205–218.CrossRefGoogle Scholar
Devi, EL, Avasthe, RK, Meetei, CC, Dutta, SK, Das, SK, Singh, AR, Bhutia, TL, Kumar, A, Saha, S and Mishra, VK (2022) Miracle maize lines of North-Eastern region that can fix atmospheric nitrogen. CAU Farm Mazagine, July – September, p. 15.Google Scholar
Earl, DA and Vonholdt, BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetic Resources 4, 359–361.CrossRefGoogle Scholar
Evanno, G, Regnaut, S and Goudet, J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology 14, 2611–2620.CrossRefGoogle ScholarPubMed
Govindaraj, M, Vetriventhan, M and Srinivasan, M (2015) Importance of genetic diversity assessment in crop plants and its recent advances: an overview of its analytical perspectives. Genetics Research International 2015, 431487.CrossRefGoogle ScholarPubMed
Hasan, N, Choudhary, S, Naaz, N, Sharma, N and Laskar, RA (2021) Recent advancements in molecular marker-assisted selection and applications in plant breeding programmes. Journal of Genetic Engineering and Biotechnology 19, 128. doi: 10.1186/s43141-021-00231-1CrossRefGoogle Scholar
Hung, TN, Huyen, TN, Loc, NV and Cuong, BM (2012) The application of SSR molecular indicator to assess the purity and genetic diversity of waxy corn inbred lines. Journal of ISSASS (International Society for Southeast Asian Agricultural Sciences) 18, 45–54.Google Scholar
Islam, MA, Alam, MS, Maniruzzaman, M and Haque, MS (2023) Microsatellite marker-based genetic diversity assessment among exotic and native maize inbred lines of Bangladesh. Saudi Journal of Biological Sciences 30, 103715.CrossRefGoogle ScholarPubMed
Jones, ES, Sullivan, H, Bhattramakki, D and Smith, JSC (2007) A comparison of simple sequence repeat and single nucleotide polymorphism marker technologies for the genotypic analysis of maize (Zea mays L. Theoretical and Applied Genetics 115, 361–371. https://doi.org/10.1007/s00122-007-0570-9CrossRefGoogle ScholarPubMed
Laosatit, K, Amkul, K, Somta, P, u ma Tanadul, O, Kerdsri, C, Mongkol, W, Jitlaka, C, Suriharn, K and Jompuk, C (2022) Genetic diversity of sweet corn inbred lines of public sectors in Thailand revealed by SSR markers. Crop Breeding and Applied Biotechnology 22(4), 1–8. https://doi.org/10.1590/1984-70332022v22n4a45Google Scholar
Mukri, G, Gadag, RN, Bhat, JS, Nepolean, T, Gupta, NC, Mittal S, ML N, Kumar, R and Pal, D (2022) Characterization of sub-tropical maize (Zea Mays L.) inbred lines for the variation in kernel row numbers (KRN). bioRxiv preprint. https://doi.org/10.1101/2022.09.23.509129CrossRefGoogle Scholar
Peakall, RO and Smouse, PE (2012) GenAlex 6.5: genetic analysis in excel. Population genetic software for teaching and research. Bioinformatics (Oxford, England) 28, 2537–2539.Google ScholarPubMed
Perrier, X and Jacquemoud-Collet, JP (2006) DARwin Software. Available at http://darwin.cirad.fr/darwinGoogle Scholar
Porebski, S, Bailey, LG and Baum, BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Molecular Biology Reporter 15, 8–15.CrossRefGoogle Scholar
Prasanna, BM (2012) Diversity in global maize germplasm: characterization and utilization. Journal of Biosciences 37, 843–855.CrossRefGoogle ScholarPubMed
Pritchard, JK, Stephens, M and Donnelly, P (2000) Inference of populationstructure using multilocus genotype data. Genetics 155, 945–959.CrossRefGoogle Scholar
Sbordoni, V, Allegrucci, G and Cesaroni, D (2012) Population structure. In White, WB and Culver, DC (eds), Encyclopedia of Caves, 2nd Edn. USA:Academic Press, pp. 608–618. https://doi.org/10.1016/B978-0-12-383832-2.00090-6CrossRefGoogle Scholar
Semagn, K, Magorokosho, C, Vivek, BS, Makumbi, D, Beyene, Y, Mugo, S, Prasanna, BM and Warburton, ML (2012) Molecular characterization of diverse CIMMYT maize inbred lines from eastern and Southern Africa using single nucleotide polymorphic markers. BMC Genomics 13, 113. https://doi.org/10.1186/1471-2164-13-113CrossRefGoogle ScholarPubMed
Silva, TA, Cantagalli, LB, Saavedra, J, Lopes, AD, Mangolin, CA, Pires, MF, Machado, S and Scapim, CA (2015) Population structure and genetic diversity of Brazilian popcorn germplasm inferred by microsatellite markers. Electronic Journal of Biotechnology 18, 181–187.CrossRefGoogle Scholar
Singh, N, Choudhury, DR, Singh, AK, Kumar, S, Srinivasan, K, Tyagi, RK, Singh, NK and Singh, R (2013) Comparison of SSR and SNP markers in estimation of genetic diversity and population structure of Indian rice varieties. PLoS One 8, e84136. doi: 10.1371/journal.pone.0084136CrossRefGoogle ScholarPubMed
Singh, AR, Devi, EL, Dayal, V, Saha, S, Lungmauna, , Dutta, SK, Boopathi, T and Singh, SB (2019) Diversity of landraces maize in Mizoram: prospects, challenges and opportunities. In Souvenir of National Workshop on Scientific Maize Cultivation in North East India. Aizawl, India: ICAR Research Complex for NEH Region, Umiam, pp. 98–104.Google Scholar
van Berloo, R (2008) GGT 2.0: versatile software for visualization and analysis of genetic data. Journal of Heredity 99, 232–236.CrossRefGoogle ScholarPubMed
Van Deynze, A, Zamora, P, Delaux, PM, Heitmann, C, Jayaraman, D, Rajasekar, S, Graham, D, Maeda, J, Gibson, D, Schwartz, KD, Berry, AM, Bhatnagar, S, Jospin, G, Darling, A, Jeannotte, R, Lopez, J, Weimer, BC, Eisen, JA, Shapiro, HY, Ane, JM and Bennet, AB (2018) Nitrogen fixation in a landrace of maize is supported by a mucilage-associated diazotrophic microbiota. PLoS Biology 16, e2006352. https://doi.org/10.1371/journal.pbio.2006352CrossRefGoogle Scholar
Wegary, D, Teklewold, A, Prasanna, BM, Ertiro, BT, Alachiotis, N, Negera, D, Awas, G, Abakemal, D, Ogugo, V, Gowda, M and Semagn, K (2019) Molecular diversity and selective sweeps in maize inbred lines adapted to African highlands. Scientific Reports 9, 13490. https://doi.org/10.1038/s41598-019-49861-zCrossRefGoogle ScholarPubMed
Wu, Y, Vicente, FS, Huang, K, Dhliwayo, T, Costich, DE, Semagn, K, Sudha, N, Olsen, M, Prasanna, BM, Zhang, X and Babu, R (2016) Molecular characterization of CIMMYT maize inbred lines with genotyping-by-sequencing SNPs. Theoretical and Applied Genetics 129, 753–765.CrossRefGoogle ScholarPubMed
Xia, XC, Reif, JC, Melchinger, AE, Frisch, M, Hoisington, DA, Beck, D, Pixley, K and Warburton, ML (2005) Genetic diversity among CIMMYT maize inbred lines investigated with SSR markers: II. Subtropical, tropical midaltitude, and highland maize inbred lines and their relationships with elite U.S. and European maize. Crop Science 45, 2573–2582. doi: 10.2135/cropsci2005.0246CrossRefGoogle Scholar
Yadav, OP, Hossain, F, Karjagi, CG, Kumar, B, Zaidi, PH, Jat, SL, Chawla, JS, Kaul, J, Hooda, KS, Kumar, P, Yadava, P and Dhillon, BS (2015) Genetic improvement of maize in India: retrospect and prospects. Agricultural Research 4, 325–338.Google Scholar
Yang, X, Xu, Y, Shah, T, Li, H, Han, Z, Li, J and Yan, J (2011) Comparison of SSRs and SNPs in assessment of genetic relatedness in maize. Genetica 139, 1045–1054.CrossRefGoogle ScholarPubMed
Yin, V, Sa, KJ, Lim, SE and Lee, JK (2019) Genetic diversity and association analyses of Chinese maize inbred lines using SSR markers. Plant Breeding and Biotechnology 7, 186–199.Google Scholar
Yu, RH, Wang, YL, Sun, Y and Liu, B (2012) Analysis of genetic distance by SSR in waxy maize. Genetics and Molecular Research 11, 254–260.CrossRefGoogle ScholarPubMed

Lamalakshmi Devi et al. supplementary material 1
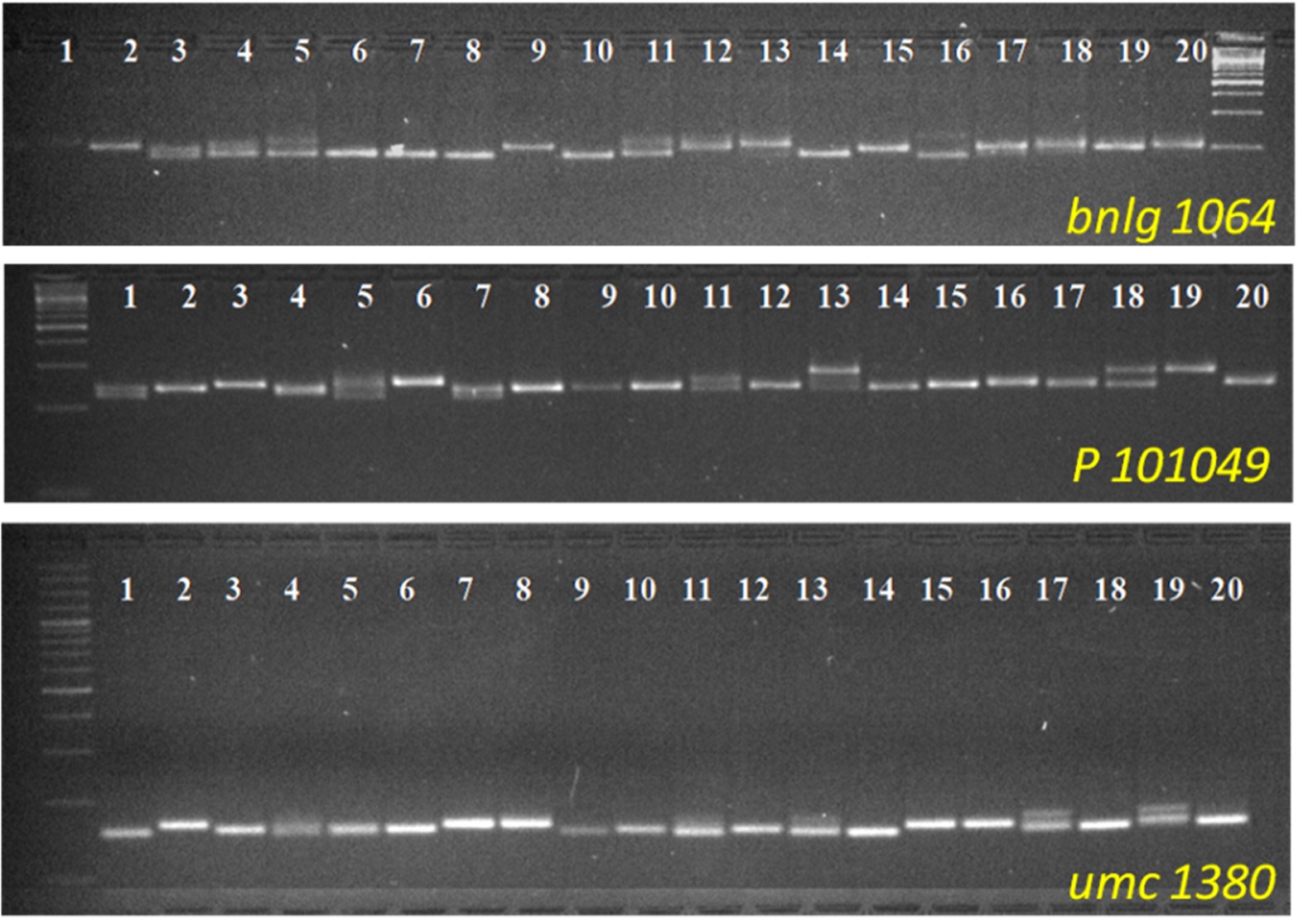
File
280.9 KB
Lamalakshmi Devi et al. supplementary material 2
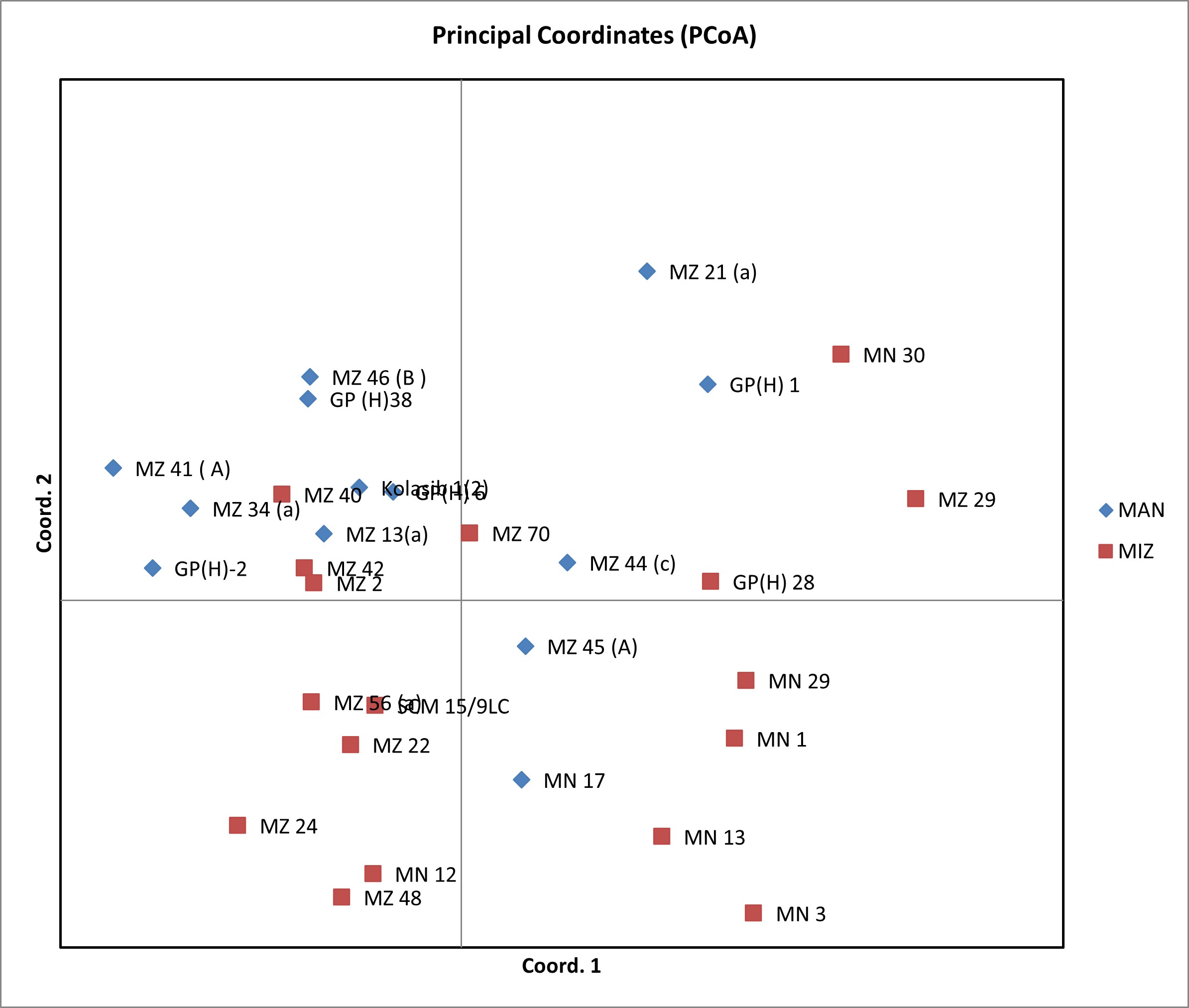
File
251.8 KB
Lamalakshmi Devi et al. supplementary material 3
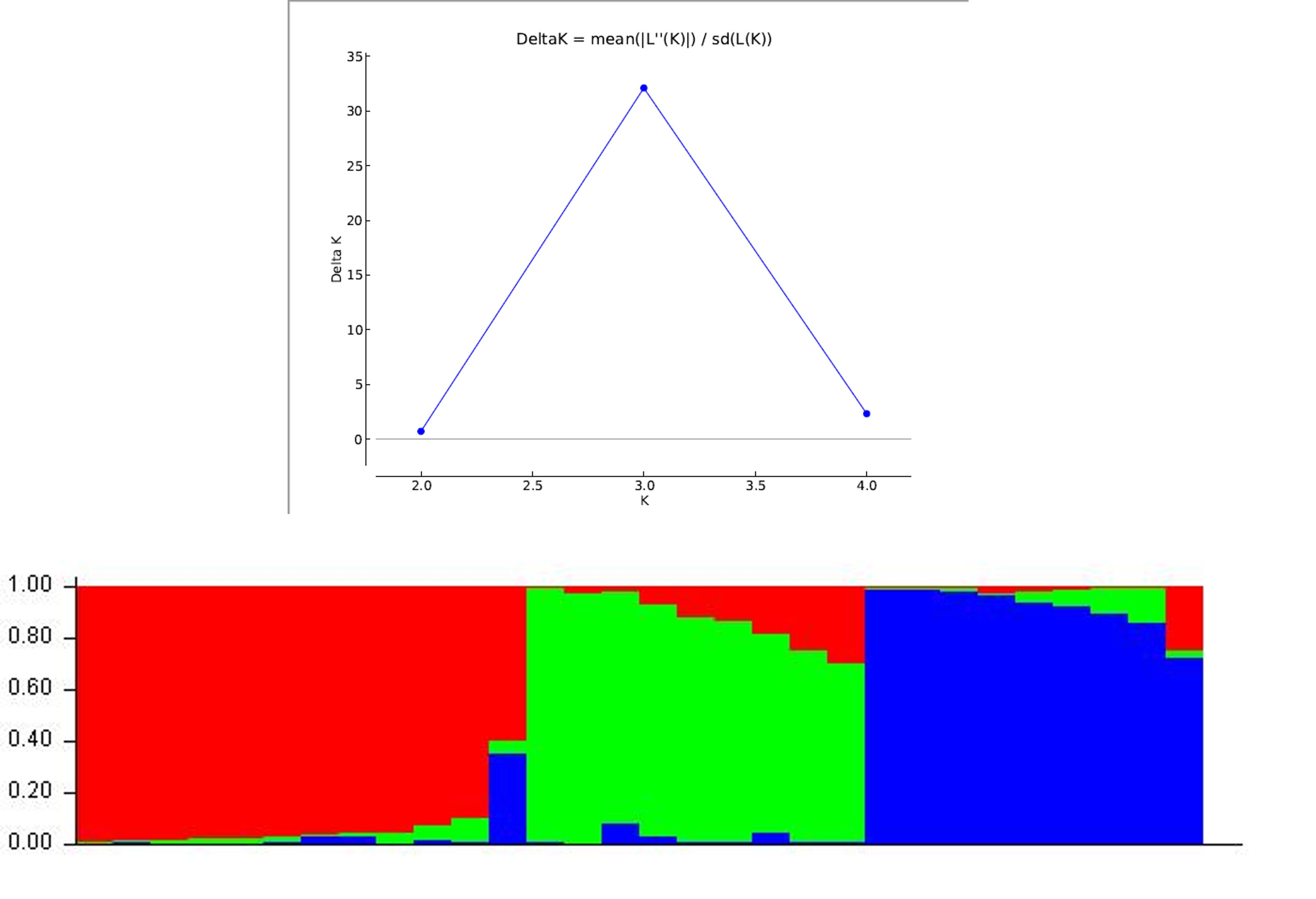
File
240.6 KB
Lamalakshmi Devi et al. supplementary material 4
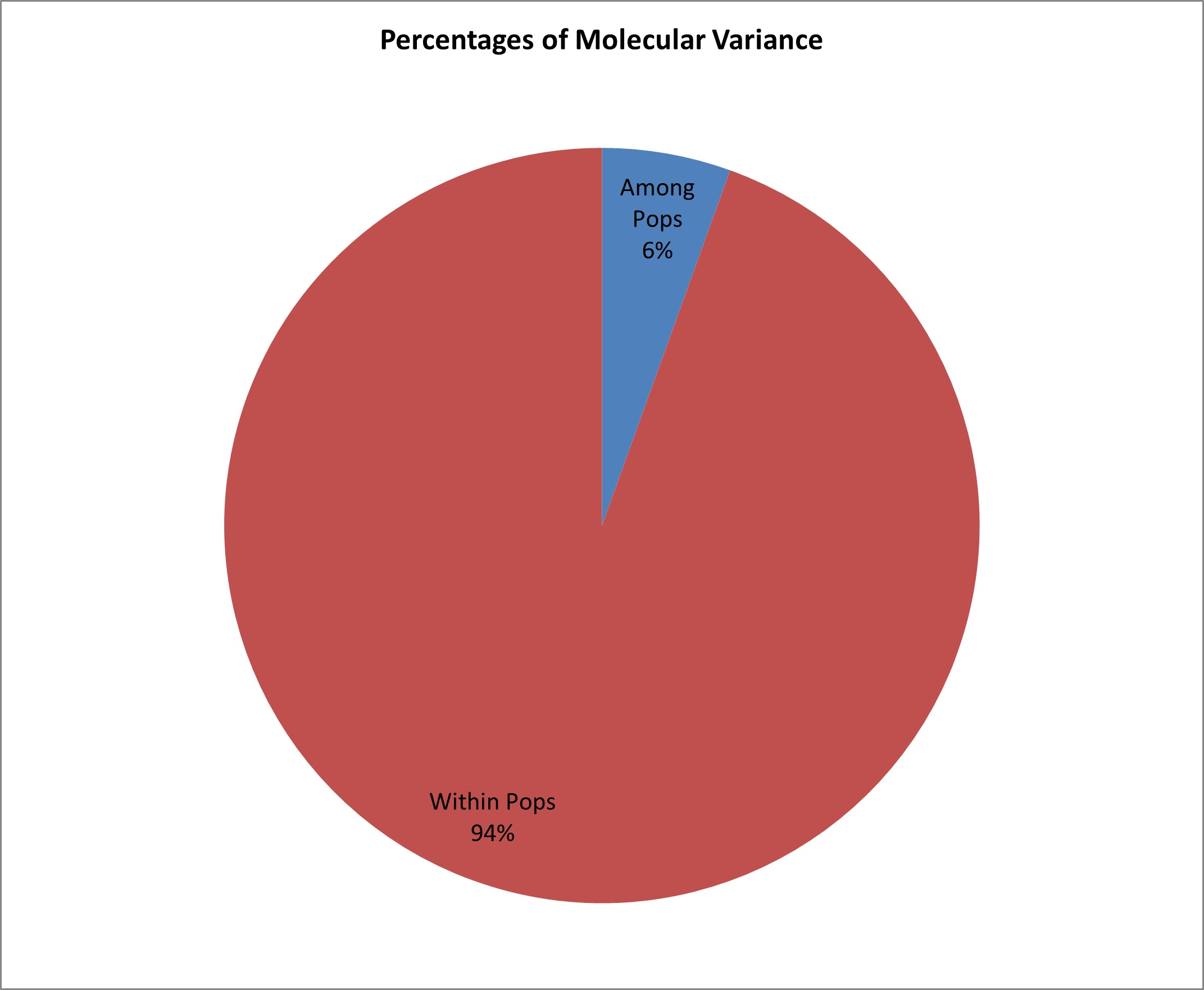
File
130.8 KB
Lamalakshmi Devi et al. supplementary material 5
File
126.8 KB
Lamalakshmi Devi et al. supplementary material 6

File
189.1 KB
Lamalakshmi Devi et al. supplementary material 7

File
28.2 KB