


1. Introduction
1.1 Protein folding and metals
To understand biological function and dysfunction of proteins on a molecular level, the folded state structures of the involved proteins are important to define. But, in addition, thermodynamic and kinetic aspects of reaching these active folded structures are highly relevant. Most proteins fold spontaneously to unique and biologically-active three-dimensional structures in vitro. Notably, these folded states are not static structures; often structural dynamics play crucial roles in protein function (Olsson & Wolf-Watz, Reference Olsson and Wolf-Watz2010). The relatively small magnitude in the overall favorable stability of folded proteins means that spontaneous unfolding occurs with an occurrence such that continuous refolding reactions are important to maintain protein biological activity in vivo. Through groundbreaking in vitro and in silico work during several decades, using for example protein engineering (Matouschek et al. Reference Matouschek, Kellis, Serrano and Fersht1989) and the energy landscape concept (Bryngelson et al. Reference Bryngelson, Onuchic, Socci and Wolynes1995), substantial progress has been made to identify driving forces and mechanisms important for protein folding. In fact, the computational methodology has advanced to the point that a wide range of protein-folded structures can be designed from scratch with atomic-level accuracy (Huang et al. Reference Huang, Boyken and Baker2016).
In general, polypeptide folding is viewed as a random search of conformational space on a more (resulting in populated intermediates) or less (resulting in two-state reaction) rugged funneled-shaped energy surface. The energy surface for many small, single-domain proteins (<100 residues) is smooth and these proteins fold by cooperative two-state equilibrium and kinetic mechanisms (Jackson, Reference Jackson1998) with the formation of the transition state being rate-limiting for folding. As the transition state is at the highest energy, and generally short-lived, its structural characteristics must be concluded indirectly. The phi (ϕ) value approach (see below) is an experimental strategy for collecting residue-specific information about interactions in the folding transition state (de los Rios et al. Reference de los Rios, Muralidhara, Wildes, Sosnick, Marqusee, Wittung-Stafshede, Plaxco and Ruczinski2006; Matouschek & Fersht, Reference Matouschek and Fersht1991; Matouschek et al. Reference Matouschek, Kellis, Serrano, Bycroft and Fersht1990). Proteins longer than 100 residues often populate transient or equilibrium intermediate structures (Matthews, Reference Matthews1993; Roder & Colon, Reference Roder and Colon1997) which parallels that these proteins have a rugged energy surface. Folding intermediates may be on-pathway (i.e. facilitating the reaction to the folded state) or off-pathway (i.e. acting as traps that may lead to misfolding or aggregation) (Privalov, Reference Privalov1996).
30% or more of all folded proteins coordinate a metal ion to obtain its specific functionality (Gray, Reference Gray2003). In addition to the folding of the polypeptide chain, metal-binding proteins must also coordinate the metal ion to reach its active state. The cellular environments where metal-binding proteins are made contain the cognate metals either as a free metal ion in solution or bound to a specific metal-delivery protein (to be discussed further in subsequent sections for copper ions). It is thus reasonable to pose the question as to when during the polypeptide folding process the metal ion binds or is delivered. In addition, it is important to reveal what effects metal coordination has on the folded state properties such as conformational dynamics, structure, and stability. Copper (Cu) is the focus of this text and is an essential transition metal that we must obtain through the food. Cu with atomic number 29 in the periodic table has an electron configuration with the high ability both to accept and donate electrons. This redox property makes Cu suitable to transfer electrons in enzymatic reactions and the biologically important oxidation states are Cu(I) (i.e. reduced Cu) and Cu(II) (i.e. oxidized Cu). In this text, Cu ions are referred to simply as Cu; only when important is the redox state given. Cu is found in the active sites of proteins that participate in many basic life functions such as in cellular respiration, antioxidant defense, neurotransmitter biosynthesis, connective-tissue biosynthesis and pigment formation which all involve electron transfer steps (Harris, Reference Harris2003; Huffman & O'Halloran, Reference Huffman and O'Halloran2001; Puig & Thiele, Reference Puig and Thiele2002). We note that coordination of copper in protein active sites was excellently reviewed in 2014 (Liu et al. Reference Liu, Chakraborty, Hosseinzadeh, Yu, Tian, Petrik, Bhagi and Lu2014; Solomonet al. Reference Solomon, Heppner, Johnston, Ginsbach, Cirera, Qayyum, Kieber-Emmons, Kjaergaard, Hadt and Tian2014).
Metal coordination has been reported to stabilize as well as destabilize the folded states of copper proteins in vitro (Sedlak et al. Reference Sedlak, Ziegler, Kosman and Wittung-Stafshede2008a; Wittung-Stafshede, Reference Wittung-Stafshede2002, Reference Wittung-Stafshede2004). Importantly, it was demonstrated in vitro that several copper proteins can retain specific metal coordination also after polypeptide unfolding (Allen, et al. Reference Allen, Badarau and Dennison2013; Wittung-Stafshede, Reference Wittung-Stafshede2002, Reference Wittung-Stafshede2004; Wittung-Stafshede et al. Reference Wittung-Stafshede, Malmstrom, Winkler and Gray1998b). Thus the metal ion can remain bound upon protein unfolding and, in reverse, it may bind to the unfolded states of proteins. Based on this knowledge, it is tempting to speculate that metals may interact with their corresponding proteins prior to polypeptide folding in vivo and thus the metal may guide or modulate polypeptide folding (Fig. 1). To probe this, folding of metal-binding proteins must be investigated together with the cognate metal for a full molecular-mechanistic understanding. Although there has been a huge number of mechanistic studies of protein folding reactions in vitro, not as much focus has been directed to folding processes for metal-binding proteins.
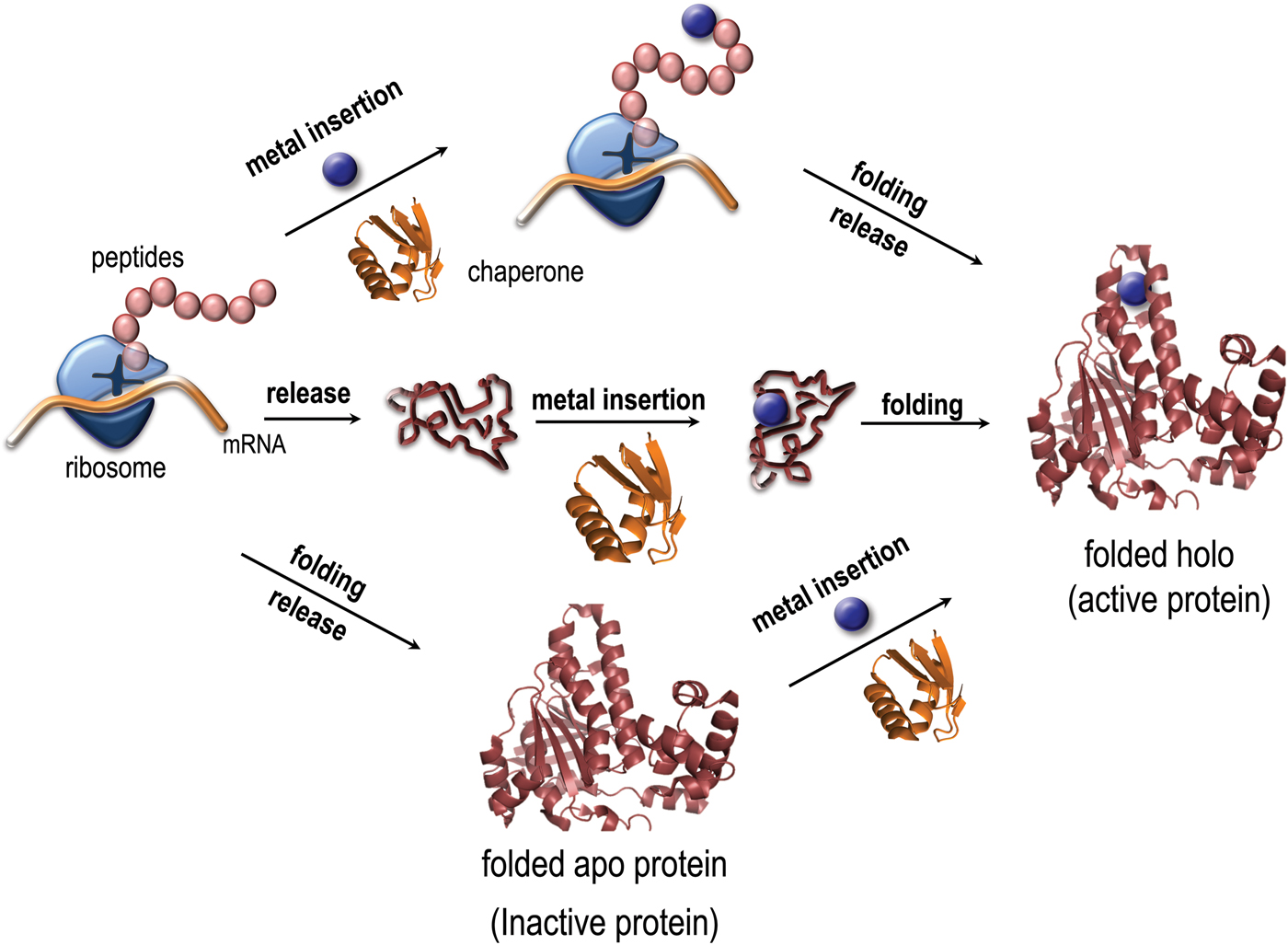
Fig. 1. Paths for folding and metal binding of copper proteins. Illustration of putative folding/binding mechanisms for copper proteins (adapted from Gomes, C. M. and P. Wittung-Stafshede (2010) Metal ion, Protein Folding, and Conformational States: An Introduction in Protein Folding and Metal Ions: Mechanisms, Biology and Disease (2010). Ed(s): C. M. Gomes and P. Wittung-Stafshede Florida, CRC Press: 3–11). The protein may be released from the ribosome followed by folding and the last step be metal insertion, or the polypeptide be released followed by metal insertion and subsequently, folding. It is also possible, that the metal is inserted at the ribosome, followed by release and folding. Intermediate mechanisms are also possible.
1.2 Scope of review
In this review, we will discuss the existing molecular-mechanistic findings on the roles of the metal in copper protein folding and stability in vitro (because of the limited number of studies, we discuss copper proteins from several organisms; Table 1). We will also describe Cu exchange between copper transport proteins, but here we limit ourselves to paths and proteins in human cells. Because there are no free Cu ions in human cells, how the metal is delivered to target Cu-dependent proteins by Cu transport proteins, often taking place via direct protein–protein interactions, becomes an integral part of the molecular understanding of copper protein folding in vivo. Our focus is on biophysical-biochemical aspects which include (but is not limited to) equilibrium and kinetic folding and unfolding reactions as well as thermodynamic and dynamic features of protein–protein and protein–metal interactions. Such information is often collected on purified protein samples in vitro using biophysical/spectroscopic methods (further described in the next section). Towards the end, we will describe (mainly dysfunctional) biological situations where aberrant copper loading promotes protein misfolding reactions that are linked to human pathology. Since the topic of this review overlaps with our research focus over many years; it is necessary to discuss many of our own papers but the aim is to summarize the key knowledge within this topic in an unbiased way.
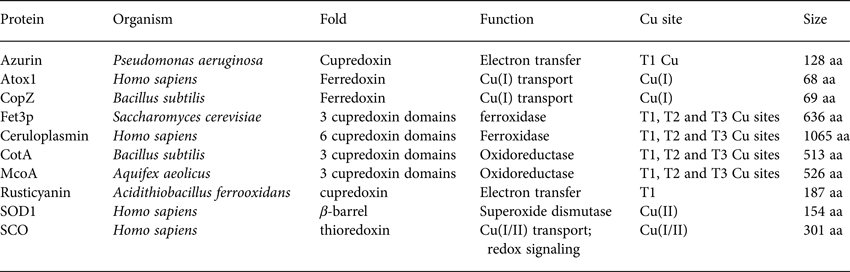
Table 1. Basic properties (name, organism, fold, function, Cu site and size) of the proteins discussed with respect to folding and stability in Section 2
1.3 Biophysical methods to study protein folding, stability and copper loading
In vitro studies of protein unfolding and refolding reactions often involve equilibrium and kinetic (stopped-flow mixing(Fersht, Reference Fersht1999)) experiments. Chemical denaturants such as urea and guanidine hydrochloride (GuHCl) or heat are used to perturb the protein structure in controlled ways (Maxwell et al. Reference Maxwell, Wildes, Zarrine-Afsar, De Los Rios, Brown, Friel, Hedberg, Horng, Bona, Miller, Vallee-Belisle, Main, Bemporad, Qiu, Teilum, Vu, Edwards, Ruczinski, Poulsen, Kragelund, Michnick, Chiti, Bai, Hagen, Serrano, Oliveberg, Raleigh, Wittung-Stafshede, Radford, Jackson, Sosnick, Marqusee, Davidson and Plaxco2005). Spectroscopic signals that report on different structural properties of the protein are then used to probe changes in conformation (Palm-Espling et al. Reference Palm-Espling, Niemiec and Wittung-Stafshede2012). Far-UV circular dichroism (CD) probes the secondary structure of proteins whereas near-UV CD probes tertiary structure and can also report on metal–protein interactions (such as Cu-cysteine coordination). Intrinsic fluorescence (excitation about 280 nm; emission 300–350 nm) probes aromatic residues, with tryptophan (Trp) dominating this signal and with the ability to report on exposure to solvent via its emission maximum wavelength. Metal binding to proteins can quench Trp fluorescence, if the metal center is close enough to the fluorophore and, thus, can be used to probe metal binding. Visible absorption is excellent for detection of Cu(II) binding to protein sites with cysteines (Cys), such as for example in blue copper proteins (vide infra), but Cu(I) does not exhibit much visible absorption when bound to proteins. However, Cu(I)-Cys coordination, as found in Cu chaperones, results in ligand-to-metal charge transfer with stronger absorption about 250 nm, which can be used to probe Cu transfer between proteins (Niemiec et al. Reference Niemiec, Weise and Wittung-Stafshede2012). In Fig. 2a–c , we show folded and unfolded visible absorption, far-UV CD and Trp fluorescence data for the blue copper protein azurin (to be further discussed below) along with an illustration of the structural parts in the protein that is responsible for the signals.

Fig. 2. Analysis of protein folding and stability in vitro. (a) Visible absorption of metal-ligand bonds reveals information on cofactor coordination (here folded, red and unfolded, blue, Cu-loaded azurin). (b) Fluorescence from aromatic residues reports on local environment, tertiary interactions and solvent exposure (here folded, red and unfolded, blue, apo-azurin). (c) Far-UV CD reports on the secondary structure content of proteins (here folded, red, and unfolded, blue, Cu-azurin). (d) Energy diagram for a protein folding with a two-state mechanism (folded and unfolded states are separated by a high energy barrier called the transition state). (e) Two-state equilibrium unfolding curve (fraction folded on y axis; chemical denaturant on x-axis). (f) A semi-logarithmic so-called Chevron plot of observed rate constants as a function of chemical denaturant for a two-state kinetic folding/unfolding reaction (here, data for apo-azurin). Reproduced with permission from (Palm-Espling, Niemiec et al. Reference Niemiec, Weise and Wittung-Stafshede2012).
In general, when probing folding reactions of proteins (with and without metal), one wants to combine a number of experimental detection methods to assess the folding/unfolding mechanism. In a two-state process (Fig. 2d ), the reaction appears the same regardless of the detection method and the resulting so-called equilibrium unfolding curve will appear as a sigmoidal two-state transition (Fig. 2e ) that can be fitted to give a free energy of unfolding and transition-midpoint denaturant concentration. The beauty of spectroscopic methods is that they rather easily can be combined with rapid mixing devices that can be used to trigger folding/unfolding with millisecond time resolution. Most often kinetics of folding (low denaturant) and unfolding (high denaturant) processes are probed as a function of denaturant concentration using a stopped-flow mixing device (Fig. 3a ) equipped with fluorescence or CD detection. The logarithms of the detected rate constants are plotted as a function of denaturant concentration to generate a so-called Chevron plot, where folding and unfolding rate constants (and thus also equilibrium folding free energy) extrapolated to zero denaturant can be determined (Fig. 2f ). The slopes of the two arms of the Chevron plot also contain information on the transition state placement along the folding reaction coordinate.

Fig. 3. Analysis of folding transition state structure. (a) Scheme of stopped-flow mixing set up. Protein unfolded in high denaturant is mixed with buffer to reach a lower denaturant concentration such that the conditions now favor folding. Structural change is probed by spectroscopic changes as a function of time. (b) Energy diagram for a protein folding with a two-state mechanism (folded, unfolded and transition states indicated as U, F and TS) showing changes in transition state and folded state energies for the extreme ϕ-values of 1 (blue) and 0 (red). For a ϕ-value of 1 (blue), the energetic change of the transition state (i.e. effects on folding rates; ΔΔGTS−U) matches that of the folded state (i.e. effect on folded state stability; ΔΔGF−U), implying that the mutated residue makes native-like contacts already in the folding transition state. For a ϕ-value of 0 (red), there is no effect on folding rate constants (i.e. ΔΔGTS−U = 0) as the residue mutated only make native-like contacts late in the folding reaction or only in the folded state. ϕ-values may also be fractional values between 0 and 1, and may then imply partial structure formed in the TS.
An important experimental strategy for obtaining residue-specific information on interactions in the folding transition state is the so-called ϕ-value approach (de los Rios et al. Reference de los Rios, Muralidhara, Wildes, Sosnick, Marqusee, Wittung-Stafshede, Plaxco and Ruczinski2006; Matouschek & Fersht, Reference Matouschek and Fersht1991; Matouschek et al. Reference Matouschek, Kellis, Serrano, Bycroft and Fersht1990). In such experiments, the transition state is probed by measuring the kinetic and thermodynamic effects of hydrophobic-to-alanine mutations introduced in different regions of the protein. The ϕ-values represent the change in stability of the transition state accompanying the mutation of a residue relative to the effect of the same mutation on the stability of the native state, assuming there is no effect on the unfolded state by the mutation (illustrated in Fig. 3b ). A ϕ equal to 1 suggests that the corresponding residue makes interactions that contribute equally to the stability of the transition and folded states (i.e. native-like interactions in the transition state). In contrast, a ϕ equal to 0 indicates that the corresponding residue forms very few interactions with other residues in the transition state. Fractional ϕ-values may be interpreted as residues forming a partial structure in the folding transition state. Several small proteins that fold by two-state kinetic mechanisms have been subjects of mutagenesis to obtain a picture of the transition state structure for folding with residue-specific resolution. Such in vitro studies have suggested two classes of transition states for folding: diffuse transition states, in which most side chains have similar relatively low ϕ-values, and polarized transition states that display distinct substructures with very high ϕ-values.
For equilibrium measurements, in addition to spectroscopic methods, calorimetry is useful as it reports on the heat change of the reaction, which does not require any probe molecule (such as a fluorophore). Here differential scanning calorimetry is used to probe thermal unfolding whereas isothermal titration calorimetry (ITC) is used to study interactions between a protein and a ligand, such as a metal ion or a partner protein, at a fixed condition/temperature (that can be changed) (examples in Fig. 4a, b ). Another way to probe metal–protein interactions or metal-mediated protein–protein interactions, which also gives kinetic information, is surface plasmon resonance measurements (e.g. Biacore). However, here one species has to be attached to a surface and is not in true solution equilibrium; moreover, this method depends on mass changes so binding of small ligands, such as a free Cu ion, is borderline to what is possible to detect. Size exclusion chromatography (SEC), being a trivial method during protein purification, has turned out to be very useful when it comes to Cu-dependent protein–protein interactions. Because some of the Cu chaperones and Cu-binding domains elute differently in SEC, despite similar sizes, the proteins can be separated by SEC and, using dual wavelength absorption detection, apparent thermodynamic parameters for Cu transfer can be estimated.

Fig. 4. Biophysical methods to probe copper protein stability, metal-protein interactions and protein/metal site properties. (a) Example of typical DSC trace showing protein unfolding. (b) Example of ITC data for titration of ligand to protein. (c) HSQC NMR spectra of 15N-labeled human Atox1 (apo, black; Cu(I)-loaded, red). (d) Typical EPR spectrum of blue copper protein (here, spinach plastocyanin (Xue et al. Reference Xue, Okvist, Hansson and Young1998) at 140 K).
Of course, many additional biophysical, biochemical and analytical methods are used to complete specific investigations, for example, metal analysis by ion-coupled plasma mass spectrometry (ICP-MS) is crucial to directly prove in what sample (after separation of species) the Cu ions are. Solution NMR is an important method used in many of the studies described in this text (Smith et al. Reference Smith, Zhang, Pielak and Li2015). NMR can fill up a review on its own; here, we only note a few key points. Using 15N-labeled protein, two-dimensional heteronuclear single quantum correlation (HSQC) experiments can give residue specific information and thus reveal folded versus unfolded as well as, for metal-binding proteins, the specific residues affected by metal binding. This approach is straightforward for apo-forms of copper proteins and for Cu(I)-loaded proteins since Cu(I) is diamagnetic (examples in Fig. 4c ). Using strategically-designed NMR experiments, one may probe metal transfer between proteins/peptides and metal-induced folding/unfolding. Oxidized Cu is paramagnetic and broadens NMR signals due to the long electron spin relaxation time of Cu(II), and in the past, this was thought to preclude the study of Cu(II) ligands in for example blue copper proteins. In recent years, the adaption of new multidimensional NMR techniques to paramagnetic proteins provide new possibilities. For example, tailored pulse sequences like super-WEFT allow direct observation of the hyperfine-shifted signals of Cu(II) in blue copper proteins (Kalverda et al. Reference Kalverda, Salgado, Dennison and Canters1996; Vila et al. Reference Vila, Ramirez, Di Bilio, Mizoguchi, Richards and Gray1997). It was also revealed that signals corresponding to metal ligands of Cu(II) can be assigned by performing saturation transfer experiments in a sample of approximately a one-to-one ratio of oxidized and reduced forms of blue copper proteins. This concept relies on the fact the electron-self exchange rate between the Cu redox states at equilibrium is of the right order of magnitude to allow transfer of magnetization between a nucleus in the oxidized species and the same nucleus in the reduced species, when the former is subjected to a continuous wave irradiation in a 1D NMR experiment (Bertini et al. Reference Bertini, Fernández, Karlsson, Leckner, Luchinat, Malmström, Nersissian, Pierattelli, Shipp, Valentine and Vila2000).
A key method to characterize electronic and geometric properties of active sites in folded Cu proteins is electron paramagnetic resonance (EPR). EPR can be used to probe Cu(II) sites or mixed Cu(I)/Cu(II) sites in proteins (an example of EPR signal for type 1 copper protein in Fig. 4d ). Blue copper proteins, binuclear copper proteins, and multi-copper oxidases all have distinct EPR spectral features relative to normal cupric complexes (Solomon et al. Reference Solomon, Baldwin and Lowery1992), which can reveal detailed information about coordination environment and electronics of copper active sites (Culpepper et al. Reference Culpepper, Cutsail, Gunderson, Hoffman and Rosenzweig2014). In recent years, pulsed EPR experiments (e.g. double electron–electron resonance, DEER) have emerged that can measure nanometer distances between two attached paramagnetic centers as well as observe dynamics of protein chains (Shenberger et al. Reference Shenberger, Shimshi and Ruthstein2015). Thus, in addition to probing a paramagnetic metal site in a metal-binding protein, EPR approaches are nowadays used to investigate distances in metal-free proteins using spin labels attached to strategically-placed cysteine residues. We note that as a complement to in vitro experiments, in silico molecular dynamic (MD) simulations using all-atom models of the proteins as well as quantum mechanical (QM) calculations on Cu sites have proven useful (Li et al. Reference Li, Wang, Zhang and Wang2015).
For all in vitro folding, stability and copper exchange experiments, pure protein samples are of uttermost importance. Thus, a lot of effort has been spent on developing good purification schemes for each protein variant and assuring the chemical/structural identity of each purified protein. For metal exchange between copper proteins, one must consider inorganic chemical aspects of Cu redox chemistry. Cu(I) is not soluble and will rapidly be oxidized to Cu(II) if no reducing agent or anaerobic conditions are present. Special care has to be taken to keep conditions anaerobic and one should keep in mind that reducing-agent molecules themselves may interact with the Cu(I) ions, also when bound to proteins.
2. Copper protein folding and stability in vitro
2.1 Azurin
Pseudomonas aeruginosa azurin is an excellent model for basic biophysical folding/binding studies (Fig. 5a ). It is a small (128 residues) blue-copper (i.e. intense absorption at about 630 nm, see Fig. 2a ) protein that is believed to facilitate electron transfer in de-nitrification and/or respiration chains (Adman, Reference Adman1991). Azurin has one α-helix and eight β-strands that fold into a β-barrel structure arranged in a double-wound Greek-key topology (Adman, Reference Adman1991; Nar et al. Reference Nar, Messerschmidt, Huber, van de Kamp and Canters1992). In Pseudomonas aeruginosa azurin, the redox-active copper (Cu(I)/Cu(II)) coordinates two histidine imidazoles (His46 and His117), one cysteine thiolate (Cys112) and two weaker axial ligands, sulfur of methionine (Met121) and carbonyl of glycine (Gly45), in a trigonal bipyramidal geometry, Fig. 5. The protein structure appears to define the geometry of the metal site, leading to an unusual Cu(II) coordination in azurin, and also in other blue-copper proteins (Wittung-Stafshede et al. Reference Wittung-Stafshede, Hill, Gomez, Di Bilio, Karlsson, Leckner, Winkler, Gray and Malmstrom1998a). P. aeruginosa azurin can bind different transition metals, such as zinc (Zn), in the active site in vitro. High-resolution crystal structures of azurin have demonstrated that the three-dimensional structure is identical with and without a metal (Cu or Zn) ion in the active site (Nar et al. Reference Nar, Messerschmidt, Huber, van de Kamp and Canters1991, Reference Nar, Messerschmidt, Huber, van de Kamp and Canters1992). Thus, the metal is not needed for obtaining a folded structure, and metal insertion may be viewed as a ‘lock and key’ mechanism.

Fig. 5. Ribbon illustrations of Cu proteins discussed with respect to in vitro folding. Cu ions are shown as blue spheres. Also shown are enlargements of Cu sites with involved coordination residues noted (for C and D, the T1, T2 and T3 Cu sites are similar and here shown only for Fet3p). (a) P. aeruginosa azurin, 3FSA, (b) human Atox1, 1TL4, (c) human ceruloplasmin, 4ENZ and (d). yeast Fet3p, 1ZPU.
From equilibrium-unfolding experiments of Cu(II) and Cu(I) forms of azurin, two-state like transitions were observed for both metal forms, but azurin with oxidized Cu was more stable than azurin with reduced Cu. Surprisingly, it was discovered that the metal (either Cu or Zn) remained bound to the polypeptide after unfolding (Leckner et al. Reference Leckner, Wittung, Bonander, Karlsson and Malmstrom1997). Based on the difference in thermodynamic stability of the two redox forms of Cu azurin, the reduction potential of Cu in unfolded azurin was predicted to be 0·13 V higher than that for Cu in folded azurin (Leckner et al. Reference Leckner, Wittung, Bonander, Karlsson and Malmstrom1997). Using cyclic-voltammetry experiments on folded and unfolded Cu azurin, this prediction was later on confirmed (20°C, pH 7) (Wittung-Stafshede et al. Reference Wittung-Stafshede, Hill, Gomez, Di Bilio, Karlsson, Leckner, Winkler, Gray and Malmstrom1998a). The high Cu reduction potential in unfolded azurin was rationalized by a favorable trigonal Cu(I)-coordination. Upon combining extended X-ray absorption fine structure (EXAFS), single-site azurin mutants and small model peptides, we managed to elucidate that Cys112, His117 and Met121 are the three Cu ligands in unfolded azurin (Marks et al. Reference Marks, Pozdnyakova, Guidry and Wittung-Stafshede2004; Pozdnyakova et al. Reference Pozdnyakova, Guidry and Wittung-Stafshede2001). Thermodynamic cycles connecting folded and unfolded apo and metal-loaded forms of azurin demonstrated that the metals stabilize folded azurin substantially. Folded Zn-, Cu(I)-, and Cu(II)-forms of azurin have thermodynamic stabilities of 39, 40, and 52 kJ/mol, respectively, whereas the stability of apo-azurin is only 29 kJ/mol (pH 7, 20°C) (Leckner et al. Reference Leckner, Wittung, Bonander, Karlsson and Malmstrom1997; Pozdnyakova & Wittung-Stafshede, Reference Pozdnyakova and Wittung-Stafshede2003; Pozdnyakova et al. Reference Pozdnyakova, Guidry and Wittung-Stafshede2002).
To investigate putative folding and loading paths towards active azurin (i.e. folded protein with Cu(II) in the metal site), we investigated the two extreme scenarios: Cu(II) binding before polypeptide folding (Path 1) and copper-binding after polypeptide folding (Path 2), Fig. 6a . The folding and unfolding kinetics for apo-azurin follow two-state behavior, showing single-exponential decays in stopped-flow mixing experiments (Pozdnyakova & Wittung-Stafshede Reference Pozdnyakova and Wittung-Stafshede2001a, Reference Pozdnyakova and Wittung-Stafshedeb, Reference Pozdnyakova and Wittung-Stafshede2003). The extrapolated folding time in the water for apo-azurin is fast (τ ~7 ms). In contrast, Cu uptake by folded apo-azurin, to govern active azurin through Path 2, is slow (i.e. τ~min-to-hours depending on protein-to-copper excess) (Pozdnyakova & Wittung-Stafshede, Reference Pozdnyakova and Wittung-Stafshede2001a, Reference Pozdnyakova and Wittung-Stafshedeb). Notably, when the polypeptide folds in the presence of Cu, the formation of active (i.e. Cu-loaded) azurin is much faster than when the Cu is added to the folded protein (Pozdnyakova & Wittung-Stafshede, Reference Pozdnyakova and Wittung-Stafshede2001a, Reference Pozdnyakova and Wittung-Stafshedeb). Thus, when Cu is present in the unfolded state, formation of active azurin follows Path 1, with rapid Cu binding in the unfolded state followed by subsequent folding of the Cu-polypeptide complex (Fig. 6a ). Inspection of the time scales shows that upon introducing Cu prior to protein folding (in contrast to after apo-protein folding), active azurin is formed more than 1000-fold faster in vitro (Pozdnyakova & Wittung-Stafshede, Reference Pozdnyakova and Wittung-Stafshede2001a, Reference Pozdnyakova and Wittung-Stafshedeb). As minimization of biosynthesis time of a functional protein is an important factor in biology, although other constraints will exist, Cu coordination before folding may be the relevant biosynthetic pathway to functional azurin in a living cell. Alternatively, since apo azurin can fold without metal, the results may imply that Cu uptake by the apo form is coupled to transient unfolding.

Fig. 6. Possible folding paths and energy landscapes for azurin. (a) Two extreme paths to folded, Cu-loaded azurin from unfolded protein and free Cu(II) ions. Path 1 (green), Cu binding before protein folding; Path 2 (red), protein folding before Cu binding (F, folded, U, unfolded). (b) Illustration showing an activation barrier that unifies the kinetic behaviors determined experimentally for apo- and zinc-forms of azurin. A small pointed feature in apo-azurin's free-energy profile (dashed curve) may account for a fixed transition state (TSapo). The presence of zinc suppressed this high-energy feature and reveals the underlying broad activation barrier (solid curve). This flat and broad barrier results in a moving transition state (TSZn) for zinc-substituted azurin as a function of denaturant concentration. (Figure B adapted from (Wilson & Wittung-Stafshede, Reference Wilson and Wittung-Stafshede2005a, Reference Wilson and Wittung-Stafshedeb)).
The folding-transition states of azurin without and with metal were analyzed by ϕ-value analysis. Here Zn was used instead of Cu as the holo-form of azurin; Zn(II) ions are redox-inert and therefore one avoids unwanted redox reactions that may take place in the presence of Cu and may impede the measurements. The first target of analysis was eight core residues found by others to be so-called ‘structural determinants’ in most of all sandwich-like proteins. Sandwich-like proteins are a large family that includes blue-copper proteins such as azurin (Kister et al. Reference Kister, Finkelstein and Gelfand2002). Interestingly, when investigated in folding of apo azurin, half of the structural determinant residues were important for the folding mechanism (i.e. high ϕ-values) whereas the other half contributed mostly to folded-state stability (Wilson & Wittung-Stafshede Reference Wilson and Wittung-Stafshede2005a, Reference Wilson and Wittung-Stafshedeb; Zong et al. Reference Zong, Wilson, Shen, Wolynes and Wittung-Stafshede2006). This observation directly implies that residues may have been conserved for mechanistic reasons, which is a topic of controversy in the folding community. To obtain a deeper understanding of apo-azurin's folding transition state, the ϕ−value analysis was extended to 18 positions covering all secondary structure elements present in folded azurin. Based on the gathered equilibrium and kinetic data for these variants, apo-azurin's folding nucleus is centered at a few core residues (e.g. Val31, Leu33 and Leu50) which have ϕ-values of 1 (Chen et al. Reference Chen, Wilson, Wu, Wittung-Stafshede and Ma2006; Wilson & Wittung-Stafshede Reference Wilson and Wittung-Stafshede2005a, Reference Wilson and Wittung-Stafshedeb). In contrast to apo-azurin, the Chevron plot with folding and unfolding rate constants versus denaturant concentration for Zn-bound azurin exhibited distinct curvature in both folding and unfolding arms (Pozdnyakova & Wittung-Stafshede, Reference Pozdnyakova and Wittung-Stafshede2003). After testing various possibilities, the curvature was assigned to transition state movement as a function of denaturant concentration. At low concentrations of denaturant, the transition state occurs early in the folding reaction and thus has little structure, whereas at high denaturant concentration it moves closer to the native structure and involves the formation of significant native-like ordering. Using the denaturant-dependent ϕ-values for the different Zn-substituted variants it was possible, therefore, to probe the growth of the transition-state structure with residue-specific resolution. From this, it was concluded that during folding of Zn azurin, delocalized interactions are formed first throughout the azurin polypeptide. These then gradually grow more native-like around a few residues that are centered in the core of the folded structure (Wilson & Wittung-Stafshede, Reference Wilson and Wittung-Stafshede2005a, Reference Wilson and Wittung-Stafshedeb).
The divergent kinetic behavior for the two forms of azurin (apo, fixed transition state, and Zn, moving transition state) was rationalized as a small alteration on a common free energy profile with a broad activation barrier (Fig. 6b ). The fixed transition state for apo-azurin was proposed to be a result of a small pointed feature projecting from the top of the otherwise broad free energy profile. Somehow, the presence of Zn suppresses this local pointed feature and the underlying smooth activation barrier appears. The reason for the high-energy feature in the apo-protein's energy landscape was later explained, using a new set of ϕ-values, by complete water removal in the apo-protein folding-transition state (corresponding to a higher energy barrier) whereas the folding-transition state of Zn-bound azurin retained weakly bound water molecules (Wilson et al. Reference Wilson, Apiyo and Wittung-Stafshede2006).
2.2 Atox1
Homo sapiens Atox1 (also discussed in 5·3) is a 68-residue Cu chaperone with a ferredoxin-like fold (Fig. 5b ), and a conserved MXCXXC motif that binds one Cu(I) via the two Cys residues (Arnesano et al. Reference Arnesano, Banci, Bertini, Ciofi-Baffoni, Molteni, Huffman and O'Halloran2002; Robinson & Winge, Reference Robinson and Winge2010; Wernimont et al. Reference Wernimont, Huffman, Lamb, O'Halloran and Rosenzweig2000). In vitro unfolding and stability parameters were compared between human Atox1 and the bacterial homolog Bacillus subtilis CopZ with and without Cu (Hussain & Wittung-Stafshede, Reference Hussain and Wittung-Stafshede2007). Equilibrium unfolding of both apo- and Cu(I)-forms of CopZ and Atox1, induced by GuHCl and thermal perturbation, were found to be reversible two-state reactions. Despite the same overall folded structure, the apo-form of Atox1 has more than 20°C higher thermal stability as compared with the apo-form of CopZ. This difference may be explained on a molecular level by variations in surface-charges between the proteins (Arnesano et al. Reference Arnesano, Banci, Bertini, Ciofi-Baffoni, Molteni, Huffman and O'Halloran2002). Despite the Cu site being located in a loop at the protein surface, Cu binding increases the thermal and chemical stability of the two proteins, with the largest effect of Cu found for CopZ. Thus, binding of Cu strengthens interactions throughout the protein structure that increases the overall stability (Hussain & Wittung-Stafshede, Reference Hussain and Wittung-Stafshede2007).
To address the stability differences between Atox1 and CopZ on a molecular level, a combination of QM molecular mechanics (QM-MM) and MD computational methods in silico was applied (Rodriguez-Granillo & Wittung-Stafshede, Reference Rodriguez-Granillo and Wittung-Stafshede2008). The QM-MM optimized Cu(I) geometries were found to differ between the two Cu-forms of the proteins despite identical Cu ligands. Cu(I) in Atox1 favors a linear Cys(S)-Cu-Cys(S) arrangement whereas it is bent with an angle close to 150° in CopZ. Both proteins become less dynamic in the presence of Cu and this is most pronounced in CopZ, which matches the in vitro stability data. Both average fluctuation and radius of gyration data demonstrate that the consequences of Cu binding extend throughout the proteins. For both proteins, a bi-phasic distribution of Cys(S)-Cys(S) distances was detected in the apo-forms that were separated by 2–4 kJ/mol barriers. It was proposed that the conformations with long Cys(S)-Cys(S) distances may play a role in Cu uptake and release (Rodriguez-Granillo & Wittung-Stafshede, Reference Rodriguez-Granillo and Wittung-Stafshede2008).
Several residues that are not directly binding to the Cu are conserved in this group of proteins. For example, Met10 in the Cu-binding loop is fully conserved and Lys60 in Atox1 is always a Lys in eukaryotes but a Tyr in prokaryotes. In the yeast Atox1 homolog, Atx1, this Lys residue was shown to be important for Cu delivery and antioxidant activity (Portnoy et al. Reference Portnoy, Rosenzweig, Rae, Huffman, O'Halloran and Culotta1999). Also, in the crystal homo-dimer structure of human Atox1, Lys60 was involved in hydrogen bonding across the dimer interface (Wernimont et al. Reference Wernimont, Huffman, Lamb, O'Halloran and Rosenzweig2000). These residues may be conserved because they play roles in Cu chaperone stability, structural dynamics or target interactions and some studies have addressed this. Cu release from wild-type Atox1 and two-point mutants (Met10Ala and Lys60Ala Atox1) was characterized using the Cu(I) chelator, BCA (bicinchonic acid) as the metal acceptor. Here, it was found that BCA removes Cu from Atox1 in a three-step process involving bimolecular formation of an initial Atox1-Cu-BCA complex followed by dissociation of Atox1 and the binding of a second BCA to generate apo-Atox1 and Cu-BCA2 (Hussain et al. Reference Hussain, Olson and Wittung-Stafshede2008). Although BCA is a small molecule, the copper-bridged protein-chelator complex may kinetically mimic a ternary chaperone-Cu-target protein adduct involved in metal transfer in vivo. It was discovered that both Atox1 mutants lose Cu more readily than wild-type Atox1 due to the more rapid displacement of the protein from the Atox1-Cu-BCA intermediate by the second BCA. In addition, unrelated to the mutations, it was noted that Cu(I) uptake from solution by BCA was much slower than the transfer of Cu from holo-Atox1 to BCA (Hussain et al. Reference Hussain, Olson and Wittung-Stafshede2008). This result suggests that Cu chaperones play a role in making Cu kinetically accessible to substrates in vivo, in addition to providing transfer target specificity.
The mutated Atox1 variants mentioned, including also Thr11Ala since position 11 is conserved as Thr in all eukaryotes and Lys60Tyr since this position is a Tyr in prokaryotes, were also subjected to MD simulations (Rodriguez-Granillo & Wittung-Stafshede, Reference Rodriguez-Granillo and Wittung-Stafshede2009a, Reference Rodriguez-Granillo and Wittung-Stafshedeb). Surprisingly, both apo- and holo-Atox1 become more rigid when either Thr11 or Lys60 was exchanged for Ala, implying that these residues introduce protein flexibility. Moreover, Lys60 and Thr11 participate in electrostatic networks that stabilize the Cu-bound form and, in the apo-form, modulate the solvent exposure of the two Cu-binding Cys residues. In contrast, an Ala substitution of Met10, which is buried in the hydrophobic core of Atox1, results in a protein with more structural dynamics than the wild-type protein. Comparable trends in rigidity, structural dynamics and interaction networks were found in the corresponding point-mutated CopZ variants (Rodriguez-Granillo & Wittung-Stafshede, Reference Rodriguez-Granillo and Wittung-Stafshede2009a, Reference Rodriguez-Granillo and Wittung-Stafshedeb), suggesting general relevance.
2.3 Ceruloplasmin and Fet3p
The roles of Cu in folding and stability of multi-domain protein structures, and with multiple Cu sites, have been elucidated in depth for two multi-copper oxidases (MCOs): human ceruloplasmin and yeast Fet3p (Fig. 5c, d ). Unfolding reactions in vitro are irreversible for both these proteins but nonetheless one may extract a great deal of information from such experiments as will be explained below. More than 1000 proteins have been identified as MCOs based on the multiple copies of the cupredoxin motifs (i.e. same fold as in azurin) they contain (Kosman, Reference Kosman2002). Most of the known MCO proteins harbor three cupredoxin domains (but notably the human version has six) and they contain four Cu ions distributed in three distinct metal sites (Kosman, Reference Kosman2002).
Homo sapiens ceruloplasmin (CP) plays a key role in iron metabolism in humans due to its ability to oxidize Fe(II) to Fe(III) which allows for subsequent incorporation of Fe(III) into apo-transferrin (Hellman & Gitlin, Reference Hellman and Gitlin2002). CP is a single polypeptide chain arranged into six cupredoxin domains with one type-1 (T1, blue copper) Cu in each of domains 2, 4 and 6; the remaining Cu ions form a catalytic tri-nuclear cluster, one type-2 (T2) Cu and two Cu ions in a type-3 (T3) site, at the interface between domains 1 and 6 (Fig. 5). Urea-induced unfolding of holo- and apo-forms of CP has been investigated by a range of biophysical probes (pH 7, 20 °C) (Sedlak & Wittung-Stafshede, Reference Sedlak and Wittung-Stafshede2007). The resulting data showed that holo-CP unfolds in a multi-step process at these conditions with at least one equilibrium intermediate species populated. Formation of the intermediate was reversible and correlated with decreased secondary structure (far-UV CD), exposure of aromatic residues (TRp fluorescence), loss of two coppers (blue color) and reduced (but not absent) oxidase activity. Further additions of urea triggered complete unfolding of the protein and loss of all copper ions. Attempts to refold this species resulted in an inactive apo-protein with molten-globule structural features. The apo-form of CP also unfolds in a multi-step reaction and for this form, correct refolding was again possible from the intermediate but not from the fully unfolded state. Taken together, the observations show that CP unfolding involves equilibrium intermediates and the copper ions are removed in steps (Sedlak & Wittung-Stafshede, Reference Sedlak and Wittung-Stafshede2007), depicted in Fig. 7. When the catalytic Cu sites (T2 and T3) is finally destroyed, protein refolding is not possible at neutral pH. If one reverses the unfolding process and consider CP refolding, it implies a mechanistic role for the tri-nuclear Cu cluster as a necessary nucleation point, perhaps aligning domains 1 and 6 spatially.
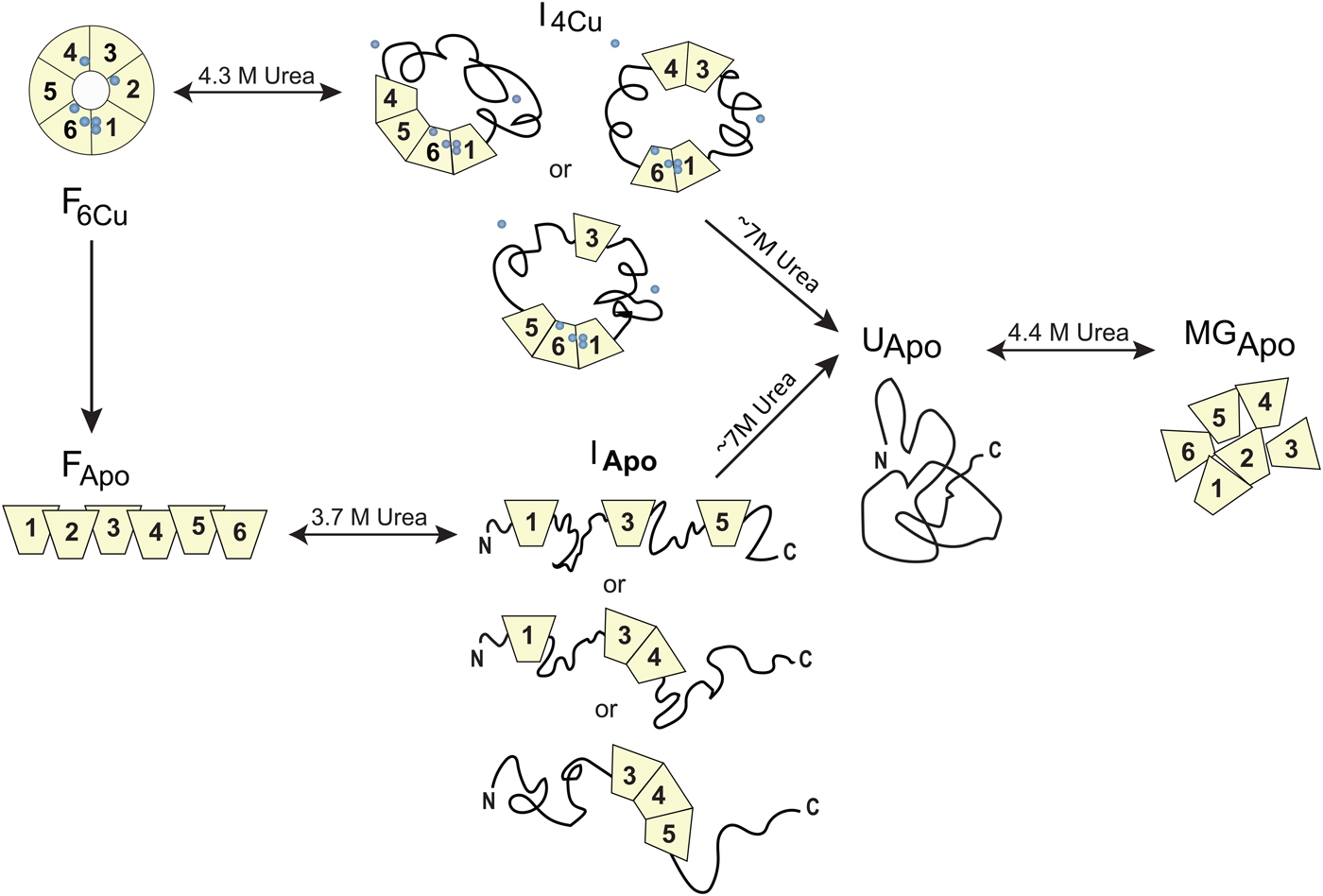
Fig. 7. Chemically-induced unfolding of holo- and apo-CP. Folded holo-CP containing six bound coppers (circles), F6Cu, unfolds partially in a reversible transition with a midpoint at ~4·3 M urea to an intermediate form (I4Cu) in which it has lost two Cu and about half of the oxidase activity. The catalytic cluster and therefore domains 1 and 6 are likely intact in the intermediate. Further increase in urea concentration leads to full CP unfolding accompanied by loss of all coppers, UApo. Analogously, apo-CP, FApo, undergoes a reversible urea-induced transition with a midpoint at ~3·7 M to an intermediate state (IApo), followed by an irreversible transition to UApo upon addition of more urea. The final unfolded state of apo-CP is similar to that obtained when starting from holo-CP. Upon attempts to refold, UApo undergoes a reversible off-pathway transition to a molten globule-like state, termed MGApo. Double-headed arrows indicate reversibility; single-headed arrows depict transitions that only go in one direction. Figure adapted from (Sedlak & Wittung-Stafshede, Reference Sedlak and Wittung-Stafshede2007).
Both apo- and holo-forms of CP undergo irreversible thermal reactions to denatured states with a significant residual structure based on far-UV CD. The spectroscopic signals for the thermally-denatured species are similar to the dead-end molten globule found in the urea experiments. The role of the Cu ions in thermal stability of CP was therefore assessed upon probing the thermal unfolding process as a function of scan rate (Sedlak et al. Reference Sedlak, Zoldak and Wittung-Stafshede2008b). For identical scan rates of heating, the apparent thermal midpoint appears at 15–20 °C higher temperatures for the holo- as compared with the apo-form of CP. The thermal transitions for both CP forms were the best fit by a mechanistic model involving two consecutive, irreversible steps (N→I→D) (Sedlak et al. Reference Sedlak, Zoldak and Wittung-Stafshede2008b). The thermal holo-intermediate, I, has lost two Cu ions (including one blue T1 Cu) and secondary structure in at least one cupredoxin domain; however, the tri-nuclear Cu cluster remains intact as this species is functional in oxidase activity. Thus, the urea-induced and thermal intermediates appear similar. The activation parameters obtained from fits to the thermal transitions versus scan rate date allowed assessment of the kinetic stability of apo- and holo-forms of CP at a physiological temperature of 37°C (Sedlak et al. Reference Sedlak, Zoldak and Wittung-Stafshede2008b). From this analysis, it emerged that native CP (i.e. with 6 coppers) is rather unstable and converts to I in less than a day at 37 °C. Nonetheless, the I form remains intact for over 2 weeks and may thus be a biologically-relevant state of CP in vivo. In the absence of all coppers, CP is more unstable: apo-CP is completely unfolded in less than 2 days at 37 °C. The low kinetic stability of apo-CP may correlate with its rapid degradation in vivo in various disease conditions where Cu loading is blocked.
Saccharomyces cerevisiae Fet3p has been used to study the role of each Cu site in unfolding/stability of an MCO system. Like typical MCOs, S. cerevisiae Fet3p contains 3 cupredoxin domains, and four Cu ions in three metal sites (T1 Cu in domain 3; T2 Cu and the binuclear T3 Cu site at the interface between domains 1 and 3) (Fig. 5). In contrast to CP, a collection of de-metallated forms of Fet3p has been prepared and electronic properties of each Cu site have been characterized (Kosman, Reference Kosman2002; Palmer et al. Reference Palmer, Quintanar, Severance, Wang, Kosman and Solomon2002). The partially-metallated Fet3p forms are named T1D, T2D and T1D/T2D (where D means depleted), in which the T1, T2, or both the T1 and T2 Cu ions, respectively, are absent due to specific mutations. Thermal unfolding processes of the Fet3p variants were monitored by spectroscopic and calorimetric methods in vitro at pH 7 (Sedlak et al. Reference Sedlak, Ziegler, Kosman and Wittung-Stafshede2008a). In analogy to CP, thermal unfolding processes of apo- and all the different Cu-loaded forms of Fet3p are irreversible. It was found that the domains in apo-Fet3p unfold sequentially with Tm values of 45, 62 and 72 °C (1 K/min). Presence of the T3 Cu (i.e. the T1D/T2D variant) imposes strain in the apo-structure that results in coupling of the unfolding of the three domains and low overall stability (Tm of 50 °C; 1 K/min). The inclusion of both T3 and T2 Cu ions (i.e. the T1D variant) increases overall Fet3p stability by ~5 °C but the unfolding process remains coupled in a single step. The introduction also the T1 Cu, producing fully-loaded holo-Fet3p, stabilizes domain 3 and this uncouples the unfolding transitions of the domains. For the wild-type holo-form, unfolding of domain 2 occurs first along with Cu-site perturbations (Tm 50–55 °C; 1 K/min), followed by unfolding of the remaining domains 1 and 3 (~65–70 °C; 1 K/min). That unfolding of domains 1 and 3 are coupled is most likely due to the T3 coppers ‘stitching’ these domains together. The data articulate the importance of the T3 Cu site in holding the Fet3p trimeric structure together (Sedlak et al. Reference Sedlak, Ziegler, Kosman and Wittung-Stafshede2008a) which may be a general feature of MCO proteins as the same conclusion was made for CP. Recent urea-induced unfolding experiments of Fet3p Cu-site variants, globally analyzed, support the proposed multi-step unfolding mechanism also when denaturant is used as the method to perturb the system (Sedlák et al. Reference Sedlák, Žoldák and Wittung-Stafshede2018). The closed structural arrangement of cupredoxin domains is required for Fet3p function but, nonetheless, results in loss of overall protein stability as compared with the nonfunctional but folded apo-form of Fet3p. Fet3p, thus, is an example of a biological situation in which trade-offs between stability and structure is facilitated by metal ions to obtain the desired functionality.
2.4 Other copper proteins
Only for a few other copper proteins (CotA, McoA, rusticyanin, SOD1 and SCO) have in vitro folding and stability data been reported in the literature. Of these additional proteins, CotA and McoA are MCO proteins like CP and Fet3p and rusticyanin is a blue-copper protein like azurin.
Quite an extensive folding and stability studies have been made on the laccase protein CotA from Bacillus subtilis. Similar to the other MCO proteins (e.g. Fet3p), CotA has three cupredoxin domains and accommodates 4 Cu ions in T1 and T2/T3 Cu sites (Enguita et al. Reference Enguita, Martins, Henriques and Carrondo2003). Recombinant CotA can be produced in Escherichiacoli in copper-supplemented media under micro-aerobic conditions. However, when the protein was expressed at fully aerobic conditions which resulted in lower cellular Cu levels during growth, the catalytic efficiency of the purified CotA was noticeably reduced and alterations of the T2/T3 Cu center geometry were detected by EPR. Based on this, it was speculated that when intracellular levels of Cu are limited, CotA fails to fold into its normal (and functional) conformation. This, therefore, indicated that early Cu coordination is required for proper folding and Cu loading of CotA in vivo (Durao et al. Reference Durao, Chen, Silva, Soares, Pereira, Todorovic, Hildebrandt, Bento, Lindley and Martins2008). This notation correlates with the in vitro findings on CP and Fet3p, described earlier, where refolding was abolished after Cu site destruction. The importance of the T1 Cu for CotA enzymatic activity and thermodynamic stability was probed using point-mutated variants (Durao et al. Reference Durao, Bento, Fernandes, Melo, Lindley and Martins2006). Replacement of the axial Cu ligand Met50 by non-ligating residues (e.g. Leu and Phe) leads to a 0·1 V increase in the Cu reduction potential relative to that of wild-type CotA. The T1 mutations did not perturb the overall geometry of the T1 Cu site and they did not affect global protein structure but, still, the catalytic activity (using a range of substrates) of the T1 mutants was severely compromised. In addition, equilibrium unfolding of CotA was affected by the mutations such that, in contrast to wild-type CotA, the unfolding processes of these mutants involved intermediates (Durao et al. Reference Durao, Bento, Fernandes, Melo, Lindley and Martins2006).
Aquifex aeolicus McoA is closely related to CotA and, as expected for a protein from a thermophilic organism, it resists high temperatures. Unfolding of partially Cu-loaded McoA did not take place until at or above 110 °C in vitro (Fernandes et al. Reference Fernandes, Martins and Melo2009). In the fully Cu-loaded form, McoA remains folded even at 130 °C, which indicates a strong stabilizing effect of the Cu ions. Regardless of its high thermal stability, McoA is surprisingly sensitive to chemical denaturation. Concentrations less than 0·2 M of the chemical denaturant GuHCl caused depletion of Cu from the tri-nuclear Cu center and, as a result, loss of enzyme activity. Nonetheless, kinetic unfolding measurements indicated that presence of Cu still stabilized McoA via Cu-mediated decrease of the unfolding rate (Fernandes et al. Reference Fernandes, Martins and Melo2009). The time-resolved unfolding experiments revealed a rather uncommon observation, that was further confirmed by light scattering and SEC, in which McoA aggregates upon unfolding triggered by GuHCl. Kinetic partitioning between aggregation and unfolding was shown to lead to low heat capacity change for McoA unfolding, which was used to explain the flat temperature dependence of the protein stability.
Like azurin, Acidithiobacillus ferrooxidans rusticyanin also retains the Cu ion upon unfolding in vitro. Because this bacterium lives in media that are very acidic, A. ferrooxidans rusticyanin is extraordinarily stable at low pH values. Through structural and thermodynamic experiments, it was shown that the folded rusticyanin scaffold not only increases the affinity for Cu in both redox states, but it also hampers drastic changes in Cu redox properties when the pH is modulated (2·5–7). Thus, the folded structure acts as a local pH buffer for the Cu active site (Alcaraz et al. Reference Alcaraz, Gomez, Ramirez, Calvente, Andreu and Donaire2007). NMR titrations of apo, Cu(I) and Cu(II) forms of rusticyanin with GuHCl revealed direct evidence of copper coordination in both oxidation states to rusticyanin in its completely unfolded state. The NMR experiments also showed that upon unfolding, one metal ligand detaches from the Cu while the other three Cu ligands remain bound to Cu in the unfolded state of rusticyanin (Alcaraz et al. Reference Alcaraz, Jimenez, Moratal and Donaire2005). Analysis of dynamic and solvent exchange properties of the unfolded protein provided clues as to the secondary structural elements involved in starting the folding process. In similarity to the results for azurin, where Cu binding to the unfolded protein is fast but Cu binding to the folded protein is slow, the same findings were made for Cu binding to rusticyanin and the authors suggested a folding/binding mechanism for active rusticyanin that involved Cu binding prior to protein folding (Alcaraz et al. Reference Alcaraz, Jimenez, Moratal and Donaire2005).
Homo sapiens Cu/Zn superoxide dismutase 1 (SOD1) catalyzes the conversion of superoxide to hydrogen peroxide and is important for antioxidant defense. The protein contains a catalytic Cu and a structural Zn ion and is a homo-dimer held together by hydrophobic interactions and an inter-protein disulfide bridge (Battistoniet al. Reference Battistoni, Folcarelli, Cervoni, Polizio, Desideri, Giartosio and Rotilio1998). In vitro equilibrium folding of SOD1 includes monomer folding, metal binding and dimer formation; the metal-loaded form is dramatically more stable than the apo-form towards chemical perturbation and acid-induced denaturation (Lynch & Colon, Reference Lynch and Colon2006). NMR studies have revealed that Cu and Zn stabilize the β-barrel fold by restriction of local unfolding (Assfalg et al. Reference Assfalg, Banci, Bertini, Turano and Vasos2003). Zn binding to the unfolded state modulates the entire folding energy landscape (Kayatekin et al. Reference Kayatekin, Zitzewitz and Matthews2008), and it was shown that transient binding of Zn to the Cu-binding site residues, which are part of the folding nucleus, speeds up SOD1 folding (Leinartaite et al. Reference Leinartaite, Saraboji, Nordlund, Logan and Oliveberg2010). After global folding has commenced, the Zn transfers to the Zn-binding site, which adopts an ordered structure only late in the folding process. Zn adheres to a highly dynamic on/off coordination at the Zn site; thus, if a dissociated Zn ion is prevented from SOD1 rebinding, the lifetime of the protein is dramatically reduced (Leinartaite et al. Reference Leinartaite, Saraboji, Nordlund, Logan and Oliveberg2010).
The last protein to be discussed in this section is SCO that participate in the assembly of the CuA center in cytochrome c oxidase (COX) (Horng et al. Reference Horng, Leary, Cobine, Young, George, Shoubridge and Winge2005). SCO has a thioredoxin fold and three Cu-binding residues: a CXXXC motif with two Cys and a His residues about 100 residues further towards the C-terminus. SCO has been proposed to directly deliver Cu to the CuA site, as well as maintain the appropriate redox state of Cys side-chains, during assembly of CuA in COX. Bacillus subtilis SCO binds Cu in both redox states but has a million-fold higher affinity for Cu(II) than for Cu(I) (Davidson & Hill, Reference Davidson and Hill2009). Chemical and thermal unfolding experiments with apo- and Cu(II)-forms of SCO have demonstrated that apo SCO is rather unstable and exhibits a two-state unfolding reaction whereas the Cu(II) form of SCO remains folded in up to 9 M urea (Lai et al. Reference Lai, Yam, Andrews and Hill2011). The effect of copper on SCO refolding is complicated to analyze in vitro due to rapid redox reactions between copper and the cysteines in the unfolded state of SCO. Nonetheless, when apo-SCO is refolded in the presence of Cu some of the protein population converts to folded holo-protein. Based on this, it was proposed that the presence of copper early in the folding process is essential for SCO attaining its functional Cu-loaded state (Lai et al. Reference Lai, Yam, Andrews and Hill2011).We will come back to SOD1 (Sections 4 and 5) and SCO (section 5), when Cu transport is discussed.
Despite the low number of proteins investigated, it appears clear from above that Cu ions can affect protein folding reactions in vitro in many delicate ways. We speculate that Cu ions may also be able to act as transient ‘chemical chaperones’ during folding of proteins not binding Cu in the final folded state. However, this idea has not yet been tested.
3. Protein folding in cell environments
3.1 Macromolecular crowding
In living systems, proteins fold inside cells which are environments very different from that of a dilute buffer solution most often used in in vitro experiments. The cell compartments (cytoplasm, ER, nucleus etc.) are full of other proteins, membranes and DNA; the level and heterogeneity of biomolecules may vary depending on the compartment. It is estimated that up to 40% of the available volume in a cell is occupied by other biomolecules which correspond to a concentration of about 200–400 mg/ml of macromolecules (Ellis & Minton, Reference Ellis and Minton2003). The crowded environment results in increased viscosity, excluded volume effects and the amplified opportunity for specific as well as non-specific inter–molecular interactions (Christiansen et al. Reference Christiansen, Wang, Samiotakis, Cheung and Wittung-Stafshede2010; Hall and Dobson Reference Hall and Dobson2006; Mikaelsson et al. Reference Mikaelsson, Aden, Johansson and Wittung-Stafshede2013, Reference Mikaelsson, Aden, Wittung-Stafshede and Johansson2014; Minton Reference Minton2005a, Reference Mintonb; Stagg et al. Reference Stagg, Christiansen and Wittung-Stafshede2011; Zhanget al. Reference Zhang, Wu, Chen and Liang2012). These environmental factors are not accounted for in the fundamental studies of protein folding mechanisms executed during the last decades, and they were not accounted for in the studies presented above in Section 3. The question thus arises as to how these effects - present when polypeptides normally fold in vivo - modulate protein folding reactions? To obtain a complete understanding of protein folding in vivo, regardless of a metal cofactor or not, we need to pinpoint the thermodynamic, kinetic and chemical effects of the cellular environment.
10–15 years ago, scientists started to add macromolecular crowding agents to protein folding experiments in order to mimic the excluded volume effects of cells. Because it was realized that inert sugar-based polymers such as Ficoll 70 (70 kDa size) and dextran (of various defined sizes, 4–200 kDa) are soluble at very high concentrations (300–400 mg/ml; like in the cell) and, importantly, are spectroscopically silent, there was an expansion of this type of studies for a number of years. Using several protein systems, it was found that the excluded volume effect, due to macromolecular crowding in the physiological range, stabilized most proteins by a few kJ/mol and it promoted compaction of the proteins’ unfolded states (Christiansen et al. Reference Christiansen, Wang, Samiotakis, Cheung and Wittung-Stafshede2010, Reference Christiansen, Wang, Cheung and Wittung-Stafshede2013; Hall & Dobson, Reference Hall and Dobson2006). Both these effects were expected based on excluded volume theory pioneered by Minton (Minton, Reference Minton2000; Minton, Reference Minton2005a, Reference Mintonb). Unexpectedly, for a few small proteins with low native-state stability, the presence of crowding agents affected the folded state structure (Perham et al. Reference Perham, Stagg and Wittung-Stafshede2007; Stagg et al. Reference Stagg, Zhang, Cheung and Wittung-Stafshede2007). This was most striking for VlsE, a football-shaped protein that first got bent followed by a collapse to a spherical structure with increasing macromolecular crowding levels in vitro (Homouz et al. Reference Homouz, Perham, Samiotakis, Cheung and Wittung-Stafshede2008).
In contrast, it was determined that the stabilizing effect of macromolecular crowding for apo-azurin arose due to an accelerated folding rate constant (Christiansen & Wittung-Stafshede, Reference Christiansen and Wittung-Stafshede2013). This result agrees with the excluded volume effect being explained by compaction of the unfolded state without effects on the folded state structure of apo-azurin (Christiansen & Wittung-Stafshede, Reference Christiansen and Wittung-Stafshede2014). One may imagine that crowding-induced compaction of the unfolded state may affect Cu binding to unfolded azurin; therefore, crowding is expected to modulate the folding landscape of Cu-azurin. However, this has not been tested as the complication of the small Cu(II) ions going inside the hollow (to small ions) macromolecular crowding agent structures have so far precluded quantitative experiments in vitro. The only Cu-loaded copper protein studied in the presence of macromolecular crowding agents is Fet3p where macromolecular crowding agents were found to modulate catalytic parameters (Pozdnyakova & Wittung-Stafshede, Reference Pozdnyakova and Wittung-Stafshede2010). At low amounts of macromolecular crowding, we detected increases in both of KM (weaker substrate binding) and kcat (improved catalytic efficiency) whereas, at higher crowding levels, both these parameters were reduced. Presence of crowding agents did not affect the structural content of folded Fet3p but it increases the protein's resistance to a thermal perturbation such that the first thermal unfolding step gradually shifts to higher temperatures (up to 3–4 °C) with an increased level of crowding agent. Considering the proposed mechanism of Fet3p unfolding, this implies that domain 2 becomes stabilized by macromolecular crowding. Taken together, the observations on Fet3p suggest that excluded-volume effects result in the ordering of the substrate-binding site and reduction of internal dynamics (Pozdnyakova & Wittung-Stafshede, Reference Pozdnyakova and Wittung-Stafshede2010).
Even if synthetic macromolecular crowding agents provide mostly excluded-volume, steric effects (for example, true in the case of apoazurin), in some cases these polymers also interact with the target protein electrostatically/enthalpically (Benton et al. Reference Benton, Smith, Young and Pielak2012; Jiao et al. Reference Jiao, Li, Chen, Minton and Liang2010; Wang et al. Reference Wang, Sarkar, Smith, Krois and Pielak2012). For more realistic conditions, researchers have started to exploit direct in-cell experiments via NMR (Robinson et al. Reference Robinson, Reardon and Spicer2012; Smith et al. Reference Smith, Zhang, Pielak and Li2015) and time-resolved fluorescence (Dhar et al. Reference Dhar, Girdhar, Singh, Gelman, Ebbinghaus and Gruebele2011) spectroscopy. Using the latter approach, it was reported that depending on the cellular compartment it was directed to, the folding mechanism could change for the same protein, notably making the process more two-state like than found in vitro. The phenomenon of a smoother folding landscape was also reported for another protein, apoflavodoxin, as a result of excluded volume effects in vitro (Stagg et al. Reference Stagg, Christiansen and Wittung-Stafshede2011). In terms of copper proteins, in-cell NMR was used to probe the initial assembly steps of human Cu/Zn SOD1 in the cytoplasm of E. coli cells (Banciet al. Reference Banci, Barbieri, Bertini, Cantini and Luchinat2011a, Reference Banci, Bertini, Cefaro, Ciofi-Baffoni and Gallob). It was found that only one Zn ion became bound to SOD1 at this condition whereas Cu binding and disulfide bond formation did not occur in the in-cell experiments. These latter steps may require the aid of helper proteins in vivo, such as the human Cu chaperone CCS (Banci et al. Reference Banci, Barbieri, Bertini, Cantini and Luchinat2011a), see further discussion of this in Section 4·5.
3.2 Cellular compartments and copper
Proteins are made on ribosomes (most often in the cytoplasm) and are then transported to the appropriate cellular compartment or exported out of the cell for extracellular functions. It may vary when newly-made polypeptides fold; folding may occur co-translationally on the ribosome when released in the cytoplasm, or only after transport to the final destination. A number of molecular chaperones and chaperonins assist with the folding of newly made proteins (Hartl, Reference Hartl2017; Horwich, Reference Horwich2017). In all domains of life, two major pathways exist to secrete proteins across membranes (Natale et al. Reference Natale, Bruser and Driessen2008). The general secretion route, termed Sec-pathway, catalyzes the transmembrane translocation of proteins in their unfolded conformation, whereupon they fold to functional structures at the trans side of the membrane. The other secretion route is the Twin-arginine translocation, or Tat, pathway, which facilitate membrane translocation of proteins in their folded state (Natale et al. Reference Natale, Bruser and Driessen2008). In eukaryotes, the Sec-pathway is found in the ER and aids in the secretion of proteins and insertion of membrane proteins that follow the vesicle sorting route.
In addition to crowding (Christiansen et al. Reference Christiansen, Wang, Samiotakis, Cheung and Wittung-Stafshede2010), the spatial and temporal location of Cu must be considered in order to reveal how copper protein folding and metal acquisition are linked in vivo. Non-bonded (‘free’) Cu ions are toxic because of their ability to catalyze the formation of free radicals and they may also interfere with Fe-S cluster assembly (Macomber & Imlay, Reference Macomber and Imlay2009). To avoid toxicity and to overcome solubility problems of Cu(I), the intracellular concentration of Cu is stringently controlled via dedicated proteins that facilitate Cu uptake, efflux, as well as delivery to target Cu-dependent proteins (Festa & Thiele, Reference Festa and Thiele2011; O'Halloran & Culotta Reference O'Halloran and Culotta2000; Robinson & Winge Reference Robinson and Winge2010). This means that there is, in essence, no ‘free’ Cu ions in cells, although emerging evidence suggests that some Cu may be present in so-called ‘labile pools’ within subcellular compartments providing a temporal component (Dean et al. Reference Dean, Qin and Palmer2012). Taking the cellular condition into account, a full molecular-mechanistic understanding of copper protein folding in vivo includes (in addition to the actual polypeptide folding reaction) knowledge of, in what compartment and by what mechanism the Cu is delivered to the polypeptide.
In bacteria, most copper proteins are located in the bacterial periplasm or plasma membrane and therefore it was initially assumed that there was no need for cytoplasmic copper protein loading. However, secretion of copper proteins via the Tat system implies that these proteins likely acquire their metal in the cytoplasm. On the other hand, membrane-bound copper proteins, as well as soluble proteins secreted by the Sec system, fold and acquire Cu from the periplasmic compartment (Arguello et al. Reference Arguello, Raimunda and Padilla-Benavides2013). Since considerable energy is spent during protein biosynthesis, the possibility of the wrong metalation must be avoided and, thus, highly regulated Cu transport and delivery systems should have co-evolved in the periplasm. Azurin (discussed above) is a bacterial protein found in the periplasm and it is secreted by the Sec pathway based on its signal sequence. Thus, azurin is likely metallated in the periplasm but the mechanism is unknown. Several Cu chaperone proteins have been identified both in the bacterial cytoplasm and periplasm (Arguello et al. Reference Arguello, Raimunda and Padilla-Benavides2013). Furthermore, via elegant experiments in E. coli, it was shown that the metal availability of the cellular compartment (here, cytoplasm versus periplasm) in which a protein folds can override the protein's intrinsic metal binding preference (Tottey et al. Reference Tottey, Waldron, Firbank, Reale, Bessant, Sato, Cheek, Gray, Banfield, Dennison and Robinson2008).
Tyrosinase, a multi-copper oxidase responsible for melanin synthesis, is interesting as the metal loading mechanism diverts between eukaryotes and bacteria (Wang & Hebert, Reference Wang and Hebert2006). In bacteria, tyrosinase is encoded in an operon composed of genes coding for both a ‘caddie’ chaperone protein and for tyrosinase (Matoba et al. Reference Matoba, Bando, Oda, Noda, Higashikawa, Kumagai and Sugiyama2011). Tyrosinase is secreted via the Tat pathway and the ‘caddie’ protein appears to function as a Cu chaperone, forming a transient complex with apo-tyrosinase in bacteria. Based on structural work, this interaction appears to facilitate incorporation of Cu into the active site of tyrosinase followed by subsequent secretion of the functional protein (Matoba et al. Reference Matoba, Bando, Oda, Noda, Higashikawa, Kumagai and Sugiyama2011). In contrast, in humans, tyrosinase is loaded with Cu in the Golgi (via ATP7A in the secretory pathway, see below) but this process is inefficient and the protein appears to lose the Cu and become reloaded with Cu (via ATP7A again) at its final destination, i.e. in specialized organelles called melanosomes (Setty et al. Reference Setty, Tenza, Sviderskaya, Bennett, Raposo and Marks2008).
In the next section, we describe chemical-mechanistic knowledge for how copper enters human cells and is then distributed along different transport pathways for the delivery of Cu-dependent enzymes (Fig. 8). Not until Cu has reached the target polypeptide, can the question about the in vivo ‘role of metal in folding/unfolding’ be considered.
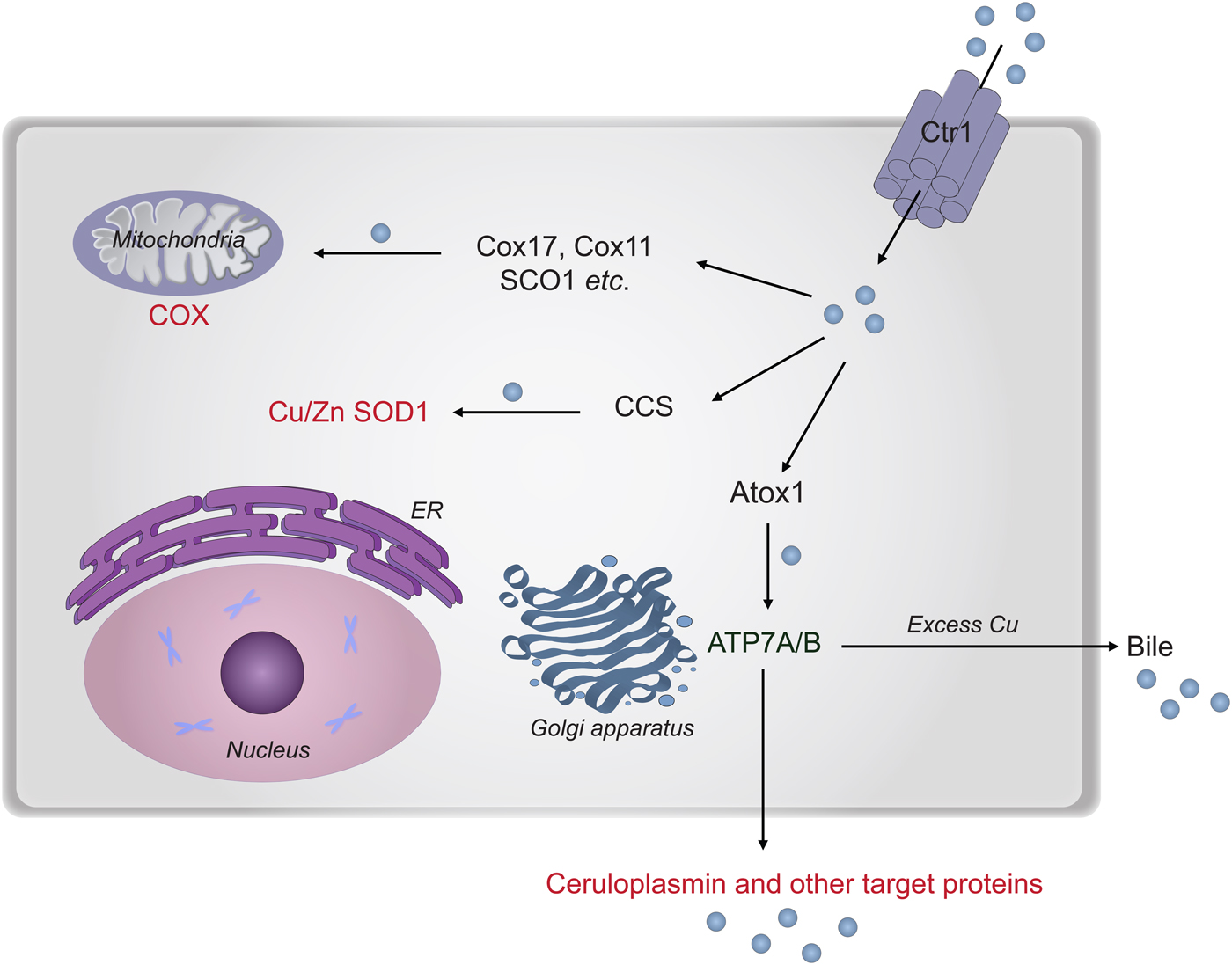
Fig. 8. Cu transport paths in the human cytoplasm. Uptake of Cu is mediated by Ctr1. Then Cu is transported by Atox1 to ATP7A/B in the Golgi network for loading of Cu-dependent enzymes in the secretory pathway. CCS transports Cu to SOD1 in the cytoplasm. Cox17 (together with Cox11, Sco1 etc.) moves Cu to the mitochondria for loading of COX.
4. Copper transport in (human) cells
4.1 Uptake of Cu into cells - Ctr1
In human cells, Cu(II) from the plasma is translocated in the reduced Cu(I) form to the cytoplasm by high-affinity membrane-spanning proteins of the Ctr1 family (Boal & Rosenzweig, Reference Boal and Rosenzweig2009; Festa & Thiele, Reference Festa and Thiele2011; Kim et al. Reference Kim, Nevitt and Thiele2008). Homo sapiens Ctr1 (Fig. 9a ) is a glycosylated trans-membrane protein, which upon assembly into a homo-trimeric structure contains a small pore that allows Cu(I) to pass into the cell down a concentration gradient (De Feo et al. Reference De Feo, Aller, Siluvai, Blackburn and Unger2009). The extracellular 67-residue N-terminus of Ctr1 contains clusters of Met and His residues capable of interacting with Cu. Model peptide studies showed that the Ctr1 N-terminus binds Cu(II) with high affinity (~10−11 M) through an amino-terminal Cu(II), Ni(II) (ATCUN) binding site. Unlike typical ATCUN-type peptides, however, the Ctr1 peptide facilitates ascorbate-dependent reduction of bound Cu(II) by virtue of an adjacent bis-His sequence. Thus, it appears that Ctr1 is capable of Cu(II) reduction by itself prior to Cu(I) import (Pushie et al. Reference Pushie, Shaw, Franz, Shearer and Haas2015).

Fig. 9. Proteins mediating Cu transport via the secretory path. (a) Schematic structure of Cu uptake protein Ctr1. (b) Schematic structure of the P1B-type ATPase ATP7B in the Golgi membrane. For A- and N-domains, 2KIJ and 2ARF were used. For the model of the six MBDs, PBD files 2N7Y, 2LQB, 2ROP, and 2EW9 were combined. TGE and SEHPL motifs are labeled in green and magenta, respectively. (c) General Post-Albers catalytic mechanism for P-type ATPases involving ATP hydrolysis and high-Cu affinity, E1, and low-Cu affinity, E2, states.
How copper is delivered to extracellular parts of Ctr1 from the blood system is unknown. The three known protein carriers of Cu in blood plasma are human serum albumin, transcuperin and CP (Shenberger et al. Reference Shenberger, Shimshi and Ruthstein2015). The first two are the main components of the exchangeable plasma pool of copper; they both bind Cu(II) at surface accessible sites exhibiting rapid on/off exchange rates. CP in contrast, which carries 70% of all plasma copper, has buried Cu sites and some perturbation of the protein structure is needed to extract the Cu ions (see section 2.3 above). Therefore it is likely that human serum albumin and transcuperin are the main Cu donators to Ctr1 although CP has also been proposed to provide Cu to Ctr1 (Ramos et al. Reference Ramos, Mar, Ishida, Vargas, Gaite, Montgomery and Linder2016). It was recently shown using an EPR spin label approach that human serum albumin (Cys34 converted to methanethiolsulfonate spin label) interacts with the first 14 amino acids of the N-terminus of Ctr1 (using peptides spin labeled at different sites), both with and without Cu ions present (Shenberger et al. Reference Shenberger, Shimshi and Ruthstein2015).
On the cytoplasmic side of the plasma membrane, a short C-terminal part of Ctr1 protrudes and NMR studies with model peptides suggest that this peptide's HCH motif binds Cu(I) and can directly deliver it to the Cu chaperone Atox1 (described below, in Section 5·2–5·3) through transient interactions (Kahra et al. Reference Kahra, Kovermann and Wittung-Stafshede2016). The affinity of Cu(I) for the HCH motif in the C-terminal Ctr1 peptide was ~10−14 M, and drastically reduced when the Cys or His residues were mutated to Ala (Kahra et al. Reference Kahra, Kovermann and Wittung-Stafshede2016). The affinity of Atox1 for Cu(I) is several orders of magnitude higher, and thus the direction of transfer from Ctr1 to Atox1 makes sense. In some similarity, another cytoplasmic Cu chaperone, CCS (described below, in Section 5·5), was proposed to be able to bind negatively-charged lipids and obtain Cu from Ctr1 through direct protein–protein interactions at the plasma membrane (Pope et al. Reference Pope, De Feo and Unger2013).
4.2 Secretory path – involved proteins
In eukaryotes, cytoplasmic Cu transport is facilitated by so-called Cu chaperones (e.g. Atox1, CCS and Cox17) (Fig. 8). In the general path, Atox1 is the Cu chaperone that transport Cu in the human cytoplasm to proteins in the Golgi, for further delivery in the secretory pathway. In this path, nascent polypeptide chains are translocated to the ER, which provides a protective folding environment, allowing for maturation events such as signal sequence cleavage, glycosylation, disulfide bond formation and chaperone binding. Eventually, the protein is packaged into vesicles that exit the ER for the Golgi, where Cu loading takes place in case of a copper-dependent protein.
Atox1 receives Cu imported by Ctr1 and then delivers it to the copper-transporting P1B-type ATPases ATP7A (i.e. Menke's disease protein) and ATP7B (i.e. Wilson disease protein) localized in the trans-Golgi network. ATP7A and ATP7B are multi-domain proteins with six cytoplasmic metal-binding domains protruding in the cytoplasm, a nucleotide-binding domain, an actuator domain, a domain for catalytic phosphorylation and eight membrane-spanning helices that is thought to include a channel for Cu (Lutsenko et al. Reference Lutsenko, LeShane and Shinde2007b) (Fig. 9b ). Each metal-binding domain, like Atox1, adopts a ferredoxin-like fold and has a surface-exposed copper-binding motif, MXCXXC (Boal & Rosenzweig, Reference Boal and Rosenzweig2009). Atox1 can interact directly with metal-binding domains of ATP7A and ATP7B and it is generally assumed that Atox1 delivers Cu to the metal-binding domains that, in turn, transfer the metal to the trans-membrane Cu channel in ATP7A/B in vivo. However, direct Cu transfer from metal-binding domains to the Cu sites in the membrane channel of ATP7A/B has not yet been demonstrated (Barry et al. Reference Barry, Shinde and Lutsenko2010). With energy from ATP hydrolysis, the ATP7A/ATP7B proteins transfer Cu to the lumen side of the trans-Golgi, where Cu is subsequently loaded onto target proteins coming from the ER. ATP7A and ATP7B are thought to follow a general reaction mechanism based on Post-Albers model, which involves shifts in copper ion affinity (between states E1 and E2) and accompanying phosphorylation/dephosphorylation (Fig. 9c ). Many human copper-dependent enzymes (e.g. blood clotting factors, tyrosinase, lysyl oxidase and CP) go via the secretory pathway and depend on either ATP7A or ATP7B for Cu delivery.
Notably, data for the Archaeoglobus fulgidus Cu transport proteins demonstrated that the A. fulgidus Cu chaperone CopZ (homologous to Atox1) could bypass the single metal-binding domain of the A. fulgidus P1B-type ATPase CopA (homologous to ATP7A/B) and be transferred directly from CopZ to the trans-membrane site in CopA (Gonzalez-Guerrero & Arguello, Reference Gonzalez-Guerrero and Arguello2008). This observation suggested that the metal-binding domains P1B-type ATPases may only play regulatory roles. Nonetheless, through yeast complementation studies it appears that the presence of at least some of the human metal-binding domains in ATP7B is essential for Cu transfer activity (Forbes et al. Reference Forbes, Hsi and Cox1999; Morin et al. Reference Morin, Gudin, Mintz and Cuillel2009). The crystal structure of the bacterial ATP7A/B homolog Legionella pneumophila CopA was reported in 2011 (Gourdon et al. Reference Gourdon, Liu, Skjorringe, Morth, Moller, Pedersen and Nissen2011) and even if it was a breakthrough, unfortunately, there was no electron density resolved for its metal-binding domain (Gourdon et al. Reference Gourdon, Liu, Skjorringe, Morth, Moller, Pedersen and Nissen2011). The CopA structure suggested a kinked helix at the membrane entry site for Cu as a putative docking site for the chaperone or a metal-binding domain. Subsequent modeling studies of ATP7B indicated that this kinked helix could indeed be a docking site for Atox1 (Gourdon et al. Reference Gourdon, Sitsel, Lykkegaard Karlsen, Birk Moller and Nissen2012) but also for the 6th metal-binding domain (Arumugam & Crouzy, Reference Arumugam and Crouzy2012). Thus, the question of where Atox1 delivers the copper ion remains unresolved. Regardless, that the metal-binding domains in ATP7B are of high importance for the function is clear since disease-causing point mutations have been found in all six metal-binding domains (Hamza et al. Reference Hamza, Schaefer, Klomp and Gitlin1999).
4.3 Secretory path – Atox1 delivery
Cu transfer from Atox1 to one of the six metal-binding domains (MBDs) in ATP7A or ATP7B has been proposed to take place through transient or long-lived copper-bridged hetero-dimers via a two-step reaction (Fig. 10) (Banci et al. Reference Banci, Bertini, Cantini, Rosenzweig and Yatsunyk2008a,Reference Banci, Bertini, Ciofi-Baffoni, Hadjiloi, Martinelli and Palumaab; Pufahl et al. Reference Pufahl, Singer, Peariso, Lin, Schmidt, Fahrni, Culotta, Penner-Hahn and O'Halloran1997; Wernimont et al. Reference Wernimont, Huffman, Lamb, O'Halloran and Rosenzweig2000):
 $${\rm Cu - Atox1} + {\rm MBD} \leftrightarrow {\rm Atox1 - Cu - MBD} \leftrightarrow {\rm Atox1} + {\rm Cu - MBD}$$
$${\rm Cu - Atox1} + {\rm MBD} \leftrightarrow {\rm Atox1 - Cu - MBD} \leftrightarrow {\rm Atox1} + {\rm Cu - MBD}$$
In this reaction, Cu(I) is thought to undergo a sequence of exchange reactions involving two- or three-coordinated Cu-Cys intermediates. This mechanism implies a diffusion-driven movement of the Cu ion from one site (Atox1) to another (MBD) on the basis of small differences in Cu binding constants (Pufahl et al. Reference Pufahl, Singer, Peariso, Lin, Schmidt, Fahrni, Culotta, Penner-Hahn and O'Halloran1997). Recent work showed that the second Cys in Atox1, Cys15, has a lower pKa value than the corresponding Cys in target MBDs, resulting in decreased nucleophilicity of Atox1's Cys15 (Badarau & Dennison, Reference Badarau and Dennison2011). The six MBDs are here (and most often in the literature) labeled from 1 to 6, with MBD1 being most N-terminal and MBD6 connecting to the subsequent membrane-spanning part in ATP7A/B. Based on NMR data, Atox1 can deliver Cu to all MBDs in ATP7B but it only forms a stable hetero-dimer complex with MBD1, MBD2 and MBD4 (Banci et al. Reference Banci, Bertini, Calderone, Della-Malva, Felli, Neri, Pavelkova and Rosato2009a, Reference Banci, Bertini, Cantini, Massagni, Migliardi and Rosatob). However, conflicting results have been reported regarding which domains form stable hetero-protein complexes with Atox1 and, thus, this likely depends on the conditions (Achila Reference Achila, Banci, Bertini, Bunce, Ciofi-Baffoni and Huffman2006; Banci et al. Reference Banci, Bertini, Cantini, Rosenzweig and Yatsunyk2008a, Reference Banci, Bertini, Ciofi-Baffoni, Hadjiloi, Martinelli and Palumaab, Reference Banci, Bertini, Ciofi-Baffoni, Janicka, Martinelli, Kozlowski and Palumaac).

Fig. 10. Cu-dependent hetero-complex of Atox1-MBD4. (a) Structural model of Atox1-MBD4 complex highlighting Cu-binding Cys residues in red. (b). Proposed two-step mechanism for Cu (blue) transfer from Atox1 (gold/yellow) to MBD4 (silver/grey) along with thermodynamic (ΔH and TΔS) values for step 1 and step 2 (shown for direction to the right) determined for the Atox1-MBD4 pair (Niemiec et al. Reference Niemiec, Weise and Wittung-Stafshede2012).
Based on MD simulations of all six ATP7B MBDs, without and with Cu in the metal site (Rodriguez-Granillo et al. Reference Rodriguez-Granillo, Crespo and Wittung-Stafshede2009), molecular features were found to vary distinctly among the six MBDs. Whereas MBD1, MBD2 and MBD6 all have defined Cu loop conformations that are stabilized by a network of interactions, MBD4 and MBD5 exhibit greater loop flexibility. In MBD4, helix α1 unwinds and rewinds continuously during the simulation. MBD3 has lowest sequence identity among the domains and it also behaves differently: its Cu loop is more rigid than the rest of the domain. Cu coordination reduces structural dynamics in all the MBD domains but MBD4. Further MD simulations of the six different Atox1-MBD hetero-protein complexes provided insights into possible inter–molecular interactions (Rodriguez-Granillo et al. Reference Rodriguez-Granillo, Crespo and Wittung-Stafshede2009). MBD4 binds most strongly to holo-Atox1 in silico, with electrostatic interactions having the greatest contribution as also expected based on the surface electrostatic potential analysis of the individual domains (Huffman & O'Halloran, Reference Huffman and O'Halloran2001).
Using MBD4 as a representative target domain for in vitro work it was found that upon mixing of Cu-Atox1 with MBD4, a Cu-dependent hetero-protein dimer (Fig. 10a ) can be detected via unique near-UV CD signals (Hussain et al. Reference Hussain, Rodriguez-Granillo and Wittung-Stafshede2009). SEC analysis revealed that the mixture is heterogeneous: there is a significant fraction of hetero-protein complex but there is also Cu-MBD4 (i.e. the transfer has been completed) and Cu-Atox1 (protein that never reacted) present. Using CD and SEC, interactions between point-mutated Atox1 variants and MBD4 were analyzed (Hussain et al. Reference Hussain, Rodriguez-Granillo and Wittung-Stafshede2009). Although there was a less hetero-protein dimer, and less Cu transfer to MBD4, when using the Thr11Ala and Met10Ala Atox1 variants, the reaction takes place. In sharp contrast, with Cu-loaded Lys60Ala Atox1, no hetero-protein complex and no Cu transfer to MBD4 was observed. This demonstrates the importance of the electrostatic attraction provided by Lys60 for hetero-protein complex formation. The same result was noted earlier in the homologous yeast system (Portnoy et al. Reference Portnoy, Rosenzweig, Rae, Huffman, O'Halloran and Culotta1999).
The energetics (enthalpy and entropy contributions) of Cu transfer from Atox1 to MBD4 via the proposed two-step reaction was investigated via a combination of SEC and calorimetry (ITC). Upon merging the equilibrium constants for both steps, a metal exchange factor, going from Atox1 to MBD4, of 10 was calculated. This favorable transport in the direction of MBD was governed by a negative enthalpy change for Cu transfer of about 10 kJ/mol (enthalpy and entropy changes for steps 1 and 2 given in Fig. 10b ) (Niemiec et al. Reference Niemiec, Weise and Wittung-Stafshede2012, Reference Niemiec, Dingeldein and Wittung-Stafshede2014). In contrast to the overall reaction, the formation of the hetero-protein dimer was governed by both favorable entropy and favorable enthalpy. This suggests that the Atox1-Cu-MBD4 complex is dynamic in nature and, moreover, that it possibly involves local protein unfolding (e.g. MBD4 helix α1 unfolding, see above) (Niemiec et al. Reference Niemiec, Weise and Wittung-Stafshede2012). The mechanism of Cu transfer was dissected using single Cys-to-Ala variants of Atox1 and MBD4 metal-binding motifs and Cu-dependent hetero-protein complexes required the presence of three cysteines (two from one protein and one from the other), with the most N-terminal Cys in each protein's metal site strictly required. Comparison of thermodynamic parameters for mutant versus wild-type reactions revealed that the Cu transfer reaction with wild-type proteins involves strong entropy-enthalpy compensation as compared with the mutant-protein Atox1-MBD4 systems. This property was explained by a dynamic inter-conversion of Cu-Cys3 coordinations in the wild-type system. Such dynamics may protect against Cu mis-ligation and bypass enthalpic traps in vivo (Niemiec et al. Reference Niemiec, Dingeldein and Wittung-Stafshede2015).
As a complement, the reaction mechanism of Cu transfer from Atox1 to MBD4 was also probed by QM/MM methods (Rodriguez-Granillo et al. Reference Rodriguez-Granillo, Crespo, Estrin and Wittung-Stafshede2010a). The QM subsystem contains the Cu ion and the four sulfur groups whereas the rest of the system (protein and solvent) corresponds to the MM system. After assessing all possible 2-, 3- and 4-coordinate Cu-intermediate species, one main reaction path emerged as most favorable. First, without activation barrier, MBD4's most N-terminal Cys binds to the Cu in Atox1 and forms a 3-coordinated intermediate. Next, another 3-coordinated intermediate forms that involve both Cys residues in MBD4 but only Cys12 in Atox1. From this species, the product is then formed upon dissociation of Atox1's Cys12 (Rodriguez-Granillo et al. Reference Rodriguez-Granillo, Crespo and Wittung-Stafshede2010b). Similar mechanisms involving 3-coordinated intermediates have been proposed based on mutational studies of the yeast Atx1-Ccc2 complex and the complex of Atox1 with ATP7A's MBD1 (Banci, Reference Banci2006; Banci et al. Reference Banci, Bertini, Calderone, Della-Malva, Felli, Neri, Pavelkova and Rosato2009a, Reference Banci, Bertini, Cantini, Massagni, Migliardi and Rosatob). In addition, these computational results agree amazingly well with the mutational in vitro work on the same Atox1-MBD4 pair (Niemiec et al. Reference Niemiec, Dingeldein and Wittung-Stafshede2015).
The Cu-binding motif in Atox1, as well as in the Cu-binding domains of ATP7A/B, consists of a MX1CXXC motif where X 1 = Thr. The same motif but with X 1 = Asp is found in metal-binding domains of bacterial zinc transporters, such as ZntA. Using MDCXXC variants of Atox1 and MBD4, it was observed that the mutants bind Cu like the wild-type proteins, but in contrast to the wild-type pair, the mutant pair favors Cu-dependent hetero-dimers over directional Cu transport from Atox1 to MBD4 (Niemiec et al. Reference Niemiec, Dingeldein and Wittung-Stafshede2014). In accord, the exchange factor for Cu transfer from Atox1 to MBD4 with Asp at X 1 in both was 0·3. Thus, Thr may be conserved in this motif to promote directional Cu transfer without formation of deep energetic sinks. Interestingly, in the absence of competing reducing agents, Zn can bind both Atox1 and MBD4 and upon mixing the proteins, an unreactive hetero-complex is the most stable species (Niemiec et al. Reference Niemiec, Dingeldein and Wittung-Stafshede2014).
4.4 Secretory path – intra-ATP7B interactions
ATP7A/B are likely to undergo significant conformational changes during the catalytic cycle that requires ATP hydrolysis and facilitates Cu transfer to the lumen (Lutsenko et al. Reference Lutsenko, Barnes, Bartee and Dmitriev2007a). Current predictions for the ATP7A/B catalysis cycle come from analogy with the calcium pump SERCA, for which there are high-resolution structures reported for several enzymatic stages (Bublitz et al. Reference Bublitz, Musgaard, Poulsen, Thogersen, Olesen, Schiott, Morth, Moller and Nissen2013). Since there is no structural information on how the six MBDs are arranged within full-length ATP7A/B, it remains unknown how these domains participate in the catalytic cycle although MBD interactions are proposed to transmit signals long-range (Gourdon et al. Reference Gourdon, Sitsel, Lykkegaard Karlsen, Birk Moller and Nissen2012). In support, even in a small construct of only domains 5 and 6 of ATP7B (MBD56), minute variations in salt and pH conditions perturbed domain-domain relative fluctuations such that the efficiency of Atox1-mediated Cu delivery to these domains was modulated (Nilsson et al. Reference Nilsson, Aden, Niemiec, Nam and Wittung-Stafshede2013). This further implies that local (temporal and spatial) fluctuations in the cellular environment may tune overall Cu pump activity via changes in domain–domain interactions.
MD simulations on three two-domain constructs of ATP7B in apo and holo forms (MBD12, MBD34 and MBD56) showed that when two MBDs are linked together, for all three two-domain pairs, the two domains exhibited a distinct pattern of correlated motions, which were domain-dependent and modulated by the presence of Cu (Rodriguez-Granillo et al. Reference Rodriguez-Granillo, Crespo and Wittung-Stafshede2010b). Also, NMR data with nano-bodies (Huang et al. Reference Huang, Nokhrin, Hassanzadeh-Ghassabeh, Yu, Yang, Barry, Tonelli, Markley, Muyldermans, Dmitriev and Lutsenko2014) as well as single-molecule FRET experiments (Keller et al. Reference Keller, Benitez, Klarin, Zhong, Goldfogel, Yang, Chen and Chen2012) have shown the presence of transient MBD domain–domain interactions in ATP7B. From NMR diffusion data on the first four ATP7B MBDs linked in one construct, MBD1-4, the addition of stoichiometric amounts of Cu resulted in faster tumbling in accord with going from an extended apo conformation to a more compact arrangement of the four domains upon Cu binding (Mondol et al. Reference Mondol, Aden and Wittung-Stafshede2016). In fact, the diffusion rate constant for Cu-loaded MBD1-4 corresponded to what is expected of a spherical protein of the size of four folded ferredoxin-like units. Dmitriev et al. recently proposed a contrasting model in which transient interactions of MBD1, MBD2 and MBD3 exist in the absence of Cu but upon Atox1-mediated Cu loading, the domains adopt a more extended conformation (Yu et al. Reference Yu, Dolgova and Dmitriev2017a, Reference Yu, Yang, Bothe, Tonelli, Nokhrin, Dolgova, Braiterman, Lutsenko and Dmitrievb). That MBD1-3 forms a regulatory unit agrees with yeast complementation experiments in which, one-by-one, the ATP7B MBDs were deleted. Upon removing MBD1 or MBD12 (i.e. part of the unit) Cu transport was reduced, whereas upon removal of all three MBDs (the whole unit, MBD1-3), normal Cu transport was restored (Ponnandai Shanmugavel et al. Reference Ponnandai Shanmugavel, Petranovic and Wittung-Stafshede2017). Further chemical-biochemical studies are desired to ultimately reveal the molecular mechanisms for how the MBDs modulate ATP7A/B activity.
Based on pull-down (a kind of affinity purification) experiments, an interaction between the ATP-binding domain (N-domain) and a construct of the six MBDs of ATP7B (MBD1-6) was proposed (Tsivkovskii et al. Reference Tsivkovskii, MacArthur and Lutsenko2001) that depended on metal loading as well as on phosphorylation (Hasan et al. Reference Hasan, Gupta, Polishchuk, Yu, Polishchuk, Dmitriev and Lutsenko2012). A similar intra–protein interaction was reported for a homologous bacterial Cu-ATPase, also using pull-down methodology (Gonzalez-Guerrero et al. Reference Gonzalez-Guerrero, Hong and Arguello2009). However, biophysical studies using 15N-labeled purified domains (N-domain mixed with MBD1-4, MBD56, or MBD1-6) did not reveal any interaction for any protein pair (Mondol et al. Reference Mondol, Aden and Wittung-Stafshede2016), although high-μM to low-mM protein concentrations were used. This suggests that the interaction identified in the pull-down experiments depends sensitively on the condition (such as pH and salt), on additional components in the lysate, or constitute a minor fraction of the total pool of proteins.
4.5 The CCS and cox17 pathways
Cu loading of human SOD1 in vivo occurs via its specific cytoplasmic Cu chaperone CCS (Wong et al. Reference Wong, Waggoner, Subramaniam, Tessarollo, Bartnikas, Culotta, Price, Rothstein and Gitlin2000) (Fig. 11a ), although the molecular mechanisms of CCS-dependent maturation of SOD1 is not fully resolved (Banci et al. Reference Banci, Bertini, Cantini, Kozyreva, Massagni, Palumaa, Rubino and Zovo2012). CCS obtains Cu from Ctr1 at the plasma membrane and it was also recently shown that SOD1 can interact with membranes in proximity to CCS (Pope et al. Reference Pope, De Feo and Unger2013). CCS is a protein with three distinct domains (Lamb et al. Reference Lamb, Torres, O'Halloran and Rosenzweig2001). Domain 1 of human CCS is similar to Atox1 with a ferredoxin-like fold and a MTCXXC motif and is thought to be the domain that receives Cu (Banci et al. Reference Banci, Bertini, Cantini, Kozyreva, Massagni, Palumaa, Rubino and Zovo2012, Reference Banci, Cantini, Kozyreva and Rubino2013). Domain 2 of CCS binds Zn and is structurally similar to SOD1; this domain is critical for CCS-SOD1 protein recognition (Lamb et al. Reference Lamb, Torres, O'Halloran and Rosenzweig2001). The third domain of CCS is only a short polypeptide segment that lacks secondary structure but it contains a CXC motif that is essential for CCS-dependent Cu-loading of SOD1 (Lamb et al. Reference Lamb, Torres, O'Halloran and Rosenzweig2001). At least in vitro, CCS and Atox1 can exchange Cu via transient complex formation, with the favored direction being Cu movement from CCS to Atox1 with an exchange factor of 6 (Petzoldt et al. Reference Petzoldt, Kahra, Kovermann, Dingeldein, Niemiec, Aden and Wittung-Stafshede2015). This may imply cross-reactivity between the two Cu chaperones in the cytoplasm, perhaps as a way to assure correct amounts of Cu going into the different paths depending on the needs.

Fig. 11. Structures of proteins mediating Cu transport to SOD1 and COX. (a) Heterodimer between yeast SOD1 (silver) and all three domains of yeast CCS (yellow, domain 1; gold, domain 2; red, domain 3) (1JK9), (b) Bovine heart COX (1V55). (c) Sinorhizobium meliloti Cox11 (1SP0) (D) Human Cox17 (2RNB), (E) Human SCO1 (2GT6). In all, Cu ions are represented in cyan and Zn in magenta.
Cu loading of COX in mitochondria (Fig. 11b ) involves several proteins, including Cox11, Cox17 and SCO1 (Fig. 11c–e ). Cox11 and SCO1 are mitochondrial inner membrane proteins thought to donate copper to the CuB and CuA sites of COX, respectively. Cox17 is a 69-residue cysteine-rich (three pairs of cysteines) protein that shuttles between the cytoplasm and mitochondria (Banci et al. Reference Banci, Bertini, Cantini, Rosenzweig and Yatsunyk2008a, Reference Banci, Bertini, Ciofi-Baffoni, Hadjiloi, Martinelli and Palumaab, Reference Banci, Bertini, Ciofi-Baffoni, Janicka, Martinelli, Kozlowski and Palumaac, Reference Banci, Bertini, Cefaro, Ciofi-Baffoni and Gallo2011b). How Cox17 obtains Cu in the cytoplasm is unclear, but likely the Cu ions come from Ctr1 via direct interactions or through an intermediate Cu transporter. Cox17 is believed to provide Cu to SCO1 and Cox11 in the inter-membrane space (IMS) of mitochondria (Cobine et al. Reference Cobine, Pierrel and Winge2006). Cox17 is imported to the IMS by the MIA pathway, which is a specific translocation pathway of metal-binding IMS proteins that couples disulfide exchange with metal transport (Mesecke et al. Reference Mesecke, Terziyska, Kozany, Baumann, Neupert, Hell and Herrmann2005; Terziyska et al. Reference Terziyska, Lutz, Kozany, Mokranjac, Mesecke, Neupert, Herrmann and Hell2005). Interactions between Cox17 and both SCO1 and Cox11 likely involve complementary electrostatic surfaces (Horng et al. Reference Horng, Cobine, Maxfield, Carr and Winge2004). In contrast to cytoplasmic Cu transfer reactions, it appears that reversible changes in the redox state of cysteines in the mitochondrial Cu transport proteins represent important steps in the pathways ultimately providing Cu to COX (Banci et al. Reference Banci, Bertini, Cantini, Rosenzweig and Yatsunyk2008a, Reference Banci, Bertini, Ciofi-Baffoni, Hadjiloi, Martinelli and Palumaab, Reference Banci, Bertini, Ciofi-Baffoni, Janicka, Martinelli, Kozlowski and Palumaac; Voronova et al. Reference Voronova, Meyer-Klaucke, Meyer, Rompel, Krebs, Kazantseva, Sillard and Palumaa2007). This makes sense as the IMS is a more oxidizing environment than the cytosol (Hu et al. Reference Hu, Dong and Outten2008) and, in accord, Cox17 exists in a partially-oxidized form with two disulfide bonds and two reduced Cys residues. Cu-loaded Cox17 transfers simultaneously a Cu(I) ion and two electrons to SCO1 when the latter is in the oxidized state, i.e. with metal-binding cysteines forming a disulfide in SCO1 (Banci et al. Reference Banci, Bertini, Cantini, Rosenzweig and Yatsunyk2008a, Reference Banci, Bertini, Cefaro, Ciofi-Baffoni and Gallob, Reference Banci, Bertini, Ciofi-Baffoni, Janicka, Martinelli, Kozlowski and Palumaac). Electron transfer-coupled metal delivery is thus a mechanism that can offer efficient Cu transfer when target metal-binding residues are oxidized.
To directly address the mechanism and kinetics of Cu loading of CuA sites in proteins, a simple model was prepared in which a CuA site was engineered into azurin as the scaffold. In its resting state, the CuA site is a mixed valence purple di-nuclear Cu site with one Cu(I) and one Cu(II) ion. Thus, upon the addition of Cu(II) in vitro, some Cu must be reduced in order to form the final metal site. Initially, using stopped-flow mixing experiments with visible absorption detection, the mechanism of Cu binding was found to involve an intermediate containing a red tetragonal Cu(II)-Cys center. This intermediate then converted to the final CuA center after Cu(I) was generated from reduction of Cu(II) by either free cysteines from nearby apo-proteins or by external reductants (Wang et al. Reference Wang, Ang and Lu1999). Subsequent studies provided further mechanistic details including, in addition to the red Cu intermediate, a novel green Cu intermediate (identified as a mononuclear Cu(Cys)2His complex) and a blue type 1 Cu intermediate prior to CuA formation (Chakraborty et al. Reference Chakraborty, Polen, Chacon, Wilson, Yu, Reed, Nilges, Blackburn and Lu2015; Wilson et al. Reference Wilson, Savelieff, Nilges, Marshall and Lu2011). Further experiments revealed pH-dependent transformations among red, green and purple Cu sites suggesting an underlying link between the cupredoxin copper sites and hinting to an evolutionary relationship between cupredoxin proteins (Savelieff et al. Reference Savelieff, Wilson, Elias, Nilges, Garner and Lu2008).
4.6 Additional mechanisms of Cu chaperones
It is becoming more and more obvious that the concept of ‘one protein – one function’ is too simple. Many proteins appear to have multiple functions and this has become clear also for the Cu chaperones. In 2008, Atox1 was reported to have additional activity in the nucleus as a Cu-dependent transcription factor (TF) of several genes (Chen et al. Reference Chen, Sudhahar, Youn, Das, Cho, Kamiya, Urao, McKinney, Surenkhuu, Hamakubo, Iwanari, Li, Christman, Shantikumar, Angelini, Emanueli, Ushio-Fukai and Fukai2015; Itoh et al. Reference Itoh, Kim, Nakagawa, Ozumi, Lessner, Aoki, Akram, McKinney, Ushio-Fukai and Fukai2008, Reference Itoh, Ozumi, Kim, Nakagawa, McKinney, Folz, Zelko, Ushio-Fukai and Fukai2009; Ozumi et al. Reference Ozumi, Sudhahar, Kim, Chen, Kohno, Finney, Vogt, McKinney, Ushio-Fukai and Fukai2012; Yamamoto et al. Reference Yamamoto, Ebisuya, Ashida, Okamoto, Yonehara and Nishida2006). We also confirmed the presence of Atox1 in the nucleus of HeLa cells but no DNA binding of Atox1 to the proposed GAAAGA promotor sequence in vitro was detected (Kahra et al. Reference Kahra, Mondol, Niemiec and Wittung-Stafshede2015). Despite this negative result, Atox1 may nonetheless regulate gene transcription via additional proteins that in turn bind DNA. Intriguingly, Atox1 contains an apparent nuclear localization sequence KKTGK within its C-terminal part and, although not discussed in the initial discovery paper of Atox1 from 1999 (Hamza et al. Reference Hamza, Schaefer, Klomp and Gitlin1999), immunofluorescence indicated that Atox1 was distributed throughout the cell, including the nucleus. Using a yeast two-hybrid screen of a large human fragment library, a number of new Atox1-interacting proteins were identified as confident hits (Ohrvik & Wittung-Stafshede, Reference Ohrvik and Wittung-Stafshede2015). Among these target proteins, several were reported as detected in the nucleus and described as DNA/RNA-binding proteins (Ohrvik & Wittung-Stafshede, Reference Ohrvik and Wittung-Stafshede2015). The new data triggers a number of speculations regarding functional consequences, but chemical-mechanistic data on these interactions are still lacking. It will of high interest to reveal if these new Atox1 interactions are dependent on Cu-loading of Atox1 and if the complex formation is coupled to Cu transfer. Intriguingly, and demanding several follow up studies on a molecular level, Atox1 was found to localize at lamellipodia edges in breast cancer cells and, by a yet unknown mechanism, promote cancer cell migration (Blockhuys & Wittung-Stafshede, Reference Blockhuys and Wittung-Stafshede2017).
Moreover, the X-linked inhibitor of apoptosis (XIAP) was recently reported as a CCS target (Brady et al. Reference Brady, Galban, Liu, Basrur, Gitlin, Elenitoba-Johnson, Wilson and Duckett2010). XIAP is a Cu-binding protein that is overexpressed in many cancers. It participates in the regulation of apoptosis such that it blocks caspases needed to induce cell death (Mufti et al. Reference Mufti, Burstein and Duckett2007). It was demonstrated that CCS delivers Cu to XIAP and XIAP, in turn, stimulates ubiquitination of CCS which resulted in increased Cu delivery to SOD1 (Brady et al. Reference Brady, Galban, Liu, Basrur, Gitlin, Elenitoba-Johnson, Wilson and Duckett2010). Cu-binding to XIAP plays a role in its regulation as when Cu is bound XIAP it adopts a conformation that is less stable and unable to inhibit caspases. Thus, upon CCS delivery of Cu to XIAP, the Cu form of XIAP is degraded and apoptosis can take place (Mufti et al. Reference Mufti, Burstein, Csomos, Graf, Wilkinson, Dick, Challa, Son, Bratton, Su, Brewer, Jakob and Duckett2006). CCS may also mediate Cu transport to the nucleus for regulation of the hypoxia-inducible factor 1 transcriptional complex, which in turn promotes expression of the vascular endothelial growth factor, a potent angiogenic factor (Qiu et al. Reference Qiu, Ding, Zhang and Kang2012). Although CCS was originally shown to be present in the nucleus as well as the cytoplasm (Casareno et al. Reference Casareno, Waggoner and Gitlin1998), mechanistic information on CCS-mediated Cu transport to the nucleus is lacking. Paralleling the discovery of additional functions for CCS and Atox1, Cox17 can form a Cu-dependent complex with a protein named Mic60 (Chojnacka et al. Reference Chojnacka, Gornicka, Oeljeklaus, Warscheid and Chacinska2015), a protein involved in establishing and maintaining the proper inner membrane architecture.
5. Copper in aberrant protein folding and metal loading
5.1 Disease-causing mutations in copper proteins
Many mutations in the genes that code for Cu transport proteins have been linked to human diseases. Mutations in ATP7A/B constitute the basis of Menke's and Wilson diseases; missense mutations in almost all domains of ATP7B have been linked to Wilson disease (WD). A range of biophysical methods was employed to try to explain the underlying mechanism for the most common WD mutation, H1069Q in the N-domain. It was discovered that this mutation did not affect domain stability or ATP binding affinity but instead it forced ATP to bind in such a way that hydrolysis was hindered (Rodriguez-Granillo et al. Reference Rodriguez-Granillo, Sedlak and Wittung-Stafshede2008). For two WD point-mutations found in the MBDs (G85 V in MBD1 and G591D in MBD6) of ATP7B, experiments on in vitro single-domain constructs showed that the mutations caused reduced thermal stability, shifting the midpoint temperature of unfolding by more than 20 °C. There was also increased structural dynamics of the folded states and increased the potential for misfolding upon perturbation found in vitro for the variants (Kumar et al. Reference Kumar, Arioz, Li, Bosaeus, Rocha and Wittung-Stafshede2017). However, because of the lack of a complete structural description of ATP7A/B, molecular-mechanistic data on the effects of most WD-causing point mutations are lacking, except for some Cu transport and trafficking studies of various constructs in cell cultures.
Mutations in CP have been linked to the rare autosomal recessive disease aceruloplasminemia, in which absence or dysfunction of CP leads to the accumulation of iron in various organs (Xu et al. Reference Xu, Pin, Gathinji, Fuchs and Harris2004). In a few cases, missense mutations linked to aceruloplasminemia have been shown to result in proteins that are retained totally (P177R) or partially (I9F) in the early secretory pathway of cells, or to mutated proteins that are secreted as inactive apo forms lacking copper (G631R and G969S). Also, other disease-causing substitutions are expected to perturb Cu loading because the mutated residues are either direct Cu ligands (His978) or positioned close to a Cu site (Ala331 and Gln692) (di Patti et al. Reference di Patti, Maio, Rizzo, De Francesco, Persichini, Colasanti, Polticelli and Musci2009). Here, a molecular understanding of CP folding and Cu uptake processes may aid in finding ways to stabilize folded structures of the mutants such that they can bind Cu.
Inherited forms of the fatal neurodegenerative disease amyotrophic lateral sclerosis (ALS) are associated with mutations in SOD1 that promote protein aggregation. More than 110 mutations in SOD1 have been reported to cause ALS. Mutant SOD1 proteins often display greater aggregation propensity than wild-type holo-SOD1 and this propensity was connected to the extent of partial unfolding and/or monomer instability (Nordlund et al. Reference Nordlund, Leinartaite, Saraboji, Aisenbrey, Grobner, Zetterstrom, Danielsson, Logan and Oliveberg2009; Rodriguez et al. Reference Rodriguez, Valentine, Eggers, Roe, Tiwari, Brown and Hayward2002). In vitro, SOD1 aggregation is a multistep reaction that involves dimer dissociation, metal loss and finally oligomerization of unfolded or partially-folded monomers (Kayatekin et al. Reference Kayatekin, Zitzewitz and Matthews2008). The Cu and Zn ions appear important players in SOD1 misfolding and aggregation, and likely their inability to bind mutant proteins or increased dissociation constants contribute to aberrant disease-causing reactions, but relevant in vivo pathways and toxic species are not yet defined.
5.2 Hijacking of Cu transport path by cancer-drug
Platinum (Pt) containing anticancer drugs have been used in cancer treatment for several decades as they trigger cell death upon DNA interaction. There are several Pt-containing anticancer drugs and derivatives with a variety of ligands around the Pt center (with cisplatin, Pt(NH3)2Cl2, being the most well-known and used compound) currently existing in clinical use and clinical trials. However, a severe drawback with these drugs is their limited efficacy due to side reactions eventually resulting in cell resistance to the compound. It has been reported that cisplatin hijacks Cu transport proteins, such as ATP7B, as a possible mechanism to become exported out of cells. Pt(II) and Cu(I) share chemical features (e.g. 5d8 and 3d10 electron configurations) that allow them to sometimes have overlapping binding sites in proteins. Both metals favor sulfur ligands, although Pt can adopt several oxidation states (0-IV), coordination geometries, and thereby a number of ligands. In contrast, Cu(I) favors linear or trigonal ligand coordinations. We and others showed that at concentrations in vitro similar to physiological conditions, Atox1 can bind cisplatin together with copper creating a di-metal site (Palm-Espling & Wittung-Stafshede, Reference Palm-Espling and Wittung-Stafshede2012; Palm-Espling et al. Reference Palm-Espling, Andersson, Bjorn, Linusson and Wittung-Stafshede2013). Based on appearing near-UV CD signals at wavelengths above 300 nm, similar to those found for metalloclusters in metallothioneins (MTs), we concluded that Pt must be close to the Cu when bound to Atox1. This would be the case if both metal ions co-occupy Atox1's metal-binding site and, in accord, modeling using density functional theory placed Pt near the Cu and interacting with the most exposed of Atox1's cysteines. Pt-Cu d8–d10 interactions have been observed in model systems and, according to theoretical calculations, these metal–metal signals are similar to d10–d10 interactions found in MTs (Palm-Espling & Wittung-Stafshede, Reference Palm-Espling and Wittung-Stafshede2012; Palm-Espling et al. Reference Palm-Espling, Andersson, Bjorn, Linusson and Wittung-Stafshede2013).
Cisplatin binding causes Atox1 unfolding and subsequent aggregation in vitro, with the extent and speed of unfolding (ranging from hours to days) depending on the platinum complex chemistry and the presence or absence of Cu in Atox1 (Palm et al. Reference Palm, Weise, Lundin, Wingsle, Nygren, Bjorn, Naredi, Wolf-Watz and Wittung-Stafshede2011; Palm-Espling & Wittung-Stafshede, Reference Palm-Espling and Wittung-Stafshede2012; Palm-Espling et al. Reference Palm-Espling, Andersson, Bjorn, Linusson and Wittung-Stafshede2013; Xi et al. Reference Xi, Guo, Tian, Wang and Liu2013). Upon connecting the chemical properties of six Pt-substances to their effects on binding, unfolding and aggregation of Atox1, it was found that both ligand type and orientation dictated the interactions with Atox1. Only substances with two good leaving groups in cis-configuration resulted in detectable interactions with Cu-Atox1. Furthermore, if a partner protein such as a metal-binding domain of ATP7B is present, the platinum drug can be transferred to the next protein from Atox1 prior to Atox1 unfolding in vitro (Palmet al. Reference Palm, Weise, Lundin, Wingsle, Nygren, Bjorn, Naredi, Wolf-Watz and Wittung-Stafshede2011; Palm-Espling & Wittung-Stafshede, Reference Palm-Espling and Wittung-Stafshede2012; Palm-Espling et al. Reference Palm-Espling, Andersson, Bjorn, Linusson and Wittung-Stafshede2013, Reference Palm-Espling, Lundin, Bjorn, Naredi and Wittung-Stafshede2014). However, when these experiments were performed in a more cell-like environment (in terms of physiological reducing agents), no such Pt transfer was observed (Galliani et al. Reference Galliani, Losacco, Lasorsa, Natile and Arnesano2014). Thus, despite the need for additional studies, part of the cellular resistance to cisplatin may be linked to the unfolding of cisplatin-Atox1 complexes prohibiting Pt transport to the nucleus.
5.3 Role of Cu in amyloid diseases
In addition to direct genetic defects in Cu homeostasis proteins that cause disease by affecting protein conformation and Cu binding, systemic and tissue imbalances of metal levels are often found in neurodegenerative disorders such as Parkinson's disease (PD), Alzheimer's disease, prion diseases and Huntington's disease (Fink, Reference Fink2006). These diseases have in common that they involve misfolding and aberrant self-assembly of proteins as the unifying molecular event. Independent of variations in primary and secondary structures, when proteins form amyloid fibrils, they all adopt the cross-β-sheet arrangement that is the hallmark of amyloid structures. However, the molecular mechanisms and initial triggers behind the self-assembly into amyloid deposits in the various neurodegenerative disorders remain unknown but an emerging idea is that metal ions (such as Cu) may play key roles (Crist et al. Reference Cristóvão, Santos and Gomes2016; Viles, Reference Viles2012).
Conformational changes resulting in aggregation of the intrinsically-disordered α-synuclein (αS) protein into amyloid fibers is directly related to PD (Galvin et al. Reference Galvin, Lee, Schmidt, Tu, Iwatsubo and Trojanowski1999; Winner et al. Reference Winner, Jappelli, Maji, Desplats, Boyer, Aigner, Hetzer, Loher, Vilar, Campioni, Tzitzilonis, Soragni, Jessberger, Mira, Consiglio, Pham, Masliah, Gage and Riek2011). Many synthetic as well as naturally-occurring molecules can modulate αS amyloid formation in vitro, for example ring-fused 2-pyridones (Horvath et al. Reference Horvath, Weise, Andersson, Chorell, Sellstedt, Bengtsson, Olofsson, Hultgren, Chapman, Wolf-Watz, Almqvist and Wittung-Stafshede2012), other amyloidogenic proteins (i.e. cross-reactivity) (Horvath & Wittung-Stafshede, Reference Horvath and Wittung-Stafshede2016), and even bacterial proteins (Chorell et al. Reference Chorell, Andersson, Evans, Jain, Gotheson, Aden, Chapman, Almqvist and Wittung-Stafshede2015), to name a few. Cu, in both redox states, can bind to αS (Camponeschi et al. Reference Camponeschi, Valensin, Tessari, Bubacco, Dell'Acqua, Casella, Monzani, Gaggelli and Valensin2013; De Ricco et al. Reference De Ricco, Valensin, Dell'Acqua, Casella, Gaggelli, Valensin, Bubacco and Mangani2015a, Reference De Ricco, Valensin, Dell'Acqua, Casella, Hureau and Fallerb) and structural features, binding sites, and affinities for the interaction between αS and Cu(II), as well as Cu(I), have been the focus of in vitro spectroscopic investigations (Binolfi et al. Reference Binolfi, Rasia, Bertoncini, Ceolin, Zweckstetter, Griesinger, Jovin and Fernandez2006, Reference Binolfi, Valiente-Gabioud, Duran, Zweckstetter, Griesinger and Fernandez2011; Camponeschi et al. Reference Camponeschi, Valensin, Tessari, Bubacco, Dell'Acqua, Casella, Monzani, Gaggelli and Valensin2013; De Ricco et al. Reference De Ricco, Valensin, Dell'Acqua, Casella, Gaggelli, Valensin, Bubacco and Mangani2015a, Reference De Ricco, Valensin, Dell'Acqua, Casella, Hureau and Fallerb) (Fig. 12). Although other divalent metals also bind to αS, Cu(II) has the highest affinity with one binding site, involving N-terminal residues and His50, exhibiting a Cu(II)-peptide dissociation constant of about 0·1 nM. Among studied metal ions, Cu(II) has the largest accelerating effect of αS amyloid formation in vitro (Davies et al. Reference Davies, Mercer, Chen and Double2016; Montes et al. Reference Montes, Rivera-Mancia, Diaz-Ruiz, Tristan-Lopez and Rios2014). (Notably, the effect of Cu(I) binding on αS amyloid formation in vitro has not been reported.) It was recently suggested that αS was able to act as a ferri-reductase, reducing iron using Cu(I)/Cu(II) as a catalytic redox center (Brown, Reference Brown2013). Notably, Cu-αS mediated redox reactions may generate toxic reactive oxygen species that can damage cell components and/or αS itself (De Ricco et al. Reference De Ricco, Valensin, Dell'Acqua, Casella, Gaggelli, Valensin, Bubacco and Mangani2015a, Reference De Ricco, Valensin, Dell'Acqua, Casella, Hureau and Fallerb). It awaits to be revealed if Cu-mediated redox reactions are related to αS function or simply is part of a cascade of deleterious processes causing disease.
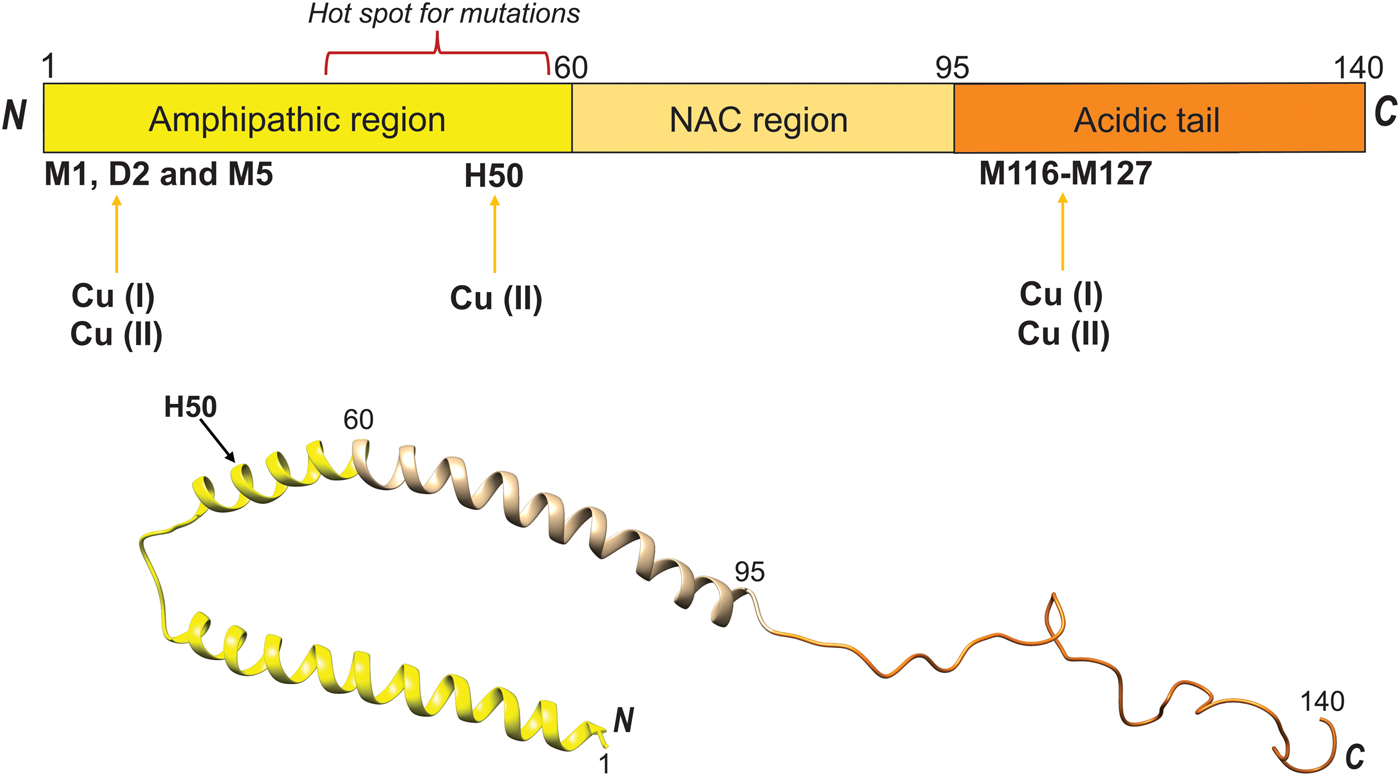
Fig. 12. Cu binding to Parkinson's disease protein αS. Cu(II)- and Cu(I)-binding sites in the 140-residue αS polypeptide are schematically shown (top), along with helical form of αS found when bound to lipid vesicles (1XQ8) (bottom).
When considering αS reactions in PD, one must also take into account that αS can undergo N-terminal acetylation in vivo. It was reported that this αS modification abolishes Cu(II) binding at the high-affinity N-terminal binding site (Moriarty et al. Reference Moriarty, Minetti, Remeta and Baum2014), but the Cu(I) binding sites remain preserved (Miotto et al. Reference Miotto, Pavese, Quintanar, Zweckstetter, Griesinger and Fernández2017). Residue-specific NMR characterization of Cu(I) interaction with acetylated αS showed that metal binding was accompanied with folding into an α-helical structure that was of larger magnitude than when Cu(I) interacted with the un-acetylated protein (Miotto et al. Reference Miotto, Pavese, Quintanar, Zweckstetter, Griesinger and Fernández2017). Induction of helical structure in the N-terminus of αS also takes place when the protein interacts with negatively-charged lipid vesicles, and the binding appears tuned at the molecular level by lipid chemistry (Kiskis et al. Reference Kiskis, Horvath, Wittung-Stafshede and Rocha2017). Although molecular details of the Cu binding sites in αS are known, it remains unknown how Cu reaches αS in vivo and in what redox state the metal is in. Is cellular stress resulting in free Cu(II) ions that then bind to αS and promote amyloid formation or, are Cu transport proteins delivering Cu(I) to αS such that (perhaps requiring Cu oxidation) protein aggregation is promoted and this then causes cell toxicity? We note that Atox1 and other Cu transporters are expressed in most neuronal cells (Davies et al. Reference Davies, Hare, Cottam, Chen, Hilgers, Halliday, Mercer and Double2013; Montes et al. Reference Montes, Rivera-Mancia, Diaz-Ruiz, Tristan-Lopez and Rios2014).
In similarity to αS, Cu(II) interactions with the amyloid-β (Aβ) peptide have been shown to impact amyloid aggregation and neurotoxicity in Alzheimer's disease (Faller et al. Reference Faller, Hureau and Berthoumieu2013). In fact, Cu interaction with Aβ has been characterized by biophysical and structural studies to the extent that researchers have started to develop new therapeutics schemes based on this knowledge (Choi et al. Reference Choi, Braymer, Nanga, Ramamoorthy and Lim2010). Aβ peptides are metabolic products generated by the proteolysis of the neuronal amyloid precursor protein, APP. Two Cu-binding domains exist within the APP, one localized in the Aβ region and the other in the N-terminus (Fig. 13, top). The enzymatic cleavage of APP occurs via either amyloidogenic or non-amyloidogenic pathways. The non-amyloidogenic pathway requires α-secretase and γ-secretase, whereas amyloidogenic processing of APP occurs by β-secretase and γ-secretase (Fig. 13, bottom) (Mathys & White, Reference Mathys and White2017). Notably, the activity of β-secretase (BACE1) in the amyloidogenic pathway is regulated by Cu. Cu(II), but also other metal ions such as Fe and Zn, play a significant role in promoting the formation of soluble Aβ oligomers. The Aβ oligomer species generated is dependent on the molar ratio of Cu(II) to Aβ: At sub-equimolar ratios, amyloid-like aggregates form that is highly stable and SDS resistant. Conversely, at supra-equimolar ratios of Cu(II) to Aβ, less stable, neurotoxic oligomers are formed. For high Cu, Cu coordination to His residues (Fig. 13) leads to the generation of His-bridged Aβ oligomers that are highly toxic (Mathys & White, Reference Mathys and White2017).
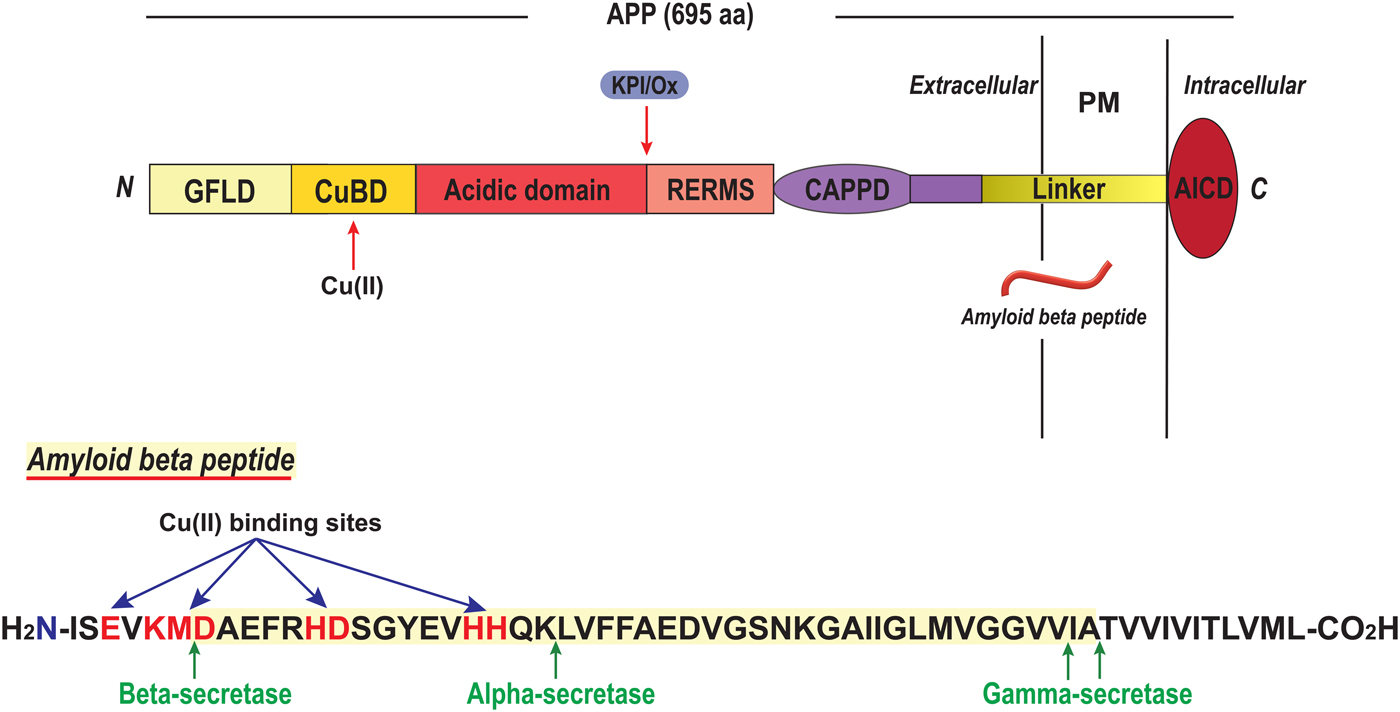
Fig. 13. Cu binding to the amyloid- β peptide, A β. Top. Cu(II) binding sites and domain arrangement of full-length APP (GFLD, growth factor-like domain; CuBD, Cu-binding domain; CAPPD, central APP domain; AICD, APP intracellular domain; RERMS, sequence segment which participates in dimerization; linker contains the Aβ peptide). Bottom. Sequence of Aβ peptide with secretase cleavage sites indicated, as well as possible Cu binding residues highlighted in red (Mathys & White, Reference Mathys and White2017).
Likewise, misfolding and aggregation of the cellular prion protein (PrP), that are key events in prion diseases, appears promoted by Cu(II). Mature PrP is a two-domain protein comprising an unstructured N-terminal region (residues 23–120) and an α-helical C-terminal domain (residues 121–231), Fig. 14. The N-terminal domain features octa-peptide repeats of the sequence PHGGGWGQ (four such repeats in the normal human protein) and these are responsible for high-affinity metal binding (Stevens et al. Reference Stevens, Walter, Rodriguez, Draper, Davies, Brown and Millhauser2009) (Fig. 14, top). Under normal conditions, PrP is monomeric and protease sensitive. On the other hand, the misfolded or scrapie form of PrP is rich in β-sheet structure, oligomeric and resistant to proteolysis. Moreover, the scrapie form can self-template its own propagation leading to amyloid plaque deposition and destruction of brain tissue (Leal et al. Reference Leal, Botelho and Gomes2012). Cu can bind to the octa-peptide repeats in the PrP N-terminus and, depending on the Cu saturation, form inter- or intra-protein Cu sites (Morante et al. Reference Morante, Gonzalez-Iglesias, Potrich, Meneghini, Meyer-Klaucke, Menestrina and Gasset2004). In addition, Cu can bind to a peptide segment just prior to the C-terminal domain involving residues between His96 and His111 (Fig. 14, bottom) (Giachin et al. Reference Giachin, Mai, Tran, Salzano, Benetti, Migliorati, Arcovito, Longa, Mancini, D'Angelo and Legname2015). In contrast to for other amyloidogenic proteins such as αS and Aβ, copper binding promotes PrP assembly towards non-amyloidogenic but toxic aggregates. These assemblies are protease-resistant and eventually become large enough such that they precipitate (Leal et al. Reference Leal, Botelho and Gomes2012). One can envisage that local conformational changes induced by Cu binding, affecting the N-terminal domain of PrP where the octa-peptide repeats are positioned (Fig. 14), prevents the formation of a β-sheet-rich amyloid core and instead directs the reaction in a non-amyloid aggregation pathway (Morante et al. Reference Morante, Gonzalez-Iglesias, Potrich, Meneghini, Meyer-Klaucke, Menestrina and Gasset2004; Stevens et al. Reference Stevens, Walter, Rodriguez, Draper, Davies, Brown and Millhauser2009). In accord, using single-molecule experiments, it was demonstrated that Cu(II) induces misfolding of PrP monomers before the assembly of misfolded monomers takes place (Yen et al. Reference Yen, Harischandra, Kanthasamy and Sivasankar2016). That the Cu-induced PrP aggregates are toxic is highlighted by the fact that copper chelation delays onset of disease in mice infected with the scrapie form of PrP. Notably, octa-peptide repeat expansion in PrP results in early-onset prion disease and age of onset was recently shown to correlate with Cu binding properties and number of repeats (Stevens et al. Reference Stevens, Walter, Rodriguez, Draper, Davies, Brown and Millhauser2009).

Fig. 14. Cu binding to the prion protein, PrP. Top. The PrP protein (residues 1–254) is schematically shown indicating the octa-peptide repeat (OPR) domain (with amino acid sequences) and the Cu binding sites (arrows). Also shown are positions for SP, signal peptide, HS, hydrophobic segment, α-helices (H1-H3), β-strands, a disulfide connecting H2 and H3, the C-terminal GPI anchor; and, protruding ovals indicate N-linked glycans (Brown, Reference Brown2001; Giachin et al. Reference Giachin, Mai, Tran, Salzano, Benetti, Migliorati, Arcovito, Longa, Mancini, D'Angelo and Legname2015). Hot-spot areas for pathological mutations are also shown (van der Kamp & Daggett, Reference van der Kamp and Daggett2009). Bottom. Model based on 1QLZ for the C-terminal part. Cu ions added based on (Walter et al. Reference Walter, Stevens, Visconte and Millhauser2007).
Taken together, Cu interactions with amyloidogenic proteins (such as, but not limited to, αS, Aβ and PrP) are clearly able to promote protein structural changes that enhance the assembly into amyloid fibers in vitro (Leal et al. Reference Leal, Botelho and Gomes2012). Such metal–protein interactions in vivo might represent a link between the pathological processes of protein aggregation, oxidative damage and neuronal cell loss (Davies et al. Reference Davies, Mercer, Chen and Double2016) and may at some conditions be the initial trigger of neurodegenerative diseases.
6. Outstanding questions around copper protein folding and metal exchange
In this review, we have attempted to describe the current knowledge around mechanisms of copper protein folding and Cu transport protein metal exchange in vitro (Fig. 1). As described, mechanistic details of the roles of copper in the folding of copper proteins in vitro have been obtained for several protein systems. This information is important when considering copper protein folding in lower organisms where Cu ions may be freely available, and in biotechnology (e.g. if a specific copper-dependent enzyme is to be overexpressed or in de novo design of new metalloenzymes (Yu et al. Reference Yu, Cangelosi, Zastrow, Tegoni, Plegaria, Tebo, Mocny, Ruckthong, Qayyum and Pecoraro2014)). For example, a newly-discovered class of copper-dependent enzymes is lytic polysaccharide monooxygenases (LPMO) that are of high importance for biomass degradation. In 2011, these enzymes were found to contain one Cu(II) ion in a special arrangement called the histidine brace where two histidines provide three nitrogen ligands, two from N-His and one from the terminal amine (Quinlan et al. Reference Quinlan, Sweeney, Lo Leggio, Otten, Poulsen, Johansen, Krogh, Jorgensen, Tovborg, Anthonsen, Tryfona, Walter, Dupree, Xu, Davies and Walton2011) (Fig. 15). Enzymes now classified as LPMOs were first annotated as either glycosyl hydrolases or as binding domains without catalytic activity until the Cu-dependence was revealed (Quinlan et al. Reference Quinlan, Sweeney, Lo Leggio, Otten, Poulsen, Johansen, Krogh, Jorgensen, Tovborg, Anthonsen, Tryfona, Walter, Dupree, Xu, Davies and Walton2011). Notably, a similar but not identical Cu coordination site occurs in a type of proteins found in bacterial plant pathogens that are thought to be Cu chaperones involved in the removal of free copper (Wijekoon et al. Reference Wijekoon, Young, Wedd and Xiao2015). This poses the question of putative (yet unknown) catalytic activity of such Cu chaperones. LPMOs have been reported to perform oxidative cleavage of several polysaccharides including the abundant but highly recalcitrant biopolymers chitin and cellulose. Enzyme activity is dependent on Cu site delivery of electrons from cofactors which may be small organic molecules (Vaaje-Kolstad et al. Reference Vaaje-Kolstad, Westereng, Horn, Liu, Zhai, Sorlie and Eijsink2010) or the enzyme cellobiose dehydrogenase (Langston et al. Reference Langston, Shaghasi, Abbate, Xu, Vlasenko and Sweeney2011). Industrial production of fuels and chemicals from cellulose holds the potential to displace petroleum-based sources and thereby reduce both economic and environmental costs of oil and gas production. To make possible such applications, the characterization of a Cu-dependent enzyme now appears at the frontline.
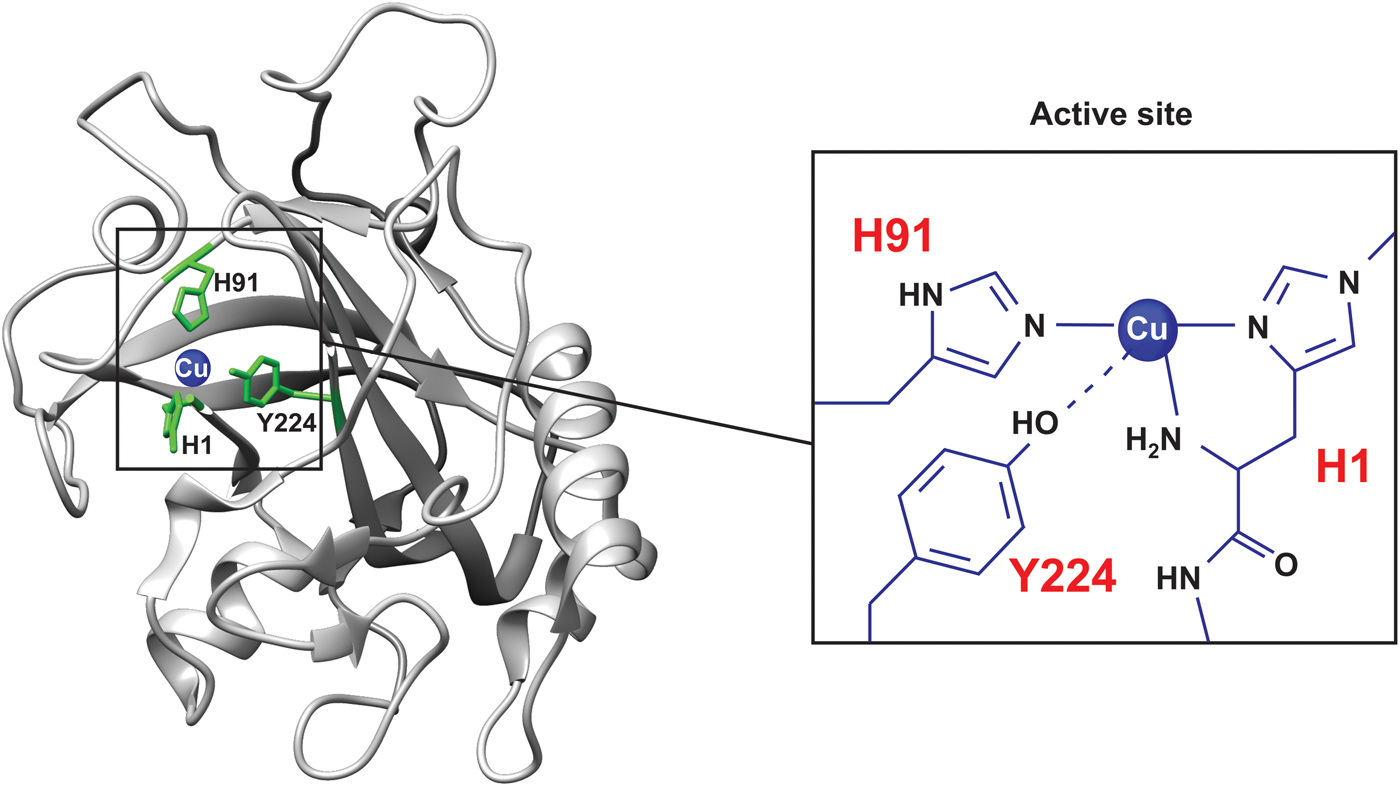
Fig. 15. LPMO with histidine brace Cu site. A starch-active lytic polysaccharide monooxygenase (LPMO) from Aspergillus Oryzae (4OPB; (Lo Leggio et al. Reference Lo Leggio, Simmons, Poulsen, Frandsen, Hemsworth, Stringer, von Freiesleben, Tovborg, Johansen, De Maria, Harris, Soong, Dupree, Tryfona, Lenfant, Henrissat, Davies and Walton2015)) with the Cu site enlarged.
It is important to emphasize that because of the marginal stability of most proteins, and their dynamic nature, most proteins will unfold (fully or in part) many times during their life time in a cell; these reactions may or may not involve transient Cu ion dissociation in the case of copper proteins. Thus, even if the metal is delivered by a transport or chaperone protein during biosynthesis, Cu reloading may take place from the free ion several times during the protein's lifetime and this may, or may not, be coupled to polypeptide un/re-folding.
When considering biosynthetic assembly of active copper proteins in human cells, one has to take into account the Cu transport pathway involved and, as this text demonstrates, the scientific community has learnt a lot in the last two decades about how Cu transport proteins facilitate Cu movement in cells. Nonetheless, there are remaining questions around copper protein folding and metal loading mechanisms. One open question is how Cu is loaded onto target Cu-dependent proteins (e.g. CP) in the trans-Golgi network. Since protein synthesis in the secretory path involves several maturation steps in the ER before the protein reaches the Golgi, most proteins likely have near-native conformations when arriving in the Golgi. ATP7A, but not ATP7B, has a long luminal peptide loop (between membrane-spanning helices 1 and 2) that was found to contain Cu-binding sites (Barry et al. Reference Barry, Otoikhian, Bhatt, Shinde, Tsivkovskii, Blackburn and Lutsenko2011). Cu binding to residues in this protruding segment of ATP7A may thus act as an intermediate step, before Cu is directly transferred to a target Cu-dependent protein or released into the vesicular space in the Golgi. Although free Cu is toxic in the cytoplasm, in the Golgi vesicles, due to the low pH, free Cu(I) ions are soluble and, thus, the Cu uptake mechanism by target proteins may simply be direct binding. There are no identified Cu chaperones in the Golgi vesicles. Moreover, one may speculate that since the pH in the Golgi lumen is acidic (Demaurex et al. Reference Demaurex, Furuya, D'Souza, Bonifacino and Grinstein1998), the passing proteins may be in partially-unfolded states, or at least in more dynamic conformations than when fully folded, when they acquire Cu here. We want to point out that the in vitro work on CP folding (reported on above) included only unfolding experiments. Today, it has become possible to produce recombinant CP (Maio et al. Reference Maio, Polticelli, De Francesco, Rizzo, Bonaccorsi di Patti and Musci2010), and thus direct in vitro folding experiments should be attainable.
Another emerging topic with many mechanistic questions unresolved is the possible roles of Cu (and copper proteins) in cell signaling (Chang, Reference Chang2015; Grubman & White, Reference Grubman and White2014; Matson Dzebo et al. Reference Matson Dzebo, Arioz and Wittung-Stafshede2016). Evidence for Cu participation in cell signal transduction has been found in the mitogen-activated protein kinase signaling pathways. In order for the kinase Mek1 to phosphorylate ERK (Turski et al. Reference Turski, Brady, Kim, Kim, Nose, Counter, Winge and Thiele2012), Mek1 has to bind two Cu ions (Turski et al. Reference Turski, Brady, Kim, Kim, Nose, Counter, Winge and Thiele2012). How Mek1 obtains Cu is unclear, but Atox1 and CCS are proposed candidate proteins. Another signaling system that is dependent on Cu is the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) proteins, which are inflammatory transcription factors. Increased Cu levels in the cytoplasm inhibit NFκB signaling via blocking of NFκB dimer formation and nuclear transport (Kenneth et al. Reference Kenneth, Hucks, Kocab, McCollom and Duckett2014). One may speculate also here that Atox1 could provide the Cu ions that mediate NFκB inhibition in the cytoplasm. In addition to pure Cu(I) transport, due to the low reduction potential of the cysteine sulfurs, Atox1 has been proposed to participate in cellular redox homeostasis (Hatori & Lutsenko, Reference Hatori and Lutsenko2013). Thus, in addition to direct Cu transfer, oxidation of cysteine sulfurs in Atox1 may be a process that releases Cu ions in the cytoplasm under certain conditions. Such redox reactions may be rescued by glutaredoxin 1 as it was found to resolve disulfides in Atox1, created by oxidative stress, and reload Cu onto Atox1 via direct protein–protein interactions in vitro (Brose et al. Reference Brose, La Fontaine, Wedd and Xiao2014). From a folding perspective, one may ask if Cu binding to these signaling proteins affect protein conformation and/or dynamics; if so, are such changes part of the mechanism by which signal transmission to the next protein takes place. Clearly, for XIAP discussed above, structural changes due to Cu binding appear to act as a regulatory switch (Mufti et al. Reference Mufti, Burstein and Duckett2007).
When we recently counted all human proteins known to bind Cu, we arrived at a set of 42 Cu-binding proteins and 12 Cu-transporting proteins, which corresponds to about 0·5% of the total human proteome (Blockhuys et al. Reference Blockhuys, Celauro, Hildesjo, Feizi, Stal, Fierro-Gonzalez and Wittung-Stafshede2017). This is an estimate as one must note that there may be more proteins to be discovered as Cu-binding proteins and some proteins defined as Cu-binding proteins today may eventually be found not to use the Cu for function. Nonetheless, the following math clearly demonstrates the many remaining unknowns there are around copper protein loading and folding reactions in humans: For the 42 identified Cu-binding proteins, the transport proteins that loads them with Cu is known for less than half (Blockhuys et al. Reference Blockhuys, Celauro, Hildesjo, Feizi, Stal, Fierro-Gonzalez and Wittung-Stafshede2017), and for only a handful of these 42 proteins has in vitro folding mechanisms (and roles of Cu binding) been explored.
7. Outlook
We find it appropriate to end this text with a quote from Richard Feynman stated in 1963: ‘…everything that living things do can be understood in terms of the jigglings and wigglings of atoms’. Biophysical knowledge of ‘jigglings and wigglings’ of copper atoms in protein folding reactions and the connected metal transport proteins (as reported on in this review) may aid in the development of new approaches to target disorders involving an imbalance in Cu metabolism as well as dysfunction of copper-dependent enzymes. Notably, these disorders include, but are not limited to, both cancer (Denoyer et al. Reference Denoyer, Masaldan, La Fontaine and Cater2015; Rigiracciolo et al. Reference Rigiracciolo, Scarpelli, Lappano, Pisano, Santolla, De Marco, Cirillo, Cappello, Dolce, Belfiore, Maggiolini and De Francesco2015) and neurodegeneration (Faller et al. Reference Faller, Hureau and Berthoumieu2013; Grubman & White, Reference Grubman and White2014). Copper binding to amyloidogenic proteins forming the basis of neurodegenerative disorders was discussed above (Section 5·3) so we focus on cancer here.
Cu is a key component of many essential enzymes and, therefore, it is not unexpected that Cu is required for characteristic phenomena involved in cancer progressions such as proliferative immortality, angiogenesis and metastasis (Denoyer et al. Reference Denoyer, Masaldan, La Fontaine and Cater2015; Hanahan & Weinberg, Reference Hanahan and Weinberg2011). In support of increased Cu demand in cancer, tissue and serum from cancer patients have increased Cu levels (Denoyer et al. Reference Denoyer, Masaldan, La Fontaine and Cater2015). Because of the recognized importance of Cu in cancer, general Cu-chelating compounds have been assessed as anti-cancer therapies (Alvarez et al. Reference Alvarez, Xue, Robinson, Canalizo-Hernandez, Marvin, Kelly, Mondragon, Penner-Hahn and O'Halloran2010; Brewer, Reference Brewer2009; Denoyer et al. Reference Denoyer, Masaldan, La Fontaine and Cater2015). For a tumor to grow larger than a few mm, angiogenesis is needed and Cu ions influence several molecular pathways that lead to a pro-angiogenic response (Rigiracciolo et al. Reference Rigiracciolo, Scarpelli, Lappano, Pisano, Santolla, De Marco, Cirillo, Cappello, Dolce, Belfiore, Maggiolini and De Francesco2015), including direct binding to and promoting expression of, angiogenic factors (Kenneth et al. Reference Kenneth, Hucks, Kocab, McCollom and Duckett2014; Pan et al. Reference Pan, Kleer, van Golen, Irani, Bottema, Bias, De Carvalho, Mesri, Robins, Dick, Brewer and Merajver2002). Cu also affects the ability of cancer cells to metastasize through the activation of metabolic and proliferative enzymes (Denoyer et al. Reference Denoyer, Masaldan, La Fontaine and Cater2015). For example, lysyl oxidase (Cu protein that is metal loaded in the secretory path) crosslinks extracellular matrix proteins and is secreted by cancer cells to create a pro-metastatic niche (Erler et al. Reference Erler, Bennewith, Cox, Lang, Bird, Koong, Le and Giaccia2009; Siddikuzzaman et al. Reference Siddikuzzaman, Grace and Guruvayoorappan2011). To form metastases, a first step is local cell invasion and migration at the primary site (Hanahan & Weinberg, Reference Hanahan and Weinberg2011) which is mediated by actin-rich plasma membrane protrusions (i.e. filopodia, lamellipodia and invadopodia). Intriguingly, Cu was reported to be enriched in such protrusions of endothelial cells (Finney et al. Reference Finney, Mandava, Ursos, Zhang, Rodi, Vogt, Legnini, Maser, Ikpatt, Olopade and Glesne2007). This observation, together with the findings mentioned above about Atox1 at lamellipodia edges in cancer cells (Blockhuys & Wittung-Stafshede, Reference Blockhuys and Wittung-Stafshede2017), suggests a role of copper and copper proteins in cancer cell migration. Because the ability of cancer cells to form distant metastases is the main cause of mortality in patients with solid tumors (Finney et al. Reference Finney, Mandava, Ursos, Zhang, Rodi, Vogt, Legnini, Maser, Ikpatt, Olopade and Glesne2007), new mechanistic knowledge of the roles of Cu-transport proteins and partner proteins in tumor cell invasion and migration processes has the potential to act as a unique scientific platform for new anti-cancer drug concepts.
There are likely more biological processes to be discovered to involve copper; for example, pioneering work has showed that Cu levels increased dramatically in phagosomes in the presence of bacteria (Wagner et al. Reference Wagner, Maser, Lai, Cai, Barry, Honer Zu Bentrup, Russell and Bermudez2005) and depletion of a particular Cu transporter (ATP7A) in cultured macrophage cells reduced their ability to kill E. coli (White et al. Reference White, Lee, Kambe, Fritsche and Petris2009). This study provided compelling support for the idea that the immune response exploits Cu toxicity towards pathogenic bacteria. However, the underlying mechanisms, including source of Cu, signaling pathways and involved Cu transport proteins, are not fully understood. In addition, there is no information on which immune cells this applies to, how many pathogens are affected by Cu in vivo, and the molecular mechanism of Cu-mediated killing. With more understanding one may imagine a powerful dual antibacterial strategy in which Cu supply to immune cells are boosted in combination with specific inhibition of the bacterial Cu transport proteins that export Cu out of the bacterial cells (Fig. 16).

Fig. 16. Illustration of how increased Cu supply to macrophages and simultaneous inhibition of bacterial Cu export may be a new dual strategy to combat bacterial infections through Cu toxicity in phagosomes.
To conclude, there is a lot of gratifying research ahead in this field. To be successful in such endeavors, a combination of biophysical, chemical and biological studies, on individual protein systems as well as with whole cells and model organisms, are desired.
Acknowledgements
Research on copper proteins in Wittung-Stafshede's laboratory is supported by the Swedish Research Council and the Knut and Alice Wallenberg foundation. We thank previous advisors for bringing us into this field of research, and all students around the world that have contributed to the work mentioned. We thank Helena Öhrvik, Örjan Hansson, Michael Kovermann, Erik Zedlak, Gabriel Zoldak, Maria Matson-Dzebo and Min Wu for help with graphical illustrations.

















