


No CrossRef data available.
Article contents
Powder X-ray diffraction intensities of corundum calculated by conventional and density functional theory methods and extracted by deconvolutional treatment on experimental data
Published online by Cambridge University Press: 05 June 2023
Abstract
Least-squares analysis on the diffraction intensity values certified for NIST SRM676a and SRM1976c α-Al2O3 (corundum) have shown that the intensities of SRM1976c can be simulated by the March-Dollase preferred orientation model along the (001)-direction. Diffraction intensities of randomly oriented corundum crystallites have been calculated from electron density data obtained by conventional and density functional theory (DFT) calculations, on the assumption of independent and similar atomic displacements for Al and O atoms. The results of DFT calculations support that the strongest peak of randomly oriented α-Al2O3 crystalline powder should be 113-reflection, though the intensities simulated by DFT calculations are not closer to NIST SRM676a intensities than those expected for a fully ionized model 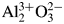 ${\rm Al}_2^{3 + } {\rm O}_3^{2-}$. Diffraction data of two types of relatively fine (nominally 2–3 μm and ca 0.3 μm) α-Al2O3 powder have been collected and processed by a deconvolutional treatment (DCT). Integrated peak intensities extracted from the DCT data by an individual peak profile fitting method also support that the 113-reflection is the strongest reflection of randomly oriented α-Al2O3 crystalline powder.
${\rm Al}_2^{3 + } {\rm O}_3^{2-}$. Diffraction data of two types of relatively fine (nominally 2–3 μm and ca 0.3 μm) α-Al2O3 powder have been collected and processed by a deconvolutional treatment (DCT). Integrated peak intensities extracted from the DCT data by an individual peak profile fitting method also support that the 113-reflection is the strongest reflection of randomly oriented α-Al2O3 crystalline powder.
Keywords
Information
- Type
- Technical Article
- Information
- Copyright
- Copyright © The Author(s), 2023. Published by Cambridge University Press on behalf of International Centre for Diffraction Data
References
REFERENCES
Creagh, D. C. 1999. International Tables for Crystallography (Vol. C, 2nd ed., pp. 242–58). Section 4.2.6. X-ray dispersion correction. Dordrecht/Boston/London, Kluwer.Google Scholar
Deutsch, M., Forster, E., Holzer, G., Hartwig, J., Hamalainen, K., Kao, C. C., Huotari, S., and Diamant, R.. 2004. “X-Ray Spectrometry of Copper: New Results on an Old Subject.” Journal of Research of the National Institute of Standards and Technology 109 (1): 75. doi:10.6028/jres.109.006.CrossRefGoogle Scholar
Dollase, W. A. 1986. “Correction of Intensities for Preferred Orientation in Powder Diffractometry: Application of the March Model.” Journal of Applied Crystallography 19 (4): 267–72. doi:10.1107/S0021889886089458.CrossRefGoogle Scholar
Giannozzi, P., Baroni, S., Bonini, N., Calandra, M., Car, R., Cavazzoni, C., Ceresoli, D., Chiarotti, G. L., Cococcioni, M., Dabo, I., Dal Corso, A., de Gironcoli, S., Fabris, S., Fratesi, G., Gebauer, R., Gerstmann, U., Gougoussis, C., Kokalj, A., Lazzeri, M., Martin-Samos, L., Marzari, N., Mauri, F., Mazzarello, R., Paolini, S., Pasquarello, A., Paulatto, L., Sbraccia, C., Scandolo, S., Sclauzero, G., Seitsonen, A. P., Smogunov, A., Umari, P., and Wentzcovitch, R. M. (2009). “QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials,” Journal of Physics: Condensed Matter 21 (39): 395502. doi:10.1088/0953-8984/21/39/395502.Google ScholarPubMed
Giannozzi, P., Andreussi, O., Brumme, T., Bunau, O., Buongiorno Nardelli, M., Calandra, M., Car, R., Cavazzoni, C., Ceresoli, D., Cococcioni, M., Colonna, N., Carnimeo, I., Dal Corso, A., de Gironcoli, S., Delugas, P., DiStasio, R. A., Ferretti, A., Floris, A., Fratesi, G., Fugallo, G., Gebauer, R., Gerstmann, U., Giustino, F., Gorni, T., Jia, J., Kawamura, M., Ko, H.-Y., Kokalj, A., Küçükbenli, E., Lazzeri, M., Marsili, M., Marzari, N., Mauri, F., Nguyen, N. L., Nguyen, H.-V., Otero-de-la-Roza, A., Paulatto, L., Poncé, S., Rocca, D., Sabatini, R., Santra, B., Schlipf, M., Seitsonen, A. P., Smogunov, A., Timrov, I., Thonhauser, T., Umari, P., Vast, N., Wu, X., and Baroni, S. (2017). “Advanced Capabilities for Materials Modelling with Quantum ESPRESSO,” Journal of Physics: Condensed Matter 29 (46): 465901. doi:10.1088/1361-648X/aa8f79.Google ScholarPubMed
Hohenberg, P., and Kohn, W.. 1964. “Inhomogeneous Electron Gas.” Physical Review 136 (3B): B864–71. doi:10.1103/PhysRev.136.B864.CrossRefGoogle Scholar
Holbrook, R. D., and Choquette, S. J.. 2021. Certificate of Analysis, Standard Reference Material 1976c, Instrument Response Standard for X-Ray Powder Diffraction. Gaithersburg, National Institute of Standards & Technology.Google Scholar
Hubbard, C. R., Evans, E. H., and Smith, D. K.. 1976. “The Reference Intensity Ratio, I/Ic, for Computer Simulated Powder Patterns.” Journal of Applied Crystallography 9 (2): 169–74. doi:10.1107/S0021889876010807.CrossRefGoogle Scholar
Ida, T. 2021a. “Equatorial Aberration for Powder Diffraction Data Collected by Continuous-Scan Integration of a Silicon Strip X-Ray Detector.” Powder Diffraction 36 (3): 169–75. doi:10.1017/S0885715621000403.CrossRefGoogle Scholar
Ida, T. 2021b. “Continuous Series of Symmetric Peak Profile Functions Determined by Standard Deviation and Kurtosis.” Powder Diffraction 36 (4): 222–32. doi:10.1017/S0885715621000567.CrossRefGoogle Scholar
Ida, T. 2022. “Convolution and Deconvolutional Treatment on Sample Transparency Aberration in Bragg–Brentano Geometry.” Powder Diffraction 37 (1): 13–21. doi:10.1017/S0885715622000021.CrossRefGoogle Scholar
Ida, T., and Toraya, H.. 2002. “Deconvolution of the Instrumental Functions in Powder X-Ray Diffractometry.” Journal of Applied Crystallography 35 (1): 58–68. doi:10.1107/S0021889801018945.CrossRefGoogle Scholar
Kaiser, D. L., and Watters, R. L. Jr. 2008. Certificate of Analysis, Standard Reference Material 676a, Alumina Powder for Quantitative Analysis by X-ray Diffraction. Gaithersburg, National Institute of Standards & Technology.Google Scholar
Kresse, G., and Joubert, D.. 1999. “From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method.” Physical Review B 59 (3): 1758–75. doi:10.1103/PhysRevB.59.1758.CrossRefGoogle Scholar
Marquardt, D. W. 1963. “An Algorithm for Least-Squares Estimation of Nonlinear Parameters.” Journal of the Society for Industrial and Applied Mathematics 11 (2): 431–41. doi:10.1137/0111030.CrossRefGoogle Scholar
Maslen, E. N., Streltsov, V. A., Streltsova, N. R., Ishizawa, N., and Satow, Y.. 1993. “Synchrotron X-Ray Study of the Electron Density in α-Al2O3.” Acta Crystallographica Section B Structural Science 49 (6): 973–80. doi:10.1107/S0108768193006901.CrossRefGoogle Scholar
Momma, K., and Izumi, F.. 2011. “VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data.” Journal of Applied Crystallography 44 (6): 1272–76. doi:10.1107/S0021889811038970.CrossRefGoogle Scholar
Ostwald, W. 1897. “Studien Über Die Bildung Und Umwandlung Fester Körper: 1. Abhandlung: Übersättigung Und Überkaltung.” Zeitschrift für Physikalische Chemie 22U (1): 289–330. doi:10.1515/zpch-1897-2233.CrossRefGoogle Scholar
Perdew, J. P., and Zunger, A.. 1981. “Self-Interaction Correction to Density-Functional Approximations for Many-Electron Systems.” Physical Review B 23 (10): 5048–79. doi:10.1103/PhysRevB.23.5048.CrossRefGoogle Scholar
Perdew, J. P., Burke, K., and Ernzerhof, M.. 1996. “Generalized Gradient Approximation Made Simple.” Physical Review Letters 77 (18): 3865–68. doi:10.1103/PhysRevLett.77.3865.CrossRefGoogle ScholarPubMed
Perdew, J. P., Burke, K., and Ernzerhof, M.. 1997. “Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)].” Physical Review Letters 78 (7): 1396–96. doi:10.1103/PhysRevLett.78.1396.CrossRefGoogle Scholar
Sun, C., and Xue, D.. 2014. “Crystal Growth and Design of Sapphire: Experimental and Calculation Studies of Anisotropic Crystal Growth upon Pulling Directions.” Crystal Growth & Design 14 (5): 2282–87. doi:10.1021/cg401867c.CrossRefGoogle Scholar
Suzuki, T. 1960. “Atomic Scattering Factor for O2−.” Acta Crystallographica 13 (3): 279. 10.1107/S0365110X60000698CrossRefGoogle Scholar
Tokonami, M. 1965. “Atomic Scattering Factor for O2−.” Acta Crystallographica 19 (3): 486. 10.1107/S0365110X65003729CrossRefGoogle Scholar
Waasmaier, D., and Kirfel, A.. 1995. “New Analytical Scattering-Factor Functions for Free Atoms and Ions.” Acta Crystallographica Section A Foundations of Crystallography 51 (3): 416–31. doi:10.1107/S0108767394013292.CrossRefGoogle Scholar