



Introduction
The synthesis of the viral DNA in human immunodeficiency virus type 1 (HIV-1) results from the reverse transcription (RTion) process, where two copies of single-stranded RNA (ssRNA) are transcribed into double-stranded DNA (dsDNA). The viral dsDNA is then trafficked to the nucleus of the target cell where it is integrated into the host cell chromatin, followed by transcription and translation processes by the cellular machinery. One of the transcription products is an mRNA that codes for the Gag polyprotein, which is the main structural protein of HIV-1, and is on its own sufficient for assembly of new viral particles in cells (Campbell and Rein, Reference Campbell and Rein1999). The Gag polyprotein consists of four major domains; the N terminus matrix (MA), the capsid (CA), the nucleocapsid (NC) and the C-terminus p6, as well as two small spacer peptides, Sp1 and Sp2 (Fig. 1a). After the viral particle leaves the infected cell, the Gag polyprotein is sequentially cleaved by the virus-encoded protease to ultimately lead to the mature MA, CA, NC and p6 proteins. In the mature virion, MA associates with the inner viral membrane, CA assembles into the conical capsid and NC coats and condenses the viral RNA (Ganser-Pornillos et al., Reference Ganser-Pornillos, Yeager and Sundquist2008; Briggs et al., Reference Briggs, Riches, Glass, Bartonova, Zanetti and Krausslich2009; Briggs and Kräusslich, Reference Briggs and Kräusslich2011; Bell and Lever, Reference Bell and Lever2013).
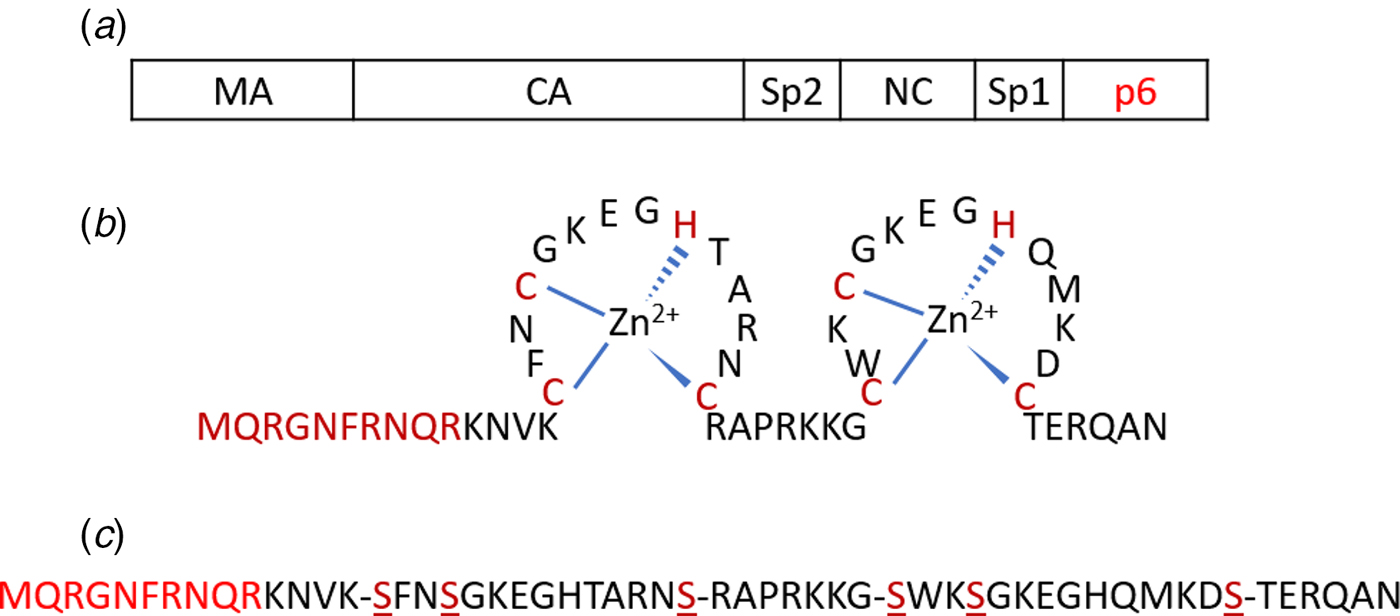
Fig. 1. (a) Schematic representation of the Gag polyprotein with major domains indicated. The p6 region highlighted in red is deleted in Δ-p6 Gag. (b) Amino acid sequence of NC(1–55) including its two-CCHC-zinc fingers. (c) Sequence of SSHS-SSHS NC(1–55) where the cysteine residues are replaced with serines.
The NC protein is a small structural protein that contains a basic N-terminal domain as well as two zinc finger motifs, separated by a short basic linker (Fig. 1b) (Darlix et al., Reference Darlix, Lapadat-Tapolsky, de Rocquigny and Roques1995). This 55-amino-acid protein acts as a nucleic acid (NA) chaperone that favours the most thermodynamically stable conformation of NAs (Rein et al., Reference Rein, Henderson and Levin1998; Levin et al., Reference Levin, Guo, Rouzina and Musier-Forsyth2005; Godet and Mély, Reference Godet and Mély2010; Darlix et al., Reference Darlix, Godet, Ivanyi-Nagy, Fossé, Mauffret and Mély2011). The NC chaperone activity relies on its ability to associate and dissociate rapidly from its NA targets, to destabilize their secondary structure and to promote the annealing of complementary sequences. The chaperone activity of NC is witnessed during the early phase of the virus life cycle, where it helps the two obligatory strand transfers in the RTion process, and also the integration of the viral DNA into the host genome. In addition, during the late phase of the virus life cycle, NC, as a part of the Gag polyprotein, plays a crucial role in the recognition and dimerization of the two copies of genomic RNA (Rein, Reference Rein2010; Abd El-Wahab et al., Reference Abd El-Wahab, Smyth, Mailler, Bernacchi, Vivet-Boudou, Hijnen, Jossinet, Mak, Paillart and Marquet2014), Gag–Gag oligomerization and Gag trafficking to the plasma membrane (El Meshri et al., Reference El Meshri, Dujardin, Godet, Richert, Boudier, Darlix, Didier, Mély and de Rocquigny2015; Freed, Reference Freed2015). The ability of NC to aggregate NAs is due to its cationic character (Darlix et al., Reference Darlix, Lapadat-Tapolsky, de Rocquigny and Roques1995; Mirambeau et al., Reference Mirambeau, Lyonnais, Coulaud, Hameau, Lafosse, Jeusset, Justome, Delain, Gorelick and Le Cam2006; Vo et al., Reference Vo, Barany, Rouzina and Musier-Forsyth2006), notably in its N-terminal domain and removal of this domain reduces the aggregation capability greatly (Stoylov et al., Reference Stoylov, Vuilleumier, Stoylova, De Rocquigny, Roques, Gérard and Mély1997; Krishnamoorthy, Reference Krishnamoorthy, Roques, Darlix and Mély2003). On the other hand, properly folded zinc fingers are critical for NA destabilization (Bernacchi et al., Reference Bernacchi, Stoylov, Piémont, Ficheux, Roques, Darlix and Mély2002; Beltz et al., Reference Beltz, Clauss, Piémont, Ficheux, Gorelick, Roques, Gabus, Darlix, de Rocquigny and Mély2005; Godet et al., Reference Godet, Ramalanjaona, Sharma, Richert, de Rocquigny, Darlix, Duportail and Mély2011; Wu et al., Reference Wu, Mitra, McCauley, Thomas, Rouzina, Musier-Forsyth, Williams and Gorelick2013). Although both double-stranded (ds) and single-stranded (ss) NAs can be bound by NC, the zinc fingers prefer ssNAs (Heath et al., Reference Heath, Derebail, Gorelick and DeStefano2003; Mirambeau et al., Reference Mirambeau, Lyonnais, Coulaud, Hameau, Lafosse, Jeusset, Justome, Delain, Gorelick and Le Cam2006; Darlix et al., Reference Darlix, Godet, Ivanyi-Nagy, Fossé, Mauffret and Mély2011).
The binding of NC to single DNA molecules has been studied thoroughly using optical tweezers, in particular by the Williams group (Williams et al., Reference Williams, Rouzina, Wenner, Gorelick, Musier-Forsyth and Bloomfield2001; Williams et al., Reference Williams, Gorelick and Musier-Forsyth2002; Cruceanu, Reference Cruceanu2006; Cruceanu et al., Reference Cruceanu, Gorelick, Musier-Forsyth, Rouzina and Williams2006; Wu et al., Reference Wu, Mitra, McCauley, Thomas, Rouzina, Musier-Forsyth, Williams and Gorelick2013, Reference Wu, Mitra, Naufer, McCauley, Gorelick, Rouzina, Musier-Forsyth and Williams2014). They used single-molecule DNA stretching to probe the NA annealing, aggregation and destabilization activities of NC. Gag (in a version where the p6 region is deleted, Δ-p6 Gag), wild-type NC, as well as different NC variants, designed to be defective in at least one of these activities, by deletion or changes of key residues in the N-terminal domain, the zinc finger and the linker regions, have been investigated. Their results show that the NC protein within the context of Gag appears to have mostly a NA binding and packaging function, while the processed forms of NC appears to act mostly as a NA chaperone. In addition, both of the two zinc fingers are required for NA destabilization and the lack of, or changes in, the zinc fingers results in significantly weaker duplex destabilization. On the other hand, by neutralizing the cationic residues, especially the N-terminal cationic residues, or by deleting the whole N-terminal domain, the ability of NC to interact with NAs is significantly decreased (Vuilleumier et al., Reference Vuilleumier, Bombarda, Morellet, Gérard, Roques and Mély1999; Beltz et al., Reference Beltz, Clauss, Piémont, Ficheux, Gorelick, Roques, Gabus, Darlix, de Rocquigny and Mély2005).
We here use a complementary single DNA molecule method, based on stretching single DNA molecules in nanofluidic channels, to study the interaction between NC, in its free form and as part of Gag, and DNA. Nanofluidic channels have during the last years emerged as a suitable tool for studying interactions between proteins and DNA (Persson and Tegenfeldt, Reference Persson and Tegenfeldt2010; Frykholm et al., Reference Frykholm, Nyberg and Westerlund2017). Single DNA molecules can be stretched in nanofluidic channels with an extension that scales linearly to its contour length (Tegenfeldt et al., Reference Tegenfeldt, Prinz, Cao, Chou, Reisner, Riehn, Wang, Cox, Sturm, Silberzan and Austin2004). In combination with fluorescence microscopy, conformational changes of DNA molecules can be studied using nanofluidic channels with cross-sectional diameters of tens to hundreds of nanometres (Levy and Craighead, Reference Levy and Craighead2010; Reisner et al., Reference Reisner, Pedersen and Austin2012; van der Maarel et al., Reference van der Maarel, Zhang and van Kan2014). Previous studies have demonstrated the use of nanofluidic channels for studying DNA-binding proteins, including proteins that compact and condense DNA (Zhang et al., Reference Zhang, Guttula, Liu, Malar, Ng, Dai, Doyle, van Kan and van der Maarel2013a; Jiang et al., Reference Jiang, Zhang, Guttula, Liu, van Kan, Lavelle, Kubiak, Malabirade, Lapp, Arluison and van der Maarel2015; Frykholm et al., Reference Frykholm, Berntsson, Claesson, de Battice, Odegrip, Stenmark and Westerlund2016; Malabirade et al., Reference Malabirade, Jiang, Kubiak, Diaz-Mendoza, Liu, van Kan, Berret, Arluison and van der Maarel2017) and proteins that form filaments on DNA (Zhang et al., Reference Zhang, Hernandez-Garcia, Jiang, Gong, Guttula, Ng, Malar, van Kan, Dai, Doyle, Vries and van der Maarel2013b; Frykholm et al., Reference Frykholm, Alizadehheidari, Fritzsche, Wigenius, Modesti, Persson and Westerlund2014; Fornander et al., Reference Fornander, Frykholm, Fritzsche, Araya, Nevin, Werner, Çakır, Persson, Garcin, Beuning, Mehlig, Modesti and Westerlund2016). In addition, DNA can be condensed inside nanofluidic channels by neutral crowding agents (Zhang et al., Reference Zhang, Shao, van Kan and van der Maarel2009) or like-charged proteins (Zhang et al., Reference Zhang, Gong, Guttula, Malar, van Kan, Doyle and van der Maarel2012). One important aspect that makes studies of single DNA molecules in nanofluidic channels unique is that the single DNA molecules are suspended free in solution. This is in stark contrast to most single DNA molecule techniques, where at least one DNA end is attached to a bead or a surface, and opens up possibilities to study reactions occurring on DNA ends.
Here, we studied the binding properties of two versions of Gag and several versions of NC, to long dsDNA (40–50 kb). Our results show that the ability to compact and condense dsDNA is mainly related to the N-terminal domain of NC. In addition, using dsDNA with short protruding ssDNA ends allowed us to probe the chaperone activity of NC by studying concatemerization and circularization of DNA with complementary ssDNA overhangs. These studies demonstrated the key role of the N-terminal domain in the annealing of ssDNA. On the other hand, compared with NC alone, the NC protein within the context of Gag shows stronger ability of compaction and condensation of dsDNA, but weaker chaperone activity. Interestingly, the chaperone activity is somewhat retained when the p6 domain is deleted, and this is the first study where Gag and Δ-p6 are directly compared using single DNA molecule techniques. Importantly, the use of nanofluidic channels allowed us to directly investigate the competition between annealing and condensation on the single DNA molecule level. In addition to the specific studies on NC, the concatemerization demonstrates the usefulness of nanofluidic channels for probing intermolecular DNA–DNA interactions on the single DNA molecule level, in particular involving DNA ends, and the example here is the first along those lines.
Materials and methods
Protein expression and purification
The different NC peptides were prepared by solid-phase peptide synthesis on a 433A synthesizer (ABI, Foster City, CA, USA), HPLC purified and characterized by ion spray mass spectrometry, as previously described (Shvadchak et al., Reference Shvadchak, Sanglier, Rocle, Villa, Haiech, Hibert, Van Dorsselaer, Mély and de Rocquigny2009). To get the zinc-bound form of NC peptides, 2.2 molar equivalents of ZnSO4 was added to the peptide and pH was raised to 7.4. Peptide concentrations were determined using an extinction coefficient of 5700 M−1 cm−1 at 280 nm.
Recombinant Gag and Δ-p6 Gag were prepared as follows:
Bacterial strains and media
All transformation steps were carried out using the Escherichia coli strains DH5a and BL21-CodonPlus, and the standard heat shock protocol. For the production of plasmid DNA in DH5a strains and the production of recombinant protein in BL21-CodonPlus strains, bacteria were cultured in LB media (1% (w/v) peptone, 0.5% (w/v) yeast extract and 0.5% NaCl). The media was supplemented with kanamycin (50 µg/ml; DH5a) or both kanamycin and chloramphenicol (50 µg/ml; BL21-CodonPlus).
Plasmid construction
The plasmid construction was adapted from McKinstry et al. (Reference McKinstry, Hijnen, Tanwar, Sparrow, Nagarajan, Pham and Mak2014), but using a cleavable TEV-His tag at the C-terminus end. The Pr55Gag-TEV-His and Pr55Gag-Δ-p6-TEV-His–pET28a expression plasmids were checked by DNA sequencing.
Large-scale protein production
Large-scale production of recombinant Pr55Gag-TEV-His and Pr55Gag-Δ-p6-TEV-His was performed by inoculating a single colony into 25 ml LB media containing antibiotics and cultured at 37 °C overnight with shaking at 200 rpm. The overnight culture was used to inoculate 1 litre of LB media containing antibiotics in a 2.5 litre glass flask. The culture was grown at 37 °C, 200 rpm, until an OD at 600 nm of approximately 0.5 was reached. Protein expression was induced with the addition of 0.2 mM IPTG, and the bacteria grown for a further 4 h at 37 °C. Bacteria were harvested by centrifugation (10 000 g; 4 °C; 15 min), and the pellet was stored at −80 °C. Bacterial pellets from the equivalent of 5 litres of culture were resuspended in 40 ml of lysis buffer (50 mM TRIS-HCl pH = 8; 1 M NaCl; 10 mM 2-mercaptoethanol; 25 mM imidazole; 1% Tween-20) supplemented with protease inhibitor cocktail and then sonicated. The suspension was supplemented with 500 units of Benzonase Nuclease (Sigma-Aldrich, St. Louis, MO, USA; E1014) and incubated for 30 min at 4 °C. DNA was sheared by repeated passage through a 23-gauge needle. The lysate was centrifuged to remove insoluble material (27 000 g; 4 °C; 45 min). The clear supernatant was filtered through a 0.45 µm syringe filter and loaded onto a Nickel column (XK16) that had previously been equilibrated with 50 mM TRIS-HCl pH = 8, 1 M NaCl, 10 mM 2-mercaptoethanol, 25 mM imidazole, 1% Tween-20 and 10% (v/v) glycerol. The column was washed with equilibration buffer and bound proteins were eluted with a 0–1000 mM imidazole gradient in equilibration buffer. Fractions containing Pr55Gag full-length or Pr55Gag Δ-p6 were pooled and concentrated using a centrifugal Ultra-15, 30 000 molecular weight cut-off membrane (Millipore, Burlington, MA, USA) and then desalted using a PD-10 column (GE Healthcare, Chicago, IL, USA). The protein was then incubated overnight at 4 °C with 1.2 kU of hexa-histidine-tagged TEV protease (Protean). To remove the cleaved His-tag, the resulting mixture was passed over a HisTrap column at 1 ml/min as above. The protein that did not bind was collected and concentrated. The last step of purification consisted of a size exclusion chromatography using a Superdex 200 (high load 16/60) column previously equilibrated in 50 mM TRIS-HCl pH 8.0, 1.0 M NaCl. Peak fractions from this column containing the protein of interest were pooled and concentrated to 1–2 mg/ml, snap frozen in liquid nitrogen and then stored at −80 °C.
Sample preparation
DNA from phage T7 (T7-DNA, MABION, Konstantynów Łódzki, Poland) or phage λ (λ-DNA, Roche, Basel, Switzerland) was pre-stained with YOYO-1 (Invitrogen, Waltham, MA, USA) at a ratio of one dye molecule per 50 base pairs. This ratio minimizes the effect of YOYO-1 on DNA conformation (Kundukad et al., Reference Kundukad, Cong, Van Der Maarel and Doyle2013; Nyberg et al., Reference Nyberg, Persson, Åkerman and Westerlund2013). Pre-stained DNA was then mixed with the wild-type or mutant Gag proteins or NC peptides and incubated at 4 °C for at least 2 h. The complexes were then introduced into the nanofluidic system and equilibrated for 60 s before image capture. The DNA concentration was 5 µM (basepairs) in all samples. 3% (v/v) β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA) was added as an oxygen scavenger to suppress oxygen radical-induced photo-damage of the DNA. The buffer used was 25 mM Tris with 30 mM NaCl and 0.2 mM MgCl2 (pH 7.5).
Nanofluidics
The single DNA molecule experiments were performed in nanochannels with a depth of 100 nm and a width of 150 nm. The devices were fabricated using advanced nanofabrication described elsewhere (Persson and Tegenfeldt, Reference Persson and Tegenfeldt2010). The channel system consists of a pair of feeding channels (micro-size), spanned by a set of parallel nanochannels. A schematic illustration of the nanofluidic chip is shown in Fig. 2a. The sample is loaded into the channel system from one of the four reservoirs that are connected to the feeding channels and moved into the nanochannels by pressure-driven (N2) flow.
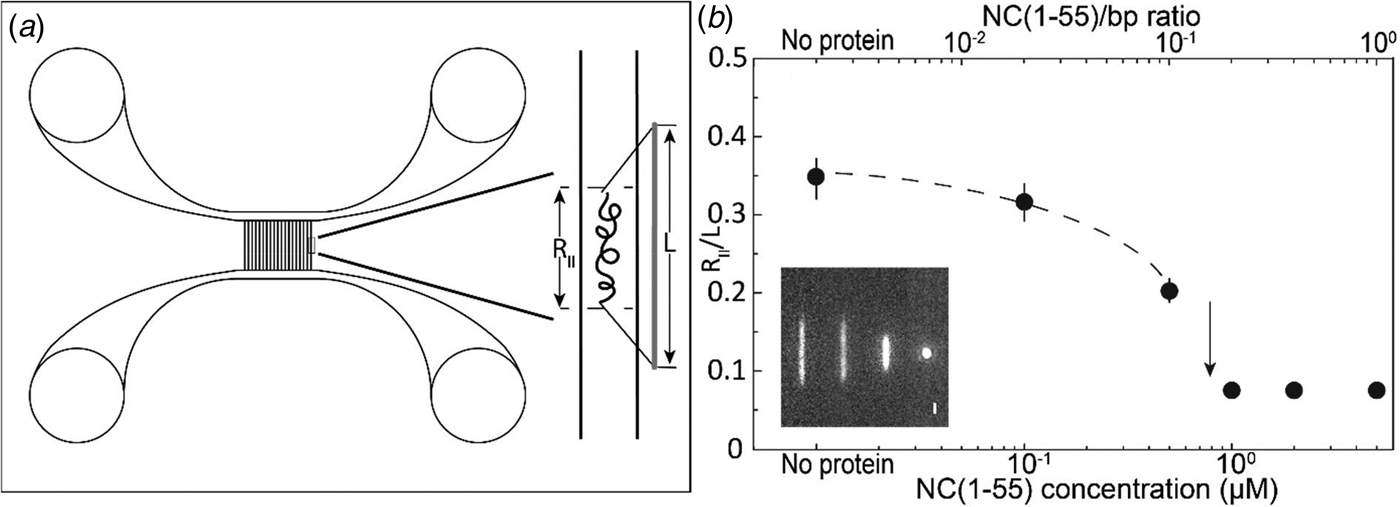
Fig. 2. (a) Schematic illustration of the nanofluidic chip design (left). The channel system consists of pairs of microchannels, spanned by an array of straight nanochannels, 500 µm long, 100 nm deep and 150 nm wide. The cartoon (right) shows DNA confined inside a nanochannel. DNA will be partially stretched along the nanochannel, with an extension R ||, shorter than its contour length L. (b) Relative extension R ||/L of T7-DNA inside 100 nm × 150 nm channels plotted versus the NC(1–55) concentration (bottom axis) and NC(1–55) to bp ratio (top axis). DNA concentration is 5 µM base pairs. The dashed line is drawn as an aid to the eye and the arrow denotes the condensation threshold. The inset is a montage of fluorescence images of T7-DNA molecules at different NC(1–55) concentrations (from left to right: 0, 0.1, 0.5 and 1 µM).
To avoid non-specific binding of protein to the negatively charged channel walls, the channels were prior to the experiments coated with a lipid bilayer comprising 99% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, Avanti) and 1% N-(fluorescein-5-thiocarbamoyl)-1,2-dihexadecanoyl-sn-Glycero-3-phosphoethanolamine, triethylammonium salt (fluorescein-DHPE, Invitrogen). The coating procedure is described elsewhere (Persson et al., Reference Persson, Fritzsche, Mir, Modesti, Westerlund and Tegenfeldt2012).
The DNA and DNA–protein complexes were imaged using an epifluorescence microscope (Zeiss AxioObserver.Z1) equipped with a Hamamatsu digital CMOS C11440-22CU camera, a 63× oil immersion TIRF objective (NA = 1.46) and a 1.6× optovar from Zeiss. Using the microscopy imaging software ZEN, 50 subsequent images were recorded with an exposure time of 200 ms. Data analysis was performed using a custom-written MATLAB-based software. Microscopy image stacks were used as input to the program. Images were first binarized by thresholding with a global average plus onefold of standard deviation. Taking advantage of the high contrast of the YOYO-stained DNA fluorescence images, regions with higher brightness were directly considered as DNA objects without additional image filtering. Finally, the lengths of the DNA molecules were extracted by identifying the longest axis of the objects and the length was measured. In total, 50–100 DNA molecules were analysed for each sample concentration. All the histograms are fit with Gaussian distributions.
Results
The goal of the study is to investigate how the binding of the NC protein, both in its isolated form and when inserted in its parent protein Gag, affects the physical properties of DNA. To do so, we mixed the proteins with pre-stained DNA (YOYO:bp ratio of 1:50) at different ratios and observed individual complexes in nanochannels with a dimension of 100 nm × 150 nm. To scrutinize the binding of the NC protein to ssDNA and dsDNA, bacteriophage T7 DNA (39 937 base pairs), which has blunt ends, and λ-DNA (48 502 base pairs), which has 12 bp-long ssDNA overhangs, were used as model DNAs in this study. In the following, the native nucleocapsid protein will be called NC(1–55) to distinguish it unambiguously from its mutants.
Compaction and condensation of T7-DNA by NC(1–55)
T7-DNA molecules at a concentration of 5 µM (base pairs) were incubated with different concentrations of NC(1–55) at 4 °C for at least 2 h. Figure 2b shows the mean extension of T7 DNA (L = 13.6 µm) along the longitudinal direction of the channel divided by the contour length (R ||/L), as a function of NC(1–55) concentration. With increasing NC(1–55) concentration, the extension of the DNA decreases. For over-threshold concentrations of NC(1–55), DNA is compacted into a condensed form, where the single DNA molecules are simply bright fluorescent blobs that can be easily distinguished from the extended form. No condensation was observed in the feeding microchannels at these concentrations, indicating that the condensation was facilitated by the nanoconfinement inside the channels, which has been observed also for other DNA-condensing proteins (Zhang et al., Reference Zhang, Guttula, Liu, Malar, Ng, Dai, Doyle, van Kan and van der Maarel2013a; Jiang et al., Reference Jiang, Zhang, Guttula, Liu, van Kan, Lavelle, Kubiak, Malabirade, Lapp, Arluison and van der Maarel2015). At higher concentrations (2 µM and above), condensation was observed also in the feeding microchannels.
NC anneals short complementary ssDNA
To further investigate the binding of NC(1–55) to ssDNA and dsDNA, we used λ-DNA, a linear dsDNA, 48.5 kilobase pairs long, where the 5′-terminal ends protrude as self-complementary single-stranded chains, 12 nucleotides long, due to the circular origin of λ-DNA. These single-stranded ends can anneal to generate circles or DNA concatemers (Sanger et al., Reference Sanger, Coulson, Hong, Hill and Petersen1982) (see schematic representation in Fig. 3c). The distribution in the extension at different concentrations of NC(1–55) with T7-DNA and λ-DNA is shown in Figs 3a and b, respectively. The larger extension R|| of λ-DNA without protein bound agrees well with its longer contour length (L = 16 µm), and for both DNAs used, the relative extension (R ||/L) without protein is ca 35% of L. With increasing concentrations of NC(1–55), a decrease in the extension of single DNA molecule was observed also for λ-DNA, but no complete condensation was observed even at 1 µM. For over-threshold concentrations (>1 µM), condensed DNA aggregates were observed in the microchannels (data not shown). Interestingly, these aggregates are too large to enter the nanochannels, indicating that they consist of more than one DNA molecule. A striking difference between λ-DNA and T7-DNA is that for λ-DNA, many DNA molecules have an extension that is much longer than naked DNA. This indicates the formation of DNA concatemers in the presence of NC(1–55). With increasing NC(1–55) concentrations, more λ-DNA concatemers with longer extension were observed (~10% at 0.1 µM, ~50% at 0.5 µM and ~60% at 1 µM), but no concatemers were observed at any protein concentration with T7-DNA. Moreover, no DNA concatemers were observed for λ-DNA in the absence of NC(1–55). This strongly indicates that the formation of concatemers is due to the annealing of the λ-DNA single-stranded overhangs, promoted by the NC(1–55) protein (Fig. 3c), in full line with the well-described ability of NC(1–55) to chaperone the annealing of complementary sequences (Beltz et al., Reference Beltz, Piémont, Schaub, Ficheux, Roques, Darlix and Mély2004; Hargittai et al., Reference Hargittai, Gorelick, Rouzina and Musier-Forsyth2004; Beltz et al., Reference Beltz, Clauss, Piémont, Ficheux, Gorelick, Roques, Gabus, Darlix, de Rocquigny and Mély2005; Godet et al., Reference Godet, De Rocquigny, Raja, Glasser, Ficheux, Darlix and Mély2006; Vo et al., Reference Vo, Barany, Rouzina and Musier-Forsyth2009). Though the overhangs are rather short (12 nucleotides), they can accommodate two NC(1–55) molecules, because it has been demonstrated on a very large number of sequences that the footprint of NC(1–55) is 5–7 nucleotides (De Guzman, Reference De Guzman1998; Fisher et al., Reference Fisher, Rein, Fivash, Urbaneja, Casas-Finet, Medaglia and Henderson1998; Vuilleumier et al., Reference Vuilleumier, Bombarda, Morellet, Gérard, Roques and Mély1999; Amarasinghe et al., Reference Amarasinghe, Zhou, Miskimon, Chancellor, McDonald, Matthews, Miller, Rouse and Summers2001; Beltz et al., Reference Beltz, Clauss, Piémont, Ficheux, Gorelick, Roques, Gabus, Darlix, de Rocquigny and Mély2005; Avilov et al., Reference Avilov, Godet, Piémont and Mély2009; Darlix et al., Reference Darlix, Godet, Ivanyi-Nagy, Fossé, Mauffret and Mély2011).
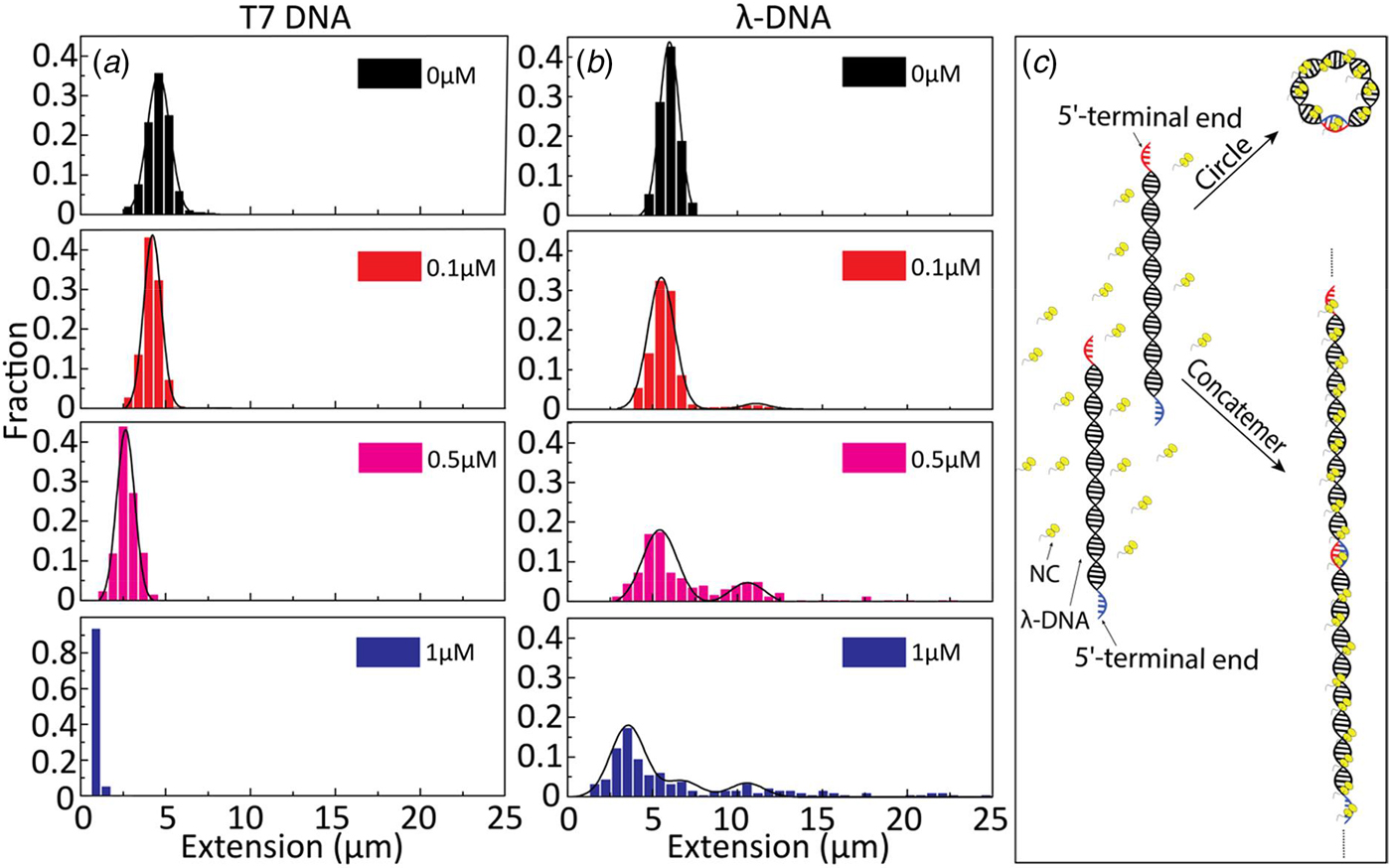
Fig. 3. Extension distribution of (a) T7-DNA molecules and (b) λ-DNA molecules at different concentrations of NC(1–55). DNA concentration is 5 µM base pairs. (c) Sketch of the process for forming circular DNA and DNA concatemers by NC(1–55). NC binds to the single-stranded ends of λ-DNA and anneals the complementary ends to form concatemers or circles.
If concatemers form as a result of the NC(1–55)-promoted annealing of complementary ssDNA overhangs, we expect that circular DNA molecules, resulting from intramolecular annealing of λ-DNA molecules, may form as well (see schematic representation in Fig. 3c). Circular DNAs are characterized by approximately half the extension and twice the emission intensity compared with linear DNAs, since they are double-folded in the channels (Alizadehheidari et al., Reference Alizadehheidari, Werner, Noble, Reiter-Schad, Nyberg, Fritzsche, Mehlig, Tegenfeldt, Ambjörnsson, Persson and Westerlund2015; Frykholm et al., Reference Frykholm, Nyberg, Lagerstedt, Noble, Fritzsche, Karami, Ambjörnsson, Sandegren and Westerlund2015). Fig. 4a shows a linear λ-DNA (right trace) together with a λ-DNA molecule with a shorter extension (~60%) and an approximately twofold increased fluorescence intensity (left trace), suggesting that it is a circular DNA molecule (Alizadehheidari et al., Reference Alizadehheidari, Werner, Noble, Reiter-Schad, Nyberg, Fritzsche, Mehlig, Tegenfeldt, Ambjörnsson, Persson and Westerlund2015).
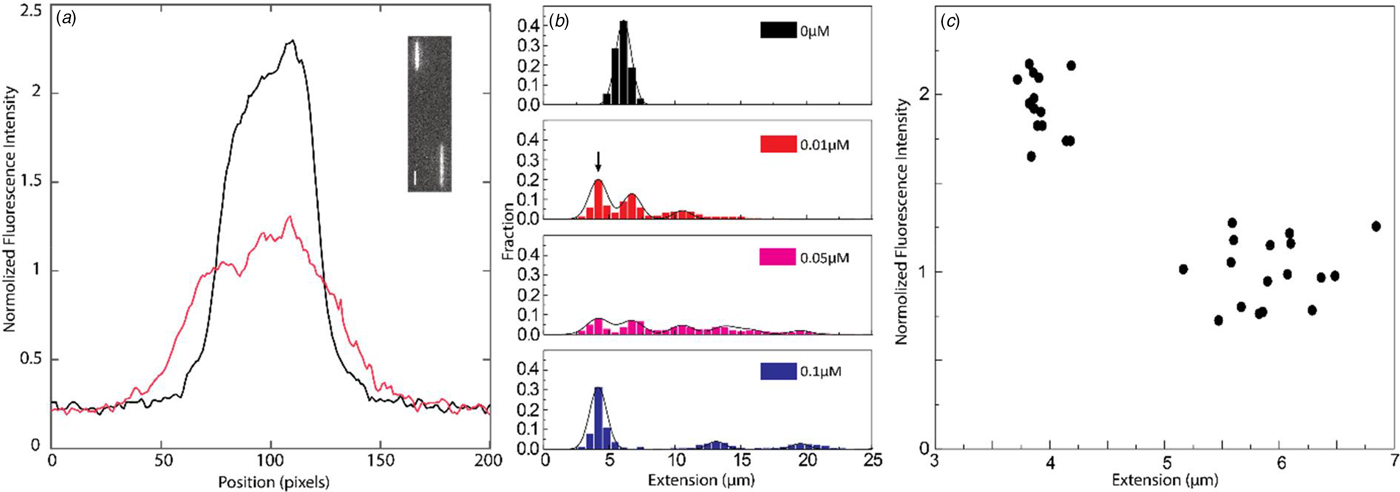
Fig. 4. (a) Intensity profile of circular (black) and linear (red) λ-DNA. Inset shows a fluorescence image of circular (left) and linear (right) λ-DNA in the presence of 0.1 µM NC(1–55) and 5 µM DNA bp. The scale bar is 1 µm. (b) Distribution in the extension of λ-DNA molecules with different concentrations of NC(1–55) at 0.5 µM DNA bp concentration. The arrow (0.01 µM) highlights the emerging peak that is interpreted as circular DNA. (c) Scatterplot of normalized fluorescence intensity versus DNA extension at 0.05 µM NC(1–55) and 0.5 µM DNA bp, highlighting a fraction of molecules with approximately double intensity and half extension.
It is difficult to distinguish circular DNA from compacted linear DNA in Fig. 3. To promote the formation of circular DNA and limit the number of concatemers, we decreased the overall DNA and protein concentration. Samples at a lower λ-DNA concentration (0.5 µM base pairs, ten times lower DNA concentration) with the same protein to DNA bp ratios as in Fig. 3 were therefore analysed (Fig. 4b). The peak at 5.5 µm, corresponding to single naked λ-DNA molecules, is split into two when NC(1–55) is added, one still at 5.5 µm and one at approximately half that extension (see arrow). The scatterplot of intensity versus extension (Fig. 4c) shows two clear clusters. The circular form has approximately double the emission intensity and half the extension of the linear form (Alizadehheidari et al., Reference Alizadehheidari, Werner, Noble, Reiter-Schad, Nyberg, Fritzsche, Mehlig, Tegenfeldt, Ambjörnsson, Persson and Westerlund2015; Frykholm et al., Reference Frykholm, Nyberg, Lagerstedt, Noble, Fritzsche, Karami, Ambjörnsson, Sandegren and Westerlund2015). This indicates that the fraction of molecules at half extension is due to the formation of circular DNA molecules. We also observe DNA concatemers in the presence of NC(1–55) at this lower total concentration. The peaks for circular, linear and concatemers of λ-DNA are observed at both 0.01 and 0.05 µM. It should be noted that at this lower DNA concentration, it is not possible to obtain data that fully represent the actual fractions of linear, circular and concatemer molecules, as was done in Figs 2 and 3. This is because the molecules are manually selected before they are inserted into the channels, which might induce a bias. The important message from Fig. 4 is instead that we can identify a fraction of DNA molecules that are circular.
Condensation and ssDNA annealing by NC(1–55) mutants
Figures 2–4 clearly demonstrate that nanofluidic channels can be used to characterize two of the main functions of the NC protein, i.e. to condense and protect NAs and to promote the formation of their thermodynamically most favoured conformation. To study the contribution of the different domains of NC(1–55) to DNA condensation and formation of DNA concatemers, several mutants were investigated. Since many studies have shown that the N-terminal domain is a major factor in the NA binding and aggregation activity of NC(1–55) (Stoylov et al., Reference Stoylov, Vuilleumier, Stoylova, De Rocquigny, Roques, Gérard and Mély1997; Fisher et al., Reference Fisher, Rein, Fivash, Urbaneja, Casas-Finet, Medaglia and Henderson1998; Vuilleumier et al., Reference Vuilleumier, Bombarda, Morellet, Gérard, Roques and Mély1999; Bernacchi et al., Reference Bernacchi, Stoylov, Piémont, Ficheux, Roques, Darlix and Mély2002; Krishnamoorthy, Reference Krishnamoorthy, Roques, Darlix and Mély2003), we first investigated the DNA condensation and concatemer formation properties of NC(11–55), a mutant where the N-terminal domain is deleted. Similar to T7-DNA with NC(1–55), compaction and eventually condensation of λ-DNA was observed with increasing concentrations of NC(11–55). However, the concentration needed for condensation was 3 µM, which is ~3 times higher than for NC(1–55), and agrees with the lower binding constant for this mutant (Beltz et al., Reference Beltz, Clauss, Piémont, Ficheux, Gorelick, Roques, Gabus, Darlix, de Rocquigny and Mély2005). Interestingly, no concatemers or circularization of λ-DNA were observed in the presence of NC(11–55) (Fig. 5a), suggesting that the first ten amino acid residues in NC(1–55) are involved in the annealing of ssDNA and concatemer formation. It is also possible that the balance between condensation and formation of concatemers is shifted when the first ten amino acids are removed, meaning that condensation happens before annealing can occur.
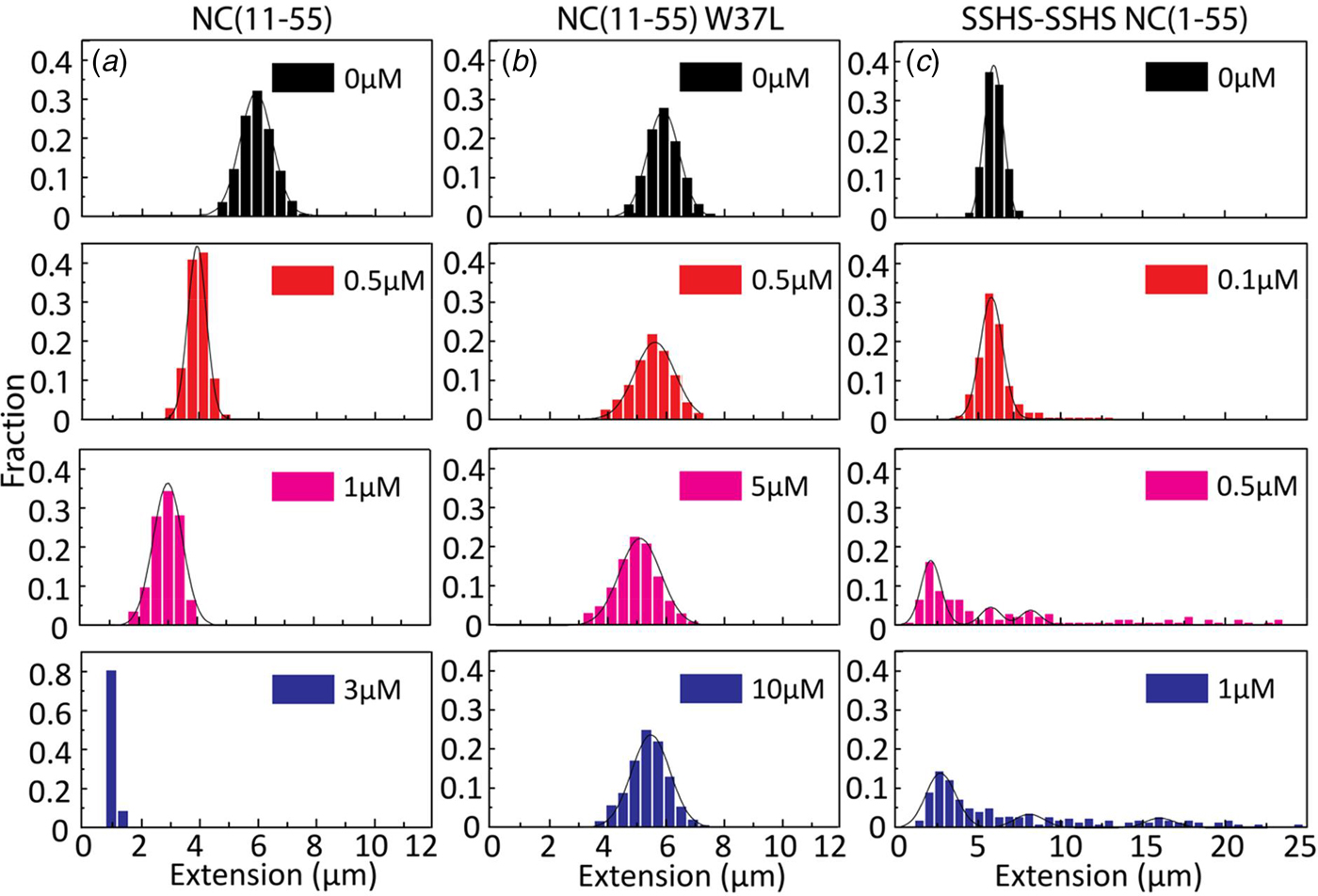
Fig. 5. Distribution in the extension of λ-DNA with different concentrations of NC(11–55) (a), NC(11–55)W37L (b) and SSHS-SSHS NC(1–55) (c). DNA concentration is 5 µM base pairs.
For the NC(11–55)W37L mutant, where the tryptophan in the second zinc finger is mutated to a leucine residue, a ~10% decrease in the DNA extension was observed at the highest protein concentration (1 µM). No DNA condensation (Fig. 5b) and no evidence for concatemer formation and hence annealing of ssDNA in the concentration interval studied were observed, in line with the much lower binding constant of this mutant (Beltz et al., Reference Beltz, Clauss, Piémont, Ficheux, Gorelick, Roques, Gabus, Darlix, de Rocquigny and Mély2005).
Finally, we examined the role of the zinc fingers in DNA condensation and concatemer formation. This mutant has lost its affinity for zinc by replacing the cysteine residues with serines. It should be noted that this mutant has a completely different secondary structure than the wild-type NC(1–55), as the zinc fingers are largely unfolded. Figure 5c shows the distribution in the extension of λ-DNA with different concentrations of the SSHS-SSHS NC(1–55) mutant. Similar to NC(1–55), DNA compaction is observed inside nanochannels, as well as formation of DNA concatemers. However, no full DNA condensation was observed inside nanochannels. For over-threshold concentration of SSHS-SSHS NC(1–55) (1 µM), condensed DNA aggregates were observed in the microchannels (data not shown).
DNA condensation and concatemer promotion by Gag and Δ-p6 Gag
To get a better understanding of the DNA-binding properties of the HIV-1 NC domain within the context of Gag, the same experiments as for NC above were performed also with Gag and its mutant Δ-p6 Gag (lacking the p6 domain at its C-terminus). Most of the studies on the NA binding and chaperone properties of Gag have been reported with the truncated Δ-p6 Gag version (Campbell and Rein, Reference Campbell and Rein1999; Cruceanu et al., Reference Cruceanu, Gorelick, Musier-Forsyth, Rouzina and Williams2006; Jones et al., Reference Jones, Datta, Rein, Rouzina and Musier-Forsyth2011; Rein et al., Reference Rein, Datta, Jones and Musier-Forsyth2011; Webb et al., Reference Webb, Jones, Parent, Rouzina and Musier-Forsyth2013). Only recently, a soluble version of full length Gag was prepared and characterized (Abd El-Wahab et al., Reference Abd El-Wahab, Smyth, Mailler, Bernacchi, Vivet-Boudou, Hijnen, Jossinet, Mak, Paillart and Marquet2014; McKinstry et al., Reference McKinstry, Hijnen, Tanwar, Sparrow, Nagarajan, Pham and Mak2014; Bernacchi et al., Reference Bernacchi, Abd El-Wahab, Dubois, Hijnen, Smyth, Mak, Marquet and Paillart2017; Tanwar et al., Reference Tanwar, Khoo, Garvey, Waddington, Leis, Hijnen, Velkov, Dumsday, McKinstry and Mak2017), but to our knowledge, its NA condensation and chaperone properties studied at the single molecule level, compared with those of the Δ-p6 Gag mutant in the same assay, have not been reported so far. The distribution in the extension at different concentrations of Gag and Δ-p6 Gag with λ-DNA is shown in Fig. 6. With increasing concentrations of both Gag and Δ-p6 Gag, a decrease in the extension of single DNA molecules was observed for λ-DNA that finally resulted in a fully condensed form. The concentration for DNA condensation (~0.2 µM) is ~5 times lower compared with NC(1–55), indicating a higher efficiency of Gag in dsDNA condensation. For over-threshold concentrations, condensed DNA aggregates were observed also in the microchannels for both Gag and Δ-p6 Gag.
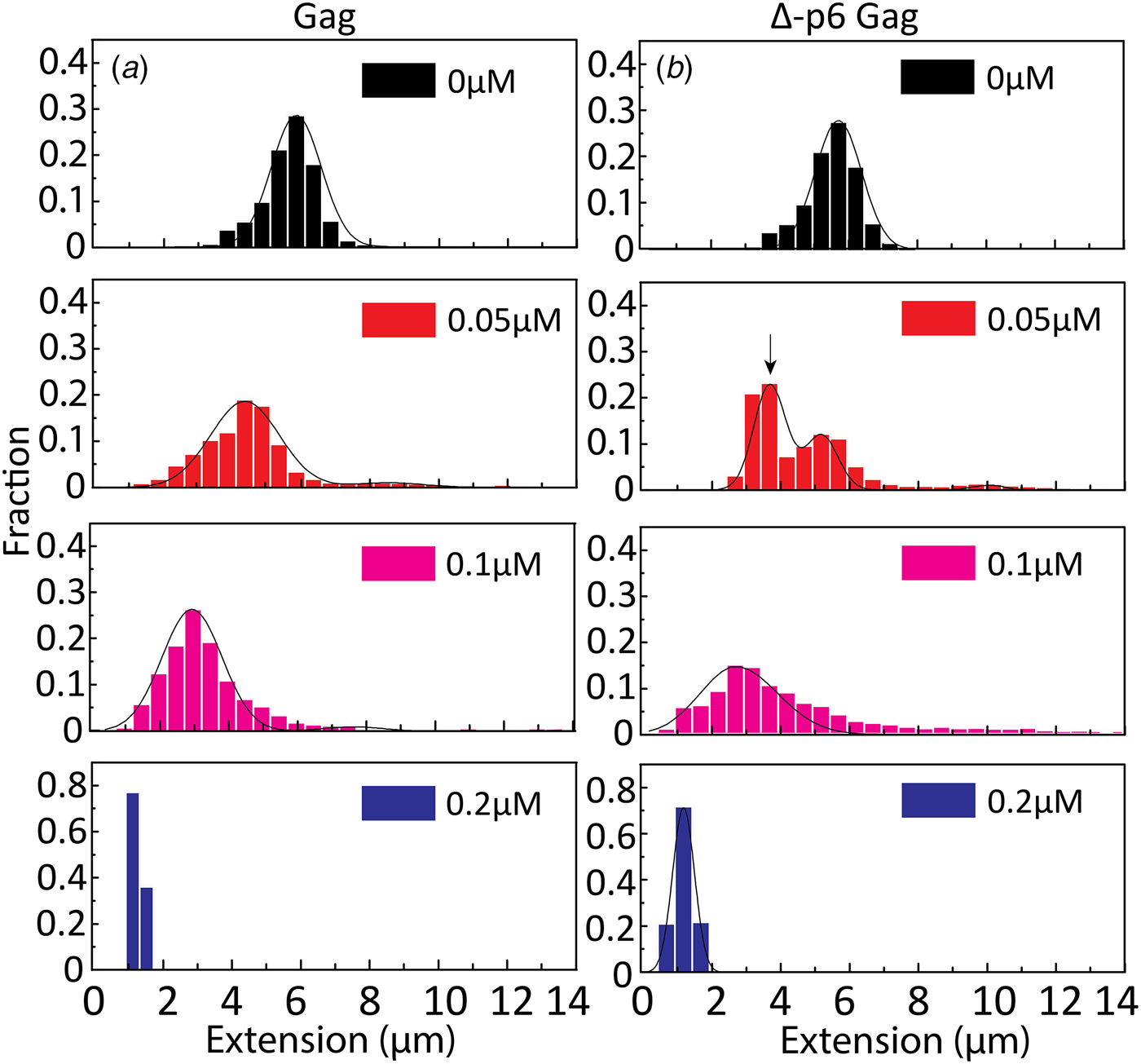
Fig. 6. Distribution in the extension of λ-DNA with Gag (a) and Δ-p6 Gag (b) at increasing protein concentrations. The arrow in (b) (0.05 µM) highlights the emerging peak that is interpreted as circular DNA. DNA concentration is 5 µM base pairs.
A very small fraction of DNA formed concatemers for Gag (<20% at both 0.05 and 0.1 µM), which is much less than with NC(1–55). Interestingly, for Δ-p6 Gag, circular DNAs (~60% of total molecules, see arrow in Fig. 6b at 0.05 µM) and more DNA concatemers (~30% at 0.1 µM) than with Gag were observed, but still less than with NC(1–55). The concentration for condensation of DNA is similar for Gag and Δ-p6 Gag, in line with their similar binding affinity for a number of NA sequences (Tanwar et al., Reference Tanwar, Khoo, Garvey, Waddington, Leis, Hijnen, Velkov, Dumsday, McKinstry and Mak2017). Interestingly, Δ-p6 Gag seems to prefer the formation of intramolecular circles instead of intermolecular concatemers, compared with NC(1–55).
Discussion
The NC protein is involved in several steps of the life cycle of the HIV-1 virus. We here use nanofluidic channels to simultaneously investigate two of the fundamental characteristics of the protein, the ability to condense and protect NAs and the chaperone ability to favour the most stable configuration of NAs. The latter was investigated via the use of a DNA construct, λ-DNA, that has a 12 bp ssDNA overhang in each end that anneal to form circles and concatemers when NC(1–55) is present.
We observed condensation of single DNA molecules as a decrease in their extension along the nanochannel. This decrease was found to depend on the NC(1–55) concentration and to result in a collapse of the DNA molecules into a condensed form at over-threshold concentrations of NC(1–55). The protein concentration required for DNA condensation inside nanofluidic channels was observed to be an order of magnitude lower than in the feeding microchannels, where the DNA is not confined. This cannot be explained by macromolecular crowding, which usually occurs with concentrations of crowding agents in the range of tens to hundreds of micromolar (Zhang et al., Reference Zhang, Shao, van Kan and van der Maarel2009). This is rather due to the confinement in the nanochannels that induces condensation, as described in earlier studies (Zhang et al., Reference Zhang, Guttula, Liu, Malar, Ng, Dai, Doyle, van Kan and van der Maarel2013a; Jiang et al., Reference Jiang, Zhang, Guttula, Liu, van Kan, Lavelle, Kubiak, Malabirade, Lapp, Arluison and van der Maarel2015; Malabirade et al., Reference Malabirade, Jiang, Kubiak, Diaz-Mendoza, Liu, van Kan, Berret, Arluison and van der Maarel2017; Guttula et al., Reference Guttula, Liu, van Kan, Arluison and van der Maarel2018).
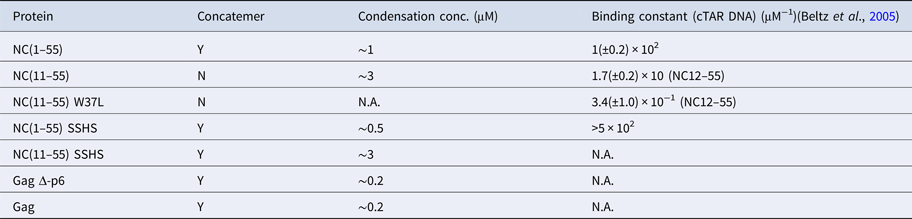
The condensation concentration varies between the different NC mutants and correlates directly with the binding constant (Table 1). For example, deleting the first ten amino acids of NC that include several positively charged residues increases the condensation concentration by >3-fold, in full line with the ~5-fold change in the reported binding constants (Beltz et al., Reference Beltz, Clauss, Piémont, Ficheux, Gorelick, Roques, Gabus, Darlix, de Rocquigny and Mély2005). Furthermore, NC(11–55)W37L, where the the tryptophan in the second zinc finger is mutated to a leucine, does not condense DNA at the concentrations investigated, in agreement with its ~30 times lower affinity (Beltz et al., Reference Beltz, Clauss, Piémont, Ficheux, Gorelick, Roques, Gabus, Darlix, de Rocquigny and Mély2005; Darlix et al., Reference Darlix, Godet, Ivanyi-Nagy, Fossé, Mauffret and Mély2011). A zinc-free mutant that is thought to be unfolded was also investigated. The slightly higher affinity of SSHS-SSHS NC(1–55) compared with NC(1–55) is again in line with the literature (Beltz et al., Reference Beltz, Clauss, Piémont, Ficheux, Gorelick, Roques, Gabus, Darlix, de Rocquigny and Mély2005) and can be explained by the unfolding of zinc fingers that allows for additional electrostatic interactions with DNA.
To probe the chaperone ability of NC, we investigated the ability of different versions of NC to anneal the 12 base pairs ssDNA overhangs of λ-DNA. While long concatemers form efficiently with the wild-type NC(1–55), no concatemers were observed for T7 DNA that has blunt ends, clearly indicating that their formation results from the annealing of the complementary single-stranded overhangs. Previous studies have shown that NC(1–55) can bind to both ssDNA and dsDNA, but there is a preference for ssDNA over dsDNA (Mirambeau et al., Reference Mirambeau, Lyonnais, Coulaud, Hameau, Lafosse, Jeusset, Justome, Delain, Gorelick and Le Cam2006). This selectivity for ssDNA can explain the formation of concatemers that is favoured over DNA compaction at low NC(1–55) concentration.
We did not observe any concatemers for the N-terminal deletion mutant NC(11–55). This could be due to the key role of the N-terminal domain in the annealing process or to a shift in the competition between the condensation and chaperone activities, so that the DNA condenses before annealing can occur. Concatemerization was also observed by the zinc-free mutant SSHS-SSHS NC(1–55), which has been shown to be an efficient NA annealer, due to its flexible and highly positively charged nature (Williams et al., Reference Williams, Rouzina, Wenner, Gorelick, Musier-Forsyth and Bloomfield2001; Godet et al., Reference Godet, Boudier, Humbert, Ivanyi-Nagy, Darlix and Mély2012).
The NC protein functions both in its mature form and as a part of the Gag protein in vivo. Both the full-length Gag and the truncated Δ-p6 Gag condensed DNA at a much lower concentration than NC(1–55). This higher condensation efficiency correlates well with their higher affinity as compared with NC(1–55) (Cruceanu, Reference Cruceanu2006; Godet et al., Reference Godet, Boudier, Humbert, Ivanyi-Nagy, Darlix and Mély2012), which is attributed to the participation of the matrix domain of Gag in the binding process (Alfadhli et al., Reference Alfadhli, McNett, Tsagli, Bächinger, Peyton and Barklis2011; Rein et al., Reference Rein, Datta, Jones and Musier-Forsyth2011). Moreover, as the full-length Gag and its truncated Δ-p6 Gag version show comparable DNA condensation, the p6 domain does not seem to play a key role in the condensation process. In sharp contrast, the DNA annealing capacity is almost completely lost with the full-length Gag, but partly retained when the p6 region is deleted, suggesting that the p6 region may hinder the annealing of ssDNA or favour DNA condensation over DNA concatemer formation. This hindrance and thus, the lower activity of the NC domain in the full-length Gag may be the result of the previously reported ability of the p6 domain to fold back and interact with the NC zinc fingers (Wang et al., Reference Wang, Naiyer, Mitra, Li, Williams, Rouzina, Gorelick, Wu and Musier-Forsyth2014). These results are in line with the tasks performed by the different proteins, where Gag is not involved in the chaperone activities that mature NC performs. Importantly, this is the first study of full Gag on the single DNA molecule level and it highlights small but significant differences compared with Δ-p6 Gag that has been used in previous single molecule studies.
It is important to relate the studies done here with other single molecule studies of NC and Gag, in particular by the Williams group. The use of optical tweezers to stretch single DNA molecules one by one gives unprecedented details on parameters like binding affinity and how many base pairs each protein covers. The chaperone activity of the protein can be characterized both when it comes to destabilizing dsDNA and promoting the formation of thermodynamically favoured structures. The nanofluidic channels work at a lower force, where DNA is not stretched to its full length, which in turn means that the DNA is exposed to forces that are more like forces in the cell. The DNA extensions studied in this paper relate to forces below 0.1 pN (Bustamante et al., Reference Bustamante, Marko, Siggia and Smith1994), which means that weak interactions easily interrupted in a tweezers setup are retained. We can directly determine the balance between DNA condensation and ssDNA annealing on long DNA molecules. We can also investigate potential intermolecular DNA–DNA interactions since many DNA molecules are present when the protein is added. The nanofluidic channels also allow measurements at higher throughput since tens of DNA molecules can be characterized at the same time and hence hundreds of molecules can be investigated for each experimental condition.
Finally, in addition to revealing biologically relevant information on the interaction between NC and Gag with DNA, the results obtained in this study highlight an important feature of the nanofluidic setup for studying DNA–protein interactions on single DNA molecules. While many single DNA molecule methods by definition involve only one molecule, we are able to study intermolecular DNA–DNA interactions between large DNA molecules. This is demonstrated by the annealing of the ssDNA overhangs by the NC protein that leads to concatemer formation. The same principles also mean that we can directly image processes on DNA ends and potentially also proteins that diffuse on DNA to find DNA ends. These two scenarios are possible to investigate since the studies are done on molecules freely suspended in solution and both ends of the DNA are free.
To conclude, we introduce nanofluidic channels to investigate the delicate balance between DNA condensation and chaperone activity of the NC protein, both in its mature form and as part of Gag. The first ten amino acids are important for both the chaperone activity and DNA condensation of NC. In Gag, the condensation is more efficient than for NC alone, while the chaperone activity is almost completely lost. When deleting the p6 region of Gag, some of the chaperone activity is retained while the condensation is not affected. Our study is the first that directly compares Gag and Δ-p6 Gag on the single DNA molecule level. Apart from revealing important information about the biophysics of NC and Gag when interacting with long DNA, our study also highlights how nanofluidic channels can be used to study reactions on DNA ends, which is not possible with most competing single DNA molecule techniques.
Table 1. Ability of NC and Gag and their mutants to form λ-DNA concatemers and concentration required to condense λ-DNA
Author ORCIDs
Fredrik Westerlund, 0000-0002-4767-4868
Acknowledgements
Dr. Rajhans Sharma is acknowledged for fruitful discussions regarding the manuscript layout and for proof-reading the manuscript.
Financial support
This project is funded by grants from the Swedish Research Council (no. 2015–5062) and the Olle Engqvist Byggmästare foundation to FW. This work was also supported by a grant from the Agence Nationale de la Recherche (ANR blanc PICO2) to YM.
Conflict of interest
None.








 Open access
Open access