


Introduction
Post-traumatic stress disorder (PTSD) is a pervasive and debilitating mental disorder in the USA with a lifetime prevalence of about 7% (Kessler et al. Reference Kessler, Berglund, Demler, Jin, Merikangas and Walters2005). PTSD has been declared ‘a life sentence’ (Abrams & Manstead, Reference Abrams and Manstead1981) based on evidence that the disorder leads to a host of physical health problems. While investigators have examined PTSD in relation to a range of health outcomes (including rheumatoid arthritis, stroke, heart disease and cancer), some of the strongest empirical research – in terms of methodology and findings – has been with cardiometabolic diseases (Lohr et al. Reference Lohr, Palmer, Eidt, Aailaboyina, Mausbach, Wolkowitz, Thorp and Jeste2015). Numerous methodologically rigorous prospective population-based observational studies have found that PTSD is associated with increased risk of incident cardiovascular disease (CVD) (Boscarino & Chang, Reference Boscarino and Chang1999; Boscarino, Reference Boscarino2006, Reference Boscarino2008; Kubzansky et al. Reference Kubzansky, Koenen, Spiro, Vokonas and Sparrow2007, Reference Kubzansky, Koenen, Jones and Eaton2009; Scherrer et al. Reference Scherrer, Chrusciel, Zeringue, Garfield, Hauptman, Lustman, Freedland, Carney, Bucholz and Owen2010; Jordan et al. Reference Jordan, Miller-Archie, Cone, Morabia and Stellman2011; Vaccarino et al. Reference Vaccarino, Goldberg, Rooks, Shah, Veledar, Faber, Votaw, Forsberg and Bremner2013; Chen et al. Reference Chen, Pan, Li, Lin, Chen, Lee, Tsai, Hsu, Huang and Tsai2015; Gradus et al. Reference Gradus, Farkas, Svensson, Ehrenstein, Lash, Milstein, Adler and Sørensen2015; Roy et al. Reference Roy, Foraker, Girton and Mansfield2015; Sumner et al. Reference Sumner, Kubzansky, Elkind, Roberts, Agnew-Blais, Chen, Cerdá, Rexrode, Rich-Edwards and Spiegelman2015; Beristianos et al. Reference Beristianos, Yaffe, Cohen and Byers2016) and type 2 diabetes (T2D) (Boyko et al. Reference Boyko, Jacobson, Smith, Ryan, Hooper, Amoroso, Gackstetter, Barrett-Connor and Smith2010; Vaccarino et al. Reference Vaccarino, Goldberg, Magruder, Forsberg, Friedman, Litz, Heagerty, Huang, Gleason and Smith2014; Roberts et al. Reference Roberts, Agnew-Blais, Spiegelman, Kubzansky, Mason, Galea, Hu, Rich-Edwards and Koenen2015), even after adjusting for depression (see Table 1).
Table 1. Prospective studies of PTSD and incident cardiovascular disease and type 2 diabetes

PTSD, Post-traumatic stress disorder; HR, hazard ratio; DSM, Diagnostic and Statistical Manual of Mental Disorders; BMI, body mass index; PCL, PTSD Checklist, n.a., not applicable; s.d., standard deviation; MI, myocardial infarction; CHD, coronary heart disease; CES-D, Center for Epidemiologic Studies Depression Scale; DIS, National Institute of Mental Health Diagnostic Interview Schedule; ICD, International Classification of Diseases; ADM, antidepressant medication; TBI, traumatic brain injury; SE, Southeast.
a From Edmondson & Cohen (Reference Edmondson and Cohen2013).
b Estimate is a standardized incidence rate.
c The HR not adjusted for depression means no adjustment for depression, dysthymia, bipolar disorder, generalized anxiety disorder, panic disorder or phobic disorder.
d Depression variable was anti-depressant medication not diagnosis of depression.
e Multivariate adjusted HR adjusting for listed covariates except where indicated otherwise.
Despite mounting evidence, PTSD is not acknowledged as a risk factor for either CVD or T2D. For example, neither PTSD nor stress is ranked in the American Heart Association 2010 Impact Goals as a risk factor that requires attention (Lloyd-Jones et al. Reference Lloyd-Jones, Hong, Labarthe, Mozaffarian, Appel, Van Horn, Greenlund, Daniels, Nichol and Tomaselli2010). Indeed, the American Heart Association explicitly states: ‘stress is not a confirmed risk factor for… heart disease, and has not been proven to cause heart disease’ (American Heart Association, 2013). In contrast, the American Diabetes Association does cite stress as a potential contributor to poor diabetes management (Godfrey et al. Reference Godfrey, Lindamer, Mostoufi and Afari2013). However, depression is the only mental disorder specifically acknowledged to contribute to diabetes. As a result, neither systematic surveillance nor treatment is provided to individuals with PTSD to reduce potential risk of developing these important diseases. Absent compelling evidence that PTSD causally contributes to CVD and T2D, the approach by cardiovascular and branches of medicine concerned with metabolic diseases, is unlikely to change. At this writing, PTSD has not been established as a causal risk factor for CVD or T2D from the perspective of the larger medical community.
This review offers a roadmap for the next steps needed in research to provide compelling evidence that PTSD causally contributes to cardiometabolic disease. Although we recognize that the term ‘cardiometabolic’ is broad, we focus on CVD and T2D due to their high importance for population health and strong extant evidence for associations of these diseases with PTSD. We argue that developments in epidemiological methods and the explosion of knowledge about the behavioral and biological effects of PTSD offer new opportunities to provide more definitive answers to questions of causality. First, given that studies of PTSD and physical health rely primarily on observational data, we discuss methods to improve causal inference using such data, with particular reference to the issues of temporality and confounding. Second, we consider recent work (also observational) linking PTSD with specific behaviors and biological processes, and evaluate whether these may plausibly serve as mechanisms by which PTSD leads to cardiometabolic disease. Third, we evaluate how looking more comprehensively into the PTSD phenotype would provide insight into whether specific aspects of PTSD phenomenology are particularly relevant to cardiometabolic disease. Finally, we discuss new areas of research that are feasible and could enhance understanding of the PTSD–cardiometabolic relationship, such as testing whether PTSD treatment alters behaviors and biomarkers on the pathways to CVD and T2D.
Improving causal inference from observational data
There are at least five possible causal structures (Fig. 1) that may explain the well-documented statistical association between PTSD and cardiometabolic disease, but only one (scenario c) includes a unidirectional causal impact of PTSD on cardiometabolic disease. Scenario a in Fig. 1 reflects confounding bias, where the relationship between PTSD and cardiometabolic disease is explained by a third variable. Because PTSD is conditional on trauma, the most commonly suggested confounder is trauma exposure. Many types of trauma exposure, from childhood maltreatment (Felitti et al. Reference Felitti, Anda, Nordenberg, Williamson, Spitz, Edwards, Koss and Marks1998; Dube et al. Reference Dube, Felitti, Dong, Giles and Anda2003; Dong et al. Reference Dong, Giles, Felitti, Dube, Williams, Chapman and Anda2004) to combat exposure (Schnurr & Spiro, Reference Schnurr and Spiro1999; Schnurr et al. Reference Schnurr, Spiro, Vielhauer, Findler and Hamblen2002), have been linked to chronic disease. Furthermore, predictors of trauma (e.g. neighborhood environment, socio-economic status), which vary by trauma type, may also bias estimates if associated with both PTSD and cardiometabolic risk and left unaccounted for by study designs or analyses. Various other factors have been proposed, including shared genetic vulnerability, lower intelligence quotient, and even maternal trauma and PTSD (Koenen et al. Reference Koenen, Moffitt, Poulton, Martin and Caspi2007; Schaefer et al. Reference Schaefer, Caspi, Belsky, Harrington, Houts, Israel, Levine, Sugden, Williams, Poulton and Moffit2015). Moreover, inflammatory biomarkers associated with increased risk of cardiometabolic disease, such as elevated C-reactive protein, have been associated with increased risk of PTSD onset prospectively (Eraly et al. Reference Eraly, Nievergelt, Maihofer, Barkauskas, Biswas, Agorastos, O'Connor and Baker2014). Thus, inflammation may represent common vulnerability for both PTSD and cardiometabolic disease. However, although C-reactive protein level is considered a useful biomarker of risk for CVD, a large cross-consortium Mendelian randomization analysis did not support a causal role of C-reactive protein in any neuropsychiatric or somatic outcome examined (Prins et al. Reference Prins, Abbasi, Wong, Vaez, Nolte, Franceschini, Stuart, Guterriez Achury, Mistry, Bradfield, Valdes, Bras, Shatunov, Lu, Han, Raychaudhuri, Bevan, Mayes, Tsoi, Evangelou, Nair, Grant, Polychronakos, Radstake, van Heel, Dunstan, Wood, Al-Chalabi, Dehghan, Hakonarson, Markus, Elder, Knight, Arking, Spector, Koeleman, van Duijn, Martin, Morris, Weersma, Wijmenga, Munroe, Perry, Pouget, Jamshidi, Snieder and Alizadeh2016).
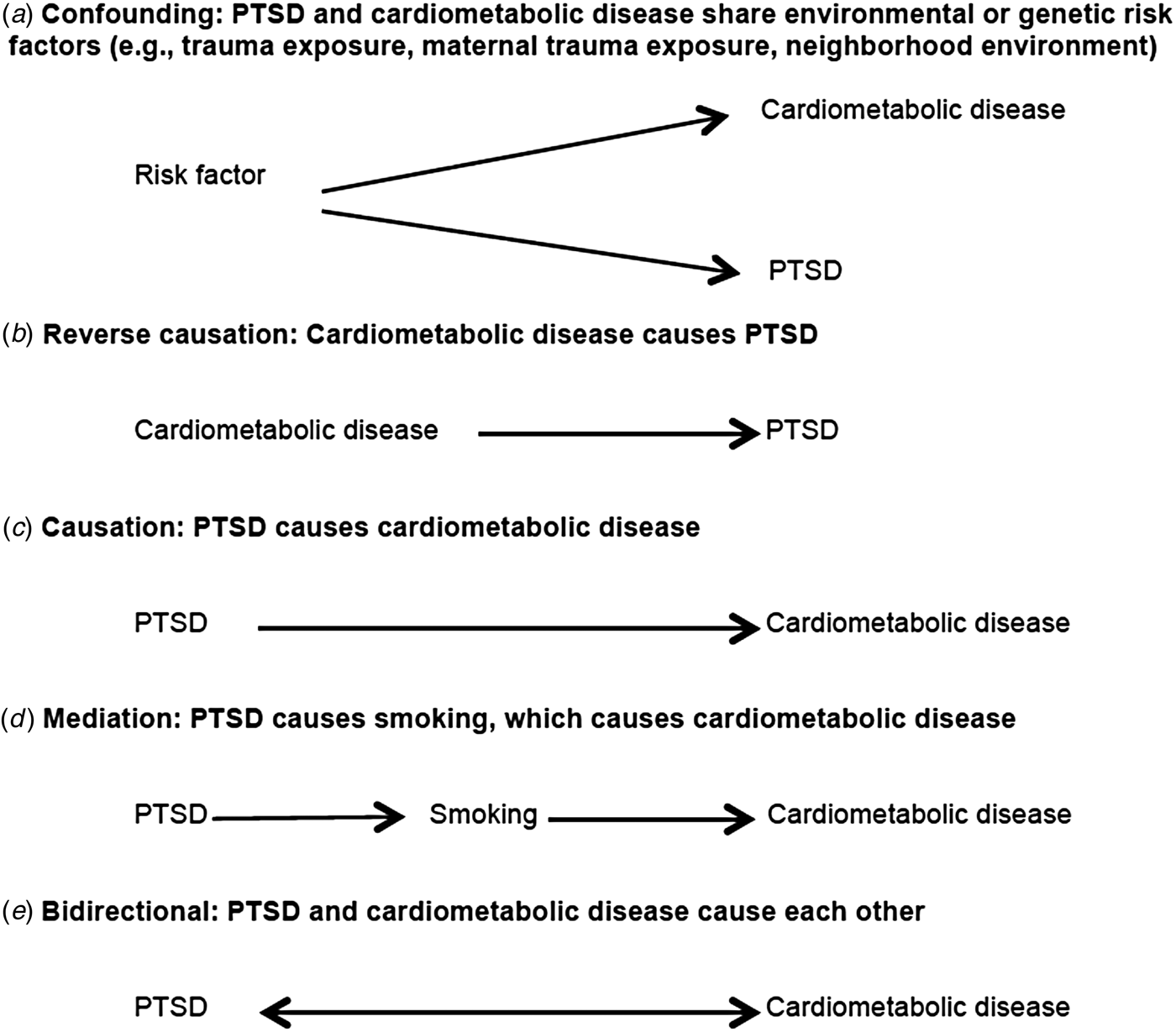
Fig. 1. Possible causal structures of the relationship between post-traumatic stress disorder (PTSD) and cardiometabolic risk.
Scenario b in Fig. 1 reflects reverse causation. For example, the accumulation of evidence summarized in meta-analyses indicates that acute life-threatening medical events, like myocardial infarction and stroke, are traumatic events that may trigger PTSD onset (Edmondson et al. Reference Edmondson, Richardson, Falzon, Davidson, Mills and Neria2012; Edmondson & Cohen, Reference Edmondson and Cohen2013). Epidemiological studies have also shown increased risk of PTSD associated with diagnosis of breast cancer and in critical illness survivors (Vin-Raviv et al. Reference Vin-Raviv, Hillyer, Hershman, Galea, Leoce, Bovbjerg, Kushi, Kroenke, Lamerato, Ambrosone, Valdimorsdottir, Jandorf, Mandelblat, Tsai and Neugut2013; Parker et al. Reference Parker, Sricharoenchai, Raparla, Schneck, Bienvenu and Needham2015). It is notable that commonly used assessments for traumatic events such as the Life Events Checklist-V include life-threatening illness or injury as a potential index trauma for PTSD assessment (Weathers et al. Reference Weathers, Blake, Schnurr, Kaloupek, Marx and Keane2013).
Scenario d in Fig. 1 reflects mediation, whereby the effect of PTSD on the outcome is explained by an intermediate variable. We discuss this in more detail in the section below titled ‘Elucidating mechanisms to better understand risk’. Finally, scenario e in Fig. 1 reflects a bidirectional relationship between PTSD and cardiometabolic outcomes. A bidirectional association is plausible given evidence for both scenario b (reverse causation) and scenario c (PTSD causes cardiometabolic disease), and is thus a potential focus for future longitudinal research.
Moreover, we see at least two potential forms of bidirectionality. PTSD could lead to increased susceptibility to disease and developing a major disease could be a trauma that leads to PTSD development. Alternatively, PTSD and cardiometabolic disease may mutually influence each other whereby people who have PTSD and then develop a major disease are at greater risk for having more severe PTSD symptoms, which lead to worse disease. To our knowledge, only one longitudinal epidemiological study has examined the first type of bidirectional relationship (El-Gabalawy et al. Reference El-Gabalawy, Mackenzie, Pietrzak and Sareen2014). This study examined the relationship between anxiety disorders, including PTSD, and physical health conditions (self-reported). The authors did not find evidence for a bidirectional relationship between PTSD and cardiometabolic disease specifically although evidence was suggestive for other physical health outcomes. With regard to the second type of bidirectionality, whereby PTSD influences cardiometabolic disease and vice versa, a recent paper using a convenience sample of veterans employed a cross-lagged analysis to examine a potential bidirectional relationship between PTSD and the metabolic syndrome (Wolf et al. Reference Wolf, Bovin, Green, Mitchell, Stoop, Barretto, Jackson, Lee, Fang, Trachtenberg, Rosen, Keane and Marx2016). The authors found evidence that PTSD severity predicted increases in metabolic syndrome severity but metabolic syndrome severity did not predict onset of PTSD. In related work, other longitudinal studies have found evidence of bidirectionality for depression with both diabetes and the metabolic syndrome (Pan et al. Reference Pan, Lucas, Sun, van Dam, Franco, Manson, Willett, Ascherio and Hu2010, Reference Pan, Keum, Okereke, Sun, Kivimaki, Rubin and Hu2012).
These alternative causal structures pose a challenge for drawing conclusions from observational data about whether PTSD truly causes cardiometabolic disease. Because randomizing individuals to developing PTSD is not plausible, we must seek to provide the most convincing possible evidence from observational studies (Vandenbroucke et al. Reference Vandenbroucke, Broadbent and Pearce2016). Prospective longitudinal studies with in-depth measurement of potential confounders are needed. Studies that are cross-sectional or have limited measures of potential confounders cannot convincingly rule out confounding or reverse causation. However, when extensive data (including repeated assessments of relevant covariates) are available and well measured, statistical adjustment for confounders occurring before trauma or PTSD through direct inclusion in conventional models should mitigate bias due to confounding. Similarly, reverse causation is unlikely to explain associations between PTSD and cardiometabolic risk in prospective studies where individuals with these diseases at baseline are excluded. Prospective studies with repeated measures of both PTSD and cardiometabolic outcomes also offer the opportunity to examine the possibility of a bidirectional relationship. Cross-lagged models from longitudinal data in population-based samples with at least three follow-up waves have demonstrated bidirectional relationships between PTSD and other factors such as trauma exposure (Lowe et al. Reference Lowe, Walsh, Uddin, Galea and Koenen2014), negative emotionality (Sadeh et al. Reference Sadeh, Miller, Wolf and Harknes2015) and quality of life over time (Giacco et al. Reference Giacco, Matanov and Priebe2013). We are not aware of published studies employing such models for PTSD and medically verified health outcomes in longitudinal epidemiological samples, but such studies would undoubtedly provide important insight.
A critical issue for prospective longitudinal studies is accounting for time-varying confounding and mediation. Confounding occurs when a common prior cause explains the relationship between an exposure and an outcome. For example, low socio-economic status has been associated with increased risk of both CVD/T2D and PTSD and could be considered a potential confounder. Mediation occurs when a third variable, on the pathway between the exposure and the outcome, explains the observed relationship. For example, in scenario d of Fig. 1, PTSD increases smoking and smoking increases risk of CVD. Thus, smoking could be a mediator of the PTSD–CVD relationship.
Marginal structural models have revolutionized the epidemiological approach to time-varying confounding of time-varying exposures (Robins et al. Reference Robins, Hernán and Brumback2000). These models have particular promise for understanding the PTSD–cardiometabolic relationship given that PTSD alters health behaviors but such behaviors may also increase risk of PTSD onset, persistence or remission. Such time-varying confounding could present a substantial threat to causal inference. For example, smoking may increase PTSD risk (van der Velden et al. Reference van der Velden, Kleber and Koenen2008) and is a causal risk factor for CVD. Additionally, other research has clearly demonstrated that PTSD is associated with increased risk of initiating smoking (Koenen et al. Reference Koenen, Hitsman, Lyons, Stroud, Niaura, McCaffery, Goldberg, Eisen, True and Tsuang2006) and difficulty quitting (Koenen et al. Reference Koenen, Stellman, Sommer and Stellman2008). The conventional method of adjusting for the confounding effects of smoking through direct inclusion in a regression model blocks the mediating pathway between PTSD and CVD, and underestimates the effect of PTSD. Furthermore, direct inclusion of health behaviors in regression models could allow unmeasured confounders of health behaviors and CVD to bias estimates (i.e. collider bias) – people with PTSD who do not smoke may have some other advantages that help them avoid health-damaging behaviors.
Having a substantial period of time elapse between PTSD assessments and incidence of cardiometabolic conditions may also help to rule out reverse causation, but such delays introduce the possibility of bias due to selective mortality and can also make it difficult to determine if there are potentially acute effects of PTSD on cardiometabolic events. With higher mortality rates among people with PTSD (Boscarino, Reference Boscarino2006, Reference Boscarino2008), individuals who survive long enough to develop conditions such as CVD may be a particularly ‘hardy’ sample. This type of selective survival could lead to underestimates of the effect of PTSD on cardiometabolic outcomes, but can at least partially be addressed with methods to account for attrition, such as inverse probability weighting (IPW). IPW weights respondents who remain in the sample based on observed covariates to create an analysed sample of survivors with the same covariate distribution as the original sample. Recent methods emphasize applications of IPW to estimate the so-called ‘survivor average causal effect’ (SACE), i.e. the effect of PTSD on cardiometabolic disease risk among people whose survival was not affected by PTSD (Tchetgen Tchetgen et al. Reference Tchetgen Tchetgen, Glymour, Shpitser and Weuve2012; Tchetgen Tchetgen, Reference Tchetgen Tchetgen2014). However, even with the most comprehensive feasible control for confounding, with observational data concerns can remain about unmeasured or residual confounding, or the possibility of prodromal disease driving associations.
Instrumental variable analysis is another powerful statistical method that may estimate unbiased causal effects of the exposure on the outcome even when there are unmeasured variables that confound the link between exposure and outcome. Instruments are factors that cause variation in the exposure of interest but do not directly affect the outcome of interest. In this context, an instrumental variable would be associated with PTSD but not cardiometabolic risk except through its association with PTSD (Greenland, Reference Greenland2000). Until recently, most instrumental variable analyses used factors such as policy changes (e.g. introduction of trauma counseling) or arbitrary institutional features (e.g. randomly assigned order of military draft) as sources of exogenous variation in exposure. Recently, applications using genetic variants as instrumental variables have also become popular. Mendelian randomization is a particular form of instrumental variable analysis that capitalizes on the fact that genotypes are assigned randomly when passed from parents to offspring; thus, population genotype distribution should be unrelated to confounders that might be relevant in observational studies (Smith & Ebrahim, Reference Smith and Ebrahim2003). Genetic variants that influence a phenotype (e.g. PTSD) can be used as a natural experiment or instrumental variable for that phenotype.
Mendelian randomization could be used to assess evidence for shared genetic vulnerability to PTSD and cardiometabolic disease, reverse causation or direct causation. For example, a bidirectional Mendelian randomization approach would assess whether genetic variants that increase cardiometabolic disease risk also predict higher risk of PTSD: if not, this is evidence against reverse causation accounting for the link between PTSD and cardiometabolic disease documented in previous studies. Although an established genetic instrument for PTSD has yet to be identified, the PTSD working group of the Psychiatric Genomics Consortium is poised to identify genetic variants associated with PTSD and is developing polygenic models of PTSD that could be used as instruments (Logue et al. Reference Logue, Amstadter, Baker, Duncan, Koenen, Liberzon, Miller, Morey, Nievergelt and Ressler2015). To our knowledge, only one study of PTSD has employed Mendelian randomization (Mustapic et al. Reference Mustapic, Maihofer, Mahata, Chen, Baker, O'Connor and Nievergelt2014).
Elucidating mechanisms to better understand risk
Two pathways are commonly identified to explain how PTSD may increase risk of cardiometabolic (or other) diseases: behavioral and biological. PTSD may provoke unhealthy behaviors and/or produce neurobiological alterations that initiate relevant pathophysiological processes (Wentworth et al. Reference Wentworth, Stein, Redwine, Xue, Taub, Clopton, Nayak and Maisel2013; Levine et al. Reference Levine, Levine and Levine2014). Research clearly demonstrating that PTSD alters known mechanisms of cardiometabolic risk would provide compelling evidence that PTSD is indeed involved in the etiology of cardiometabolic disease. However, rigorous evidence demonstrating these causal associations is limited (Kibler, Reference Kibler2009; Wentworth et al. Reference Wentworth, Stein, Redwine, Xue, Taub, Clopton, Nayak and Maisel2013).
Some of the strongest evidence to date (although mostly from observational studies) is in the domain of health behaviors or behavior-related conditions. To our knowledge, there are no randomized trials evaluating whether successful PTSD treatment leads to improved health-related behaviors. Cross-sectional work suggests that PTSD is associated with numerous behavioral risk factors and conditions for cardiometabolic disease, including cigarette smoking (Fu et al. Reference Fu, McFall, Saxon, Beckham, Carmody, Baker and Joseph2007), physical inactivity (Chwastiak et al. Reference Chwastiak, Rosenheck and Kazis2011), poor diet quality (Hall et al. Reference Hall, Hoerster and Yancy2015), obesity (Scott et al. Reference Scott, McGee, Wells and Oakley Browne2008) and insomnia (Leskin et al. Reference Leskin, Woodward, Young and Sheikh2002). Furthermore, prospective studies have found associations of PTSD with increased likelihood of initiating smoking (Koenen et al. Reference Koenen, Hitsman, Lyons, Stroud, Niaura, McCaffery, Goldberg, Eisen, True and Tsuang2006), reduced likelihood of smoking cessation (Koenen et al. Reference Koenen, Stellman, Sommer and Stellman2008), increased risk of weight gain and obesity (Perkonigg et al. Reference Perkonigg, Owashi, Stein, Kirschbaum and Wittchen2009; Kubzansky et al. Reference Kubzansky, Bordelois, Jun, Roberts, Cerda, Bluestone and Koenen2014), and medication non-adherence (Kronish et al. Reference Kronish, Lin, Cohen, Voils and Edmondson2014).
Some work has specifically analysed the extent to which health behaviors may mediate the relationship between PTSD and other cardiometabolic risk factors. For example, in one study, greater PTSD symptoms were associated with reduced heart rate variability (a measure of autonomic function that is prognostic of CVD and mortality), and three health behaviors – cigarette smoking, alcohol overuse and sleep disturbance – accounted for 94% of the shared variance between PTSD symptoms and heart rate variability (Dennis et al. Reference Dennis, Watkins, Calhoun, Oddone, Sherwood, Dennis, Rissling and Beckham2014). In our own studies examining the associations of PTSD with incident T2D and CVD among women, we found that behavior-related risk factors accounted for a sizeable portion of these relationships. For example, elevated body mass index accounted for 14% of the PTSD symptoms–T2D relationship (Roberts et al. Reference Roberts, Agnew-Blais, Spiegelman, Kubzansky, Mason, Galea, Hu, Rich-Edwards and Koenen2015), and a range of health behaviors and medical risk factors (e.g. body mass index, diet quality, smoking) explained nearly half of the association of elevated PTSD symptoms with CVD (Sumner et al. Reference Sumner, Kubzansky, Elkind, Roberts, Agnew-Blais, Chen, Cerdá, Rexrode, Rich-Edwards and Spiegelman2015). Thus, evidence increasingly suggests that behavioral risk factors, in part, mediate the PTSD–cardiometabolic disease relationship. However, behavior-related factors rarely fully attenuate associations, suggesting that other unmeasured behaviors or biological processes may be at play.
Substantial research has identified neurobiological alterations associated with high levels of PTSD symptoms (Pitman et al. Reference Pitman, Rasmusson, Koenen, Shin, Orr, Gilbertson, Milad and Liberzon2012). Thus, it is plausible that PTSD, by recurrent triggering of a biological stress response and overactivation of biological processes associated with hypervigilance, could directly alter processes involved in the pathophysiology of cardiometabolic disease. Disrupted regulation of stress hormone release by the hypothalamic–pituitary–adrenal (HPA) axis and the sympathetic–adrenal–medullary system is commonly observed in PTSD (Tsigos & Chrousos, Reference Tsigos and Chrousos2002; de Kloet et al. Reference de Kloet, Vermetten, Geuze, Kavelaars, Heijnen and Westenberg2006; Yehuda, Reference Yehuda2006; Blechert et al. Reference Blechert, Michael, Grossman, Lajtman and Wilhelm2007), and other work has demonstrated that these disruptions can lead to a cascade of deleterious effects on immune, metabolic and cardiovascular systems. Dysregulation of the HPA axis and the sympathetic nervous system can contribute to hypercoagulability, elevated cardiac reactivity, unhealthy lipid profiles, immune dysfunction, chronic inflammation and endothelial damage, all processes implicated in increasing cardiometabolic disease risk (Wentworth et al. Reference Wentworth, Stein, Redwine, Xue, Taub, Clopton, Nayak and Maisel2013; Brouwers et al. Reference Brouwers, Wolf, von Känel, Martin, Preedy and Patel2016). Most evidence to date comes from cross-sectional studies, and indicates that PTSD is associated with more unhealthy levels of biomarkers related to the processes described above. Cardiometabolic biomarkers that have been linked to PTSD include markers of chronic inflammation (e.g. C-reactive protein, interleukin-6, tumor necrosis factor-α) (Smith et al. Reference Smith, Conneely, Kilaru, Mercer, Weiss, Bradley, Tang, Gillespie, Cubells and Ressler2011; Baker et al. Reference Baker, Nievergelt and O'Connor2012; Passos et al. Reference Passos, Vasconcelos-Moreno, Costa, Kunz, Brietzke, Quevedo, Salum, Magalhaes, Kapczinski and Kauer-Sant'Anna2015; Brouwers et al. Reference Brouwers, Wolf, von Känel, Martin, Preedy and Patel2016), elevated coagulation (Von Känel et al. Reference Von Känel, Hepp, Buddeberg, Keel, Mica, Aschbacher and Schnyder2006; Reference von Känel, Abbas, Schmid, Saner, Haeberli, Stutz and Begré2010) and endothelial damage (von Känel et al. Reference von Känel, Hepp, Traber, Kraemer, Mica, Keel, Mausbach and Schnyder2008; Plantinga et al. Reference Plantinga, Bremner, Miller, Jones, Veledar, Goldberg and Vaccarino2013), as well as elevated lipids (Kagan et al. Reference Kagan, Leskin, Haas, Wilkins and Foy1999; Karlovic et al. Reference Karlovic, Buljan, Martinac and Marcinko2004; Maia et al. Reference Maia, Marmar, Mendlowicz, Metzler, Nobrega, Peres, Coutinho, Volchan and Figueira2008). These findings are congruent with other work that suggests that high distress in various forms may induce autonomic imbalance and reduce parasympathetic tone, which in turn is associated with increased risk of numerous pathological conditions, including CVD and premature mortality (Thayer & Brosschot, Reference Thayer and Brosschot2005).
Although many of the extant biomarker studies provide evidence suggestive of a link between PTSD and biological indicators of cardiometabolic risk, shortcomings of these studies limit the conclusions that can be drawn. First, many studies were conducted with small samples which are not informative if results are null (Gill et al. Reference Gill, Vythilingam and Page2008; Smith et al. Reference Smith, Conneely, Kilaru, Mercer, Weiss, Bradley, Tang, Gillespie, Cubells and Ressler2011; Pace et al. Reference Pace, Wingenfeld, Schmidt, Meinlschmidt, Hellhammer and Heim2012), although there are some exceptions (Spitzer et al. Reference Spitzer, Barnow, Völzke, Wallaschofski, John, Freyberger, Löwe and Grabe2010; Ahmadi et al. Reference Ahmadi, Hajsadeghi, Mirshkarlo, Budoff, Yehuda and Ebrahimi2011). Second, studies have not consistently reported whether participants with PTSD are free of relevant chronic diseases. Thus, the PTSD–biomarker relationship could be due to higher prevalence of chronic disease already present in the PTSD cases and not due to PTSD itself. Third, some studies have failed to assess effects of trauma exposure (Su et al. Reference Su, Jimenez, Roberts and Loucks2015) separately from PTSD (Pace et al. Reference Pace, Wingenfeld, Schmidt, Meinlschmidt, Hellhammer and Heim2012). Because there is ongoing debate as to whether trauma alone might cause biological dysregulation, it is not yet clear whether PTSD per se is driving the associations with biomarkers (Seckl & Meaney, Reference Seckl and Meaney2006; Seckl, Reference Seckl2007; Sledjeski et al. Reference Sledjeski, Speisman and Dierker2008; Yehuda et al. Reference Yehuda, Flory, Pratchett, Buxbaum, Ising and Holsboer2010; Dudley et al. Reference Dudley, Li, Kobor, Kippin and Bredy2011). Fourth, given most studies are cross-sectional, they cannot provide clear evidence of the directionality of these associations or distinguish whether: (1) PTSD induces unhealthy biomarker activity; (2) unhealthy levels of biomarkers lead to increased PTSD symptoms; or (3) apparent relationships between unhealthy levels of biomarkers and PTSD are due to shared causes (e.g. genetic factors). For example, longitudinal studies suggest that poorer physical health (LeardMann et al. Reference LeardMann, Smith, Smith, Wells and Ryan2009) and some forms of biological dysregulation linked to PTSD, including higher numbers of glucocorticoid receptors (van Zuiden et al. Reference van Zuiden, Geuze, Willemen, Vermetten, Maas, Heijnen and Kavelaars2011, Reference van Zuiden, Geuze, Willemen, Vermetten, Maas, Amarouchi, Kavelaars and Heijnen2012) and hypersensitive startle response (Pole et al. Reference Pole, Neylan, Otte, Henn-Hasse, Metzler and Marmar2009), precede and predict, rather than result from, PTSD (Rusiecki et al. Reference Rusiecki, Chen, Srikantan, Zhang, Yan, Polin and Baccarelli2012; van Zuiden et al. Reference van Zuiden, Geuze, Willemen, Vermetten, Maas, Amarouchi, Kavelaars and Heijnen2012). However, bidirectionality between PTSD and dysregulated biological processes is also possible, making it even more challenging to disentangle cause from effect. For example, bidirectionality has been observed in the association between depression and C-reactive protein (Matthews et al. Reference Matthews, Schott, Bromberger, Cyranowski, Everson-Rose and Sowers2010); such relationships may also be at play with PTSD.
Delving deeper into the PTSD phenotype within and across time
Many of the extant studies on PTSD and cardiometabolic disease have compared individuals with and without PTSD diagnoses. However, PTSD is a complex and heterogeneous phenotype, and this categorical approach does not fully capture the richness of the PTSD construct (Forbes et al. Reference Forbes, Lockwood, Elhai, Creamer, O'Donnell, Bryant, McFarlane and Silove2011; Zoellner et al. Reference Zoellner, Pruitt, Farach and Jun2014). Going forward, three approaches may help us better understand how aspects of PTSD phenomenology contribute to chronic disease risk.
First, PTSD is constructed from multiple symptom dimensions that may have distinct effects on cardiometabolic risk. Fear symptoms reflect an alarm response to perceived danger, and include re-experiencing (e.g. nightmares), active avoidance (e.g. avoiding trauma-related thoughts) and anxious arousal (e.g. hypervigilance) symptoms of PTSD (Forbes et al. Reference Forbes, Lockwood, Elhai, Creamer, O'Donnell, Bryant, McFarlane and Silove2011; Zoellner et al. Reference Zoellner, Pruitt, Farach and Jun2014). In contrast, dysphoria symptoms reflect low positive affect, and encompass symptoms of emotional numbing (e.g. restricted affect) and dysphoric arousal (e.g. concentration difficulties). These dimensions are thought to be useful for distinguishing specific v. non-specific aspects of PTSD. Whereas the dysphoria component of PTSD is shared with other psychopathology (e.g. depression), fear responses are thought to be a core feature of PTSD (Forbes et al. Reference Forbes, Parslow, Creamer, O'Donnell, Bryant, McFarlane, Silove and Shalev2010, Reference Forbes, Lockwood, Elhai, Creamer, O'Donnell, Bryant, McFarlane and Silove2011; Zoellner et al. Reference Zoellner, Pruitt, Farach and Jun2014). Indeed, a recent review called for studying fear responses to trauma to improve the specificity of PTSD assessment (Zoellner et al. Reference Zoellner, Pruitt, Farach and Jun2014).
Numerous confirmatory factor analyses indicate that fear and dysphoria symptoms form valid and distinct (albeit correlated) dimensions of PTSD (Grant et al. Reference Grant, Beck, Marques, Palyo and Clapp2008; Forbes et al. Reference Forbes, Parslow, Creamer, O'Donnell, Bryant, McFarlane, Silove and Shalev2010, Reference Forbes, Lockwood, Elhai, Creamer, O'Donnell, Bryant, McFarlane and Silove2011), and models of PTSD that differentiate between factors within the fear and dysphoria dimensions have been supported as well (Elhai et al. Reference Elhai, Biehn, Armour, Klopper, Frueh and Palmieri2011; Harpaz-Rotem et al. Reference Harpaz-Rotem, Tsai, Pietrzak and Hoff2014; Armour et al. Reference Armour, Tsai, Durham, Charak, Biehn, Elhai and Pietrzak2015). Focusing on dimensions of PTSD – rather than a single PTSD construct – may help shed light on how PTSD contributes to risk for cardiometabolic disease, as PTSD symptom dimensions may differ in the extent to which they increase chronic disease risk via biological v. behavioral mechanisms. For example, by promoting autonomic nervous system dysregulation (particularly sympathetic nervous system hyperactivity), post-traumatic fear responses may contribute to CVD risk by promoting increased blood pressure, coagulation, platelet aggregation and inflammatory cytokines (Lefkowitz et al. Reference Lefkowitz, Rockman and Koch2000; Bedi & Arora, Reference Bedi and Arora2007; Abboud et al. Reference Abboud, Harwani and Chapleau2012; Edmondson & Cohen, Reference Edmondson and Cohen2013; Wentworth et al. Reference Wentworth, Stein, Redwine, Xue, Taub, Clopton, Nayak and Maisel2013). In contrast, elevated symptoms of dysphoria after trauma may be more likely than fear symptoms to promote withdrawal and failure to engage in health-promoting behaviors (e.g. physical activity, medication adherence). However, few studies have tried to tease these domains apart. Further research examining the relative contributions of the mechanisms by which PTSD dimensions may lead to cardiometabolic disease is needed.
Second, greater appreciation of the course of PTSD over time may also improve understanding of the physical health risks associated with the disorder. Several studies suggest that there is a dose–response association between PTSD severity and risk for cardiometabolic conditions, with the most severe symptoms associated with the greatest elevation in disease risk (Kubzansky et al. Reference Kubzansky, Koenen, Spiro, Vokonas and Sparrow2007, Reference Kubzansky, Koenen, Jones and Eaton2009; Roberts et al. Reference Roberts, Agnew-Blais, Spiegelman, Kubzansky, Mason, Galea, Hu, Rich-Edwards and Koenen2015). Work that takes into consideration other aspects of PTSD presentation, including the trajectory of symptom development, is also of interest. For example, several ‘longitudinal phenotypes’ of PTSD that reflect the trajectory of PTSD symptoms over time after trauma exposure have been documented (e.g. rapid remitting, slow remitting, delayed onset, chronic/non-remitting) (Galatzer-Levy et al. Reference Galatzer-Levy, Ankri, Freedman, Israeli-Shalev, Roitman, Gilad and Shalev2013; Santiago et al. Reference Santiago, Ursano, Gray, Pynoos, Spiegel, Lewis-Fernandez, Friedman and Fullerton2013). However, studies that examine how these various PTSD trajectories relate to physical health outcomes are lacking. Moreover, how trajectories of PTSD symptoms may relate to time-varying confounding or mediation has not been examined in the context of physical health outcomes. A more nuanced understanding of how patterns of PTSD development and expression over time relate to indicators of physical health may provide greater precision in understanding what aspects of PTSD are most pernicious for health. Information generated from such research also can help health care professionals identify who should be targeted for prevention.
Third, future research should examine how persistence v. remission of PTSD influences risk. PTSD is frequently a chronic condition, with meta-analytic evidence suggesting that, on average, over half of individuals with PTSD (56%) do not remit from the disorder naturally after a mean of more than 3 years (Morina et al. Reference Morina, Wicherts, Lobbrecht and Priebe2014). Epidemiological research indicates that more than a third of individuals continue to have symptoms of PTSD 30 years after onset of the disorder (Chapman et al. Reference Chapman, Mills, Slade, McFarlane, Bryant, Creamer, Silove and Teesson2012). Although this topic has not been explored extensively, there is some evidence that greater chronicity is associated with worse cardiovascular outcomes. For example, one meta-analysis examined the association between PTSD chronicity and basal heart rate by comparing individuals who had PTSD for either more than 12 years or less than 8 years with trauma-exposed individuals without PTSD (Buckley & Kaloupek, Reference Buckley and Kaloupek2001). Effect size comparisons for elevations in heart rate were larger for individuals with PTSD for more than 12 years than for individuals with PTSD for less than 8 years.
Research examining how remitted status relates to risk for cardiometabolic disease is also needed. One recent study reported that veterans with current – but not remitted – PTSD had lower heart rate variability compared with veterans without PTSD (Shah et al. Reference Shah, Lampert, Goldberg, Veledar, Bremner and Vaccarino2013). In our study of PTSD and weight gain, while women with trauma and ongoing symptoms had significantly excess odds of overweight or obesity, those with remitted symptoms did not. If future research can confirm that individuals with remitted PTSD have lower cardiometabolic risk compared with those with current PTSD, this would suggest that treating PTSD might reduce cardiometabolic disease risk. Such findings would provide a promising foundation for supporting more widespread dissemination of empirically supported treatments for PTSD. Greater access to efficacious treatment for PTSD to improve emotional health is an important goal in its own right; demonstrating that treatment would also improve cardiometabolic health would provide extra impetus for this initiative.
New research directions
Indeed, whether effective treatment of PTSD can prevent or mitigate the likelihood of developing cardiometabolic disease is a key outstanding question in the literature. The strongest design to answer this question would be a randomized clinical trial whereby individuals are randomized to PTSD treatment or placebo, and then followed to assess whether treatment reduces likelihood of developing disease. However, such a trial would be highly expensive, unethical and time consuming, and is unlikely to be feasible at present. Thus, we propose four other methodological approaches that are more feasible and could begin to address the limitations of current knowledge highlighted here regarding causal associations and mechanisms (see Table 2). The most rigorous designs that could currently be harnessed leverage existing randomized trials of PTSD treatment. One possibility is to add assessments of cardiometabolic-related biomarkers and behaviors to new clinical trials of PTSD treatment that have not yet started (Cohen et al. Reference Cohen, Deblinger, Mannarino and Steer2004; Cloitre et al. Reference Cloitre, Stovall-McClough, Nooner, Zorbas, Cherry, Jackson, Gan and Petkova2010). Ideally, biomarker assessment would be conducted prior to and after treatment, perhaps even at repeated intervals after treatment. It would then be possible to monitor if treatment was associated with less deterioration (or ability to maintain healthier levels) of cardiometabolic risk biomarkers.
Table 2. Advantages and disadvantages of research designs for added insight into the PTSD–cardiometabolic disease relationship
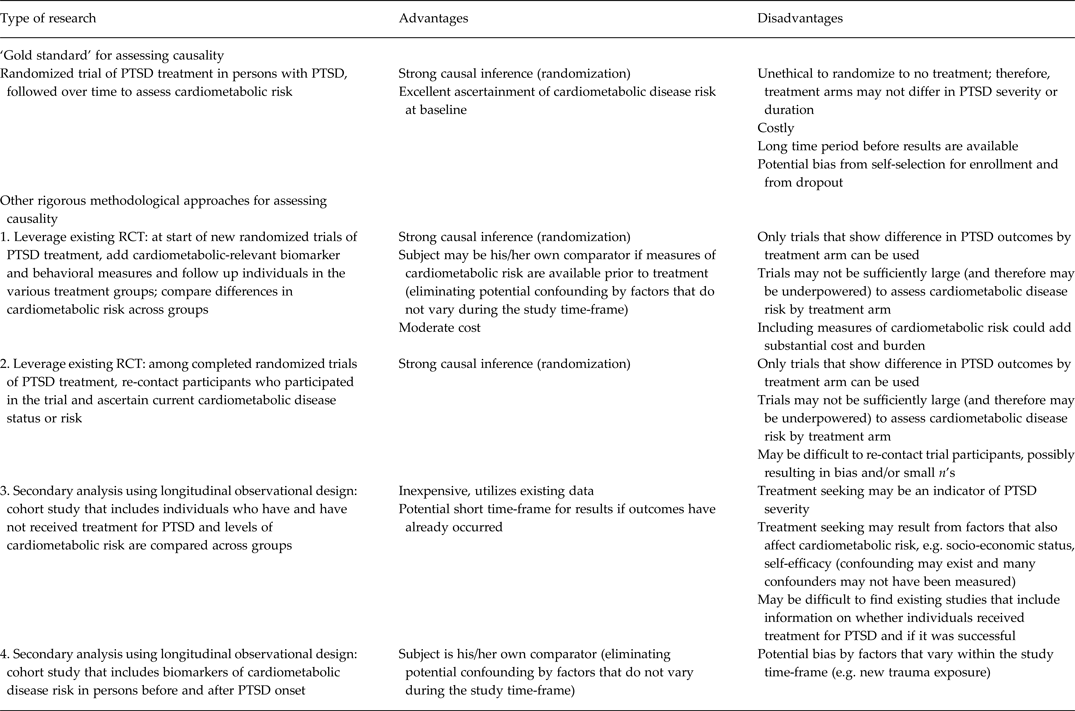
PTSD, Post-traumatic stress disorder; RCT, randomized controlled trial.
A second possibility is to re-contact patients who participated in completed PTSD treatment trials and obtain from these individuals measures of health behaviors, risk markers and physical health status. This would facilitate testing if those who were successfully treated engage in fewer unhealthy behaviors, have healthier levels of risk markers and better physical health status relative to those who did not receive treatment. In both types of studies, data from a randomized design could provide strong evidence regarding the potential efficacy of PTSD treatment on cardiometabolic risk. However, one potential challenge to this approach is differential follow-up by treatment effectiveness (i.e. individuals whose PTSD was not successfully treated are more likely to be lost to follow-up). Moreover, investigators would have had to have consent to re-contact participants included in their informed consents in order to conduct such a study.
Another set of methodological approaches leverages existing observational longitudinal cohort studies. Studies with information about whether participants were effectively treated for PTSD, as well as information on cardiometabolic disease or risk factors, could provide preliminary evidence regarding whether PTSD treatment reduces cardiometabolic risk. A drawback of this design is that such a study would be vulnerable to bias from confounding by indication. Individuals with more severe PTSD are more likely to seek treatment; thus, treatment may indicate PTSD severity. However, propensity score analyses, IPW and other statistical techniques based on covariate adjustment can mitigate such potential confounding (Klungel et al. Reference Klungel, Martens, Psaty, Grobbee, Sullivan, Stricker, Leufkens and de Boer2004).
An additional approach in this arena would leverage data from studies with measures of PTSD date of onset as well as repeated measures of biomarkers of cardiometabolic risk and relevant health behaviors. With biological and behavioral measures available before and after PTSD onset, change in risk in relation to PTSD onset can be assessed. Pooling biomarker data across multiple cohorts, as has been done in genomics, would further advance the field by increasing sample size, reducing issues of both type 1 and type 2 error, and ultimately providing more robust evidence of whether observed associations between PTSD and such markers are causal.
All four methodological approaches – measurement add-ons to new trials, re-contacting participants from past trials, observational studies comparing treated with untreated, and observational studies with measures spanning onset of PTSD – are relatively feasible. They draw on or extend data being collected from existing longitudinal or experimental studies. By obtaining cardiometabolic risk measures before and after, or at least clearly after, PTSD onset or treatment, the likelihood of reverse causation is minimized. Two of the suggested designs use individuals as their own comparator, thus eliminating possible influence of confounding factors that do not vary within individuals over the study period (Weisskopf et al. Reference Weisskopf, Kioumourtzoglou and Roberts2015). Additionally, two of the designs leverage the randomization to treatment found in most clinical trials to further improve causal inference. Together, these types of studies may establish more clearly that: (1) PTSD treatment is related to reduced risk of subsequent cardiometabolic disease; (2) increased likelihood of adverse health behaviors or biological dysregulation occurs after onset of PTSD; or (3) cardiometabolic risk behaviors or biomarkers are improved (or are less likely to deteriorate) after PTSD treatment.
Moving forward, considering how trauma exposure and the development of PTSD translate into cardiometabolic risk over the life course may also provide important insight regarding the interplay between mental and physical health. If mental health problems are an early warning sign of physical deterioration, this may alter approaches to prevention and intervention, particularly when trauma is known to have occurred. Moreover, there may be periods during which exposure to adversity or psychological distress is highly toxic in relation to physical health, or periods during which damage incurred is more difficult to reverse. One recent study found that high levels of distress in childhood, even when distress was lower during adulthood, had substantial impact on cardiometabolic health in middle adulthood (Winning et al. Reference Winning, Glymour, McCormick, Gilsanz and Kubzansky2015). Other research has similarly suggested that exposure to adversity during pregnancy and early childhood leads to damage relatively early on, and may be especially deleterious for long-term health (Ehlert, Reference Ehlert2013). Furthermore, although most studies have focused on PTSD and disease incidence in middle-aged and older adults, there is some evidence that childhood abuse is associated with higher diastolic blood pressure in adolescence (Gooding et al. Reference Gooding, Milliren, Austin, Sheridan and McLaughlin2016) and hypertension in young adulthood (Suglia et al. Reference Suglia, Clark, Boynton-Jarrett, Kressin and Koenen2014). Thus, it appears that potentially toxic health effects of PTSD are set in motion relatively early. Additional research is needed to examine how both trauma and PTSD in youth relate to myriad indicators of cardiometabolic risk in order to develop a more comprehensive understanding of when physical health risks associated with PTSD begin to manifest and how enduring these risks may be.
Future research should examine the clinical utility of screening for PTSD in assessing PTSD risk. This research would need to consider whether including PTSD, in addition to standard CVD risk factors, improves risk prediction and/or treatment matching. Increasingly cardiovascular medicine has recognized early-life origins of CVD (Lloyd-Jones et al. Reference Lloyd-Jones, Hong, Labarthe, Mozaffarian, Appel, Van Horn, Greenlund, Daniels, Nichol and Tomaselli2010). This recognition extends beyond effects of behavior to psychological factors as well. In fact, in a recent scientific statement from the American Heart Association (Goldstein et al. Reference Goldstein, Carnethon, Matthews, McIntyre, Miller, Raghuveer, Stoney, Wasiak and McCrindle2015), major depressive disorder and bipolar disorder in youth were classified as Expert Panel tier II moderate-risk conditions associated with accelerated atherosclerosis and early CVD. This appreciation that psychopathology early in life can have negative consequences for physical health in youth and adulthood may one day be expanded to include PTSD. Ultimately, this research can inform prevention and treatment resources in an age of targeted medicine.
Acknowledgements
This work was supported by the National Institutes of Health (to K.C.K., R01MH078928; to K.C.K., L.D.K. and A.W., R01MH101269; NHS II infrastructure, UM1CA176726; to J.A.S., K01HL130650; and to P.G., T32MH017119) and by the Yerby Postdoctoral Fellowship Program (to P.G.).
Declaration of Interest
None.



