


Introduction
Gaining a detailed understanding of the biological conversion of ATP to ADP is crucial for understanding the nature of energy transduction in life processes and also for practical understanding of the action of molecular motors (Boyer, Reference Boyer1997; Weber & Senior, Reference Weber and Senior1997). At present it seems that some of the details of this process are not fully clear (Abrahams et al. Reference Abrahams, Leslie, Lutter and Walker1994; Boyer, Reference Boyer1993, Reference Boyer1997; Fersht, Reference Fersht1999; Junge et al. Reference Junge, Sielaff and Engelbrecht2009; Wang & Oster, Reference Wang and Oster1998; Weber & Senior, Reference Weber and Senior1997). One of the major unresolved questions is associated with the understanding of the way the free-energy of the chemical process is converted to the conformational changes that eventually lead to torque generation and work. This challenge became even more exciting in view of the remarkable progress made in the field of single molecule studies that directly visualized the unidirectional rotation of the central stalk (subunit γ) (Adachi et al. Reference Adachi, Oiwa, Nishizaka, Furuike, Noji, Itoh, Yoshida and Kinosita2007; Okuno et al. Reference Okuno, Fujisawa, Iino, Hirono-Hara, Imamura and Noji2008; Shimo-Kon et al. Reference Shimo-Kon, Muneyuki, Sakai, Adachi, Yoshida and Kinosita2010). Among the many intriguing findings of the single molecule studies, one has been the discovery of the ATP binding and catalytic dwells (Adachi et al. Reference Adachi, Oiwa, Nishizaka, Furuike, Noji, Itoh, Yoshida and Kinosita2007) where the γ subunit rotation stops intermittently during the ATP binding and catalytic phases occurring in the α/β subunits. Another recent discovery is that the rotational process continues in the right direction even when major parts of the γ subunit are removed (Chiwata et al. Reference Chiwata, Kohori, Kawakami, Shiroguchi, Furuike, Adachi, Sutoh, Yoshida and Kinosita2014; Uchihashi et al. Reference Uchihashi, Iino, Ando and Noji2011). These findings seem to present a problem for models that have postulated steric van der Waals (vdW) as the most important driving force behind the torque generation.
Our previous work started to explore the chemical reaction in F1-ATPase (Strajbl et al. Reference Strajbl, Shurki and Warshel2003) and then proceeded to study the conformational landscape using a CG model (Mukherjee & Warshel, Reference Mukherjee and Warshel2011), and generated the rotary–chemical surface elucidating the nature of the coupling in F1-ATPase. This work has reproduced the intriguing observation of the 80°/40° substep feature in F1-ATPase and has provided a clear understanding of the electrostatics driven process of the catalytic dwell around 80° as the central stalk γ rotates through its 120° steps (Mukherjee & Warshel, Reference Mukherjee and Warshel2011). Another recent work (Mukherjee & Warshel, Reference Mukherjee and Warshel2015) has provided a more quantitative understanding of the generated torque in the system under different conditions and elucidated the nature of the free-energy landscape required for the robust rotary–chemical coupling in F1-ATPase. Furthermore, this study dissected the role of the γ-subunit in details and demonstrated the importance of the electrostatics-based mechanism underlying the torque generation process in systems with truncated γ subunits (Mukherjee & Warshel, Reference Mukherjee and Warshel2015). Although, our previous studies on F1-ATPase has elucidated the fundamental electrostatics-driven mechanism underlying the rotary–chemical coupling and torque generation processes, they have not asked whether the position of the catalytic dwell has a specific biological advantage for the efficiency of the system. Here we attempted to explore whether the position of the catalytic dwell plays a role in providing advantage to the overall torque generation and the utility of the chemical energy. It is found that the position of the dwell might not be essential for the efficiency of the torque generation in F1-ATPase.
Outlining the problem
Despite the progress in elucidating the role of key structural elements of the F1-ATPase molecular motor, we still face several challenges of generating structure/energy correlations that will tell us how the motor really works. These challenges range from the need to gain clearer understanding of the reaction mechanism and the dependence of the reaction barrier on the conformational states, as well as the determination of the steps when the chemical energy is released and, of course, finding how this energy release is coupled to the conformational changes and to the overall torque generation process.
The F1-ATPase subunit arrangements and the schematic ATPase cycle are presented in Fig. 1. The challenge is to reproduce the key observed facts from structure-based modeling approaches and then to use the corresponding computation to determine the origin of the different effects and their role in converting the chemical energy to work. Most of the theoretical studies that have attempted to resolve some of the above issues have not provided effective answers. An example is provided by Pu & Karplus (Reference Pu and Karplus2008) that attempts to explore the substeps of the γ subunit rotation and to simulate the torque generated by this rotation. This work used a CG model and targeted molecular dynamics (TMD) to rotate the γ subunit between two observed structures. Unfortunately, the TMD was done in much smaller time scale than the experimentally observed millisecond time and more importantly, the simulations were done without considering the effect of the chemical energy on γ rotation. This means that the TMD actually probed the applied external force rather than the actual torque acting on the system due to the chemical process of ATP hydrolysis. The problems faced by the approach discussed above, as well as most other approaches that use large external force to explore the inherent molecular mechanisms of biological motors, partially reflect the fact that molecular understanding of the mechano-chemical processes requires to overcome the challenge of obtaining reliable free-energy landscapes for complex systems.

Fig. 1. (a) Showing the atomistic structure of the F1-ATPase where the central stalk γ, subunits α and β are shown in red, orange and cyan, respectively, while bound nucleotides are in magenta. (b) Schematic representations of the ATP hydrolysis cycle during the 120° rotation of the γ subunit, where Scheme (I) and (II) describe two of the most likely mechanism. The stage between C to B may involve three nucleotides supporting tri-site mechanism (not shown explicitly) (Shimo-Kon et al. Reference Shimo-Kon, Muneyuki, Sakai, Adachi, Yoshida and Kinosita2010).
The above challenge has been addressed by our previous CG study (Mukherjee & Warshel, Reference Mukherjee and Warshel2011), where we calculated the rotary–chemical free-energy landscape and obtained a reasonable rotation time, which is one of the key experimental observations about the action of the system. Furthermore, the rotary–chemical surface have provided an understanding of the reason for observing the catalytic dwell around 80° along the γ rotational path of F1-ATPase. In a subsequent work (Mukherjee & Warshel, Reference Mukherjee and Warshel2015), we addressed the challenge of evaluating the actual torque obtained from the CG rotary–chemical surface (see the discussion below). However, our initial attempt to evaluate the torque has not considered the chemical barrier and only looked at the effect of releasing the chemical free energy at different possible stages of the conformational cycle.
Now, we would like to explore the relationship between the chemical free-energy landscape and the generated torque in a more rigorous way. Here we note the interesting experimental study of Watanabe et al. (Reference Watanabe2012), which explored the effect of mechanical modulation of the catalytic power of F1-ATPase. This study indicated that the barrier is reduced upon increase in the rotational angle of the γ subunit. Thus moving from γ = 0° to the catalytic dwell point (γ = 80°) results in a decrease in the barrier (although, moving past the 80° is blocked in our landscape due to very high electrostatic free energy). The substep feature of the electrostatic free-energy surface obtained in (Mukherjee & Warshel, Reference Mukherjee and Warshel2011, Reference Mukherjee and Warshel2015) is used to construct the schematic combined rotary–chemical surface shown in Fig. 2, where it is assumed that the chemical barrier has its lowest value at the point of the catalytic dwell (γ = 80°). This simplified surface and its modified versions can be used to explore whether there is any evolutionary advantage of having the lowest chemical barrier at a specific position along the γ rotational coordinate.

Fig. 2. A schematic rotary–chemical surface for the ATP hydrolysis and the 120° γ rotation of F1-ATPase. The X-axis represents the change in the α/β catalytic subunits from D1E2T3 to E1T2D3, as well as the chemical transformation from ATP to ADP. The ATP and ADP states are designated by (T) and (D), respectively. The Y axis represents the γ rotation from 0° to 120°, while the Z axis represents the free energy of the system. The least free-energy path of the rotary–chemical process is shown with a white line on the 3D surface. The insets on the top left and top right corners show the free-energy profiles for γ = 80° and the chemical barriers at all values of γ, respectively.
Methods
In order to elucidate the molecular origin of the rotary–chemical coupling, it is important to model diverse events occurring at different spatiotemporal regime while being able to capture the structure function relationship at the same time. The theoretical modeling approach should provide a quantitative understanding of the dependence of the chemical barrier on the conformational changes, and the coupling between the conformational changes of the α/β and γ subunits. It is crucial to realize that the theoretically computed torque and the rotational time must be related to the underlying molecular free-energy landscape of the whole system in order to gain a quantitative knowledge of the biological function. To advance in this direction, we need to calculate the chemical and conformational features of the overall functional surface. The dependence of the chemical surface on the α/β conformational path can be evaluated using the EVB method (Kamerlin & Warshel, Reference Kamerlin and Warshel2011) as was done in our preliminary study (Strajbl et al. Reference Strajbl, Shurki and Warshel2003). However, for the purpose of the present study it is sufficient to use experimental estimates of the chemical barrier and explore the effect of the position of the lowest barrier on the torque generation in a parametric way.
The conformational landscape has been evaluated by a CG model (Messer et al. Reference Messer, Roca, Chu, Vicatos, Kilshtain and Warshel2010) that describes the main chains by an explicit model and represents the side chains by a simplified united atom model. The model involves a unique treatment of the electrostatic energy including a self-energy term that makes it more reliable than other current models. The details of our model are given elsewhere (Dryga et al. Reference Dryga, Chakrabarty, Vicatos and Warshel2011; Messer et al. Reference Messer, Roca, Chu, Vicatos, Kilshtain and Warshel2010; Vicatos et al. Reference Vicatos, Rychkova, Mukherjee and Warshel2014). After combining the chemical and conformational surfaces into an effective rotary–chemical surface (as shown in Fig. 2), our next task is to obtain the torque from the generated landscape and this can be done in several ways, including those discussed in (Arai et al. Reference Arai, Yukawa, Iwatate, Kamiya, Watanbe, Urano and Noji2014; Panke et al. Reference Panke, Cherepanov, Gumbiowski, Engelbrecht and Junge2001; Sakaki et al. Reference Sakaki, Shimo-Kon, Adachi, Itoh, Furuike, Muneyuki, Yoshida and Kinosita2005). The first is to consider the free energy released upon rotation of the γ subunit (assuming that it is all transferred to the magnetic beads used to probe the rotation). In this case we have:
 $$\Delta {G_{{\rm Torque}}} = \int \tau d\varphi = \int {\bar \tau {\rm d}\varphi}, $$
$$\Delta {G_{{\rm Torque}}} = \int \tau d\varphi = \int {\bar \tau {\rm d}\varphi}, $$
where τ is the angular torque and
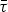 $\bar \tau $
is the average angular torque.
$\bar \tau $
is the average angular torque.
The relevant free energy can be evaluated from the potential of mean force (PMF) along the rotation coordinate, (denoted by φ), using a free-energy perturbation-type expression:
 $$\eqalign{& {{\rm e}^{ - \beta \Delta {G_{{\phi _m} \to {\phi _{m + 1}}}}}} = \cr & \displaystyle{{\mathop \int {{\rm e}^{ - \beta \left[ {{U_{X,{\phi _{m + 1}}}} - {U_{X,{\phi _m}}}} \right]}}{{\rm e}^{ - \beta \left[ {{U_{X,{\phi _m}}}} \right]}}{\rm d}X\delta \left( {\phi - {\phi _m}} \right){\rm d}\phi} \over {\mathop \int {{\rm e}^{ - \beta \left[ {{U_{X,{\phi _m}}}} \right]}}{\rm d}X\delta \left( {\phi - {\phi _m}} \right){\rm d}\phi}}} $$
$$\eqalign{& {{\rm e}^{ - \beta \Delta {G_{{\phi _m} \to {\phi _{m + 1}}}}}} = \cr & \displaystyle{{\mathop \int {{\rm e}^{ - \beta \left[ {{U_{X,{\phi _{m + 1}}}} - {U_{X,{\phi _m}}}} \right]}}{{\rm e}^{ - \beta \left[ {{U_{X,{\phi _m}}}} \right]}}{\rm d}X\delta \left( {\phi - {\phi _m}} \right){\rm d}\phi} \over {\mathop \int {{\rm e}^{ - \beta \left[ {{U_{X,{\phi _m}}}} \right]}}{\rm d}X\delta \left( {\phi - {\phi _m}} \right){\rm d}\phi}}} $$
The overall PMF can be evaluated by Monte Carlo (MC) simulations over the surface, although this can also be done by using LD simulations along with a mapping potential (Kato & Warshel, Reference Kato and Warshel2005).
Results and discussion
Our analysis of the rotary–chemical coupling starts with the evaluation of the dependence of the ATP hydrolysis activation barrier on the conformational pathway (α/β subunit changes and γ subunit rotation). This was done here in a simplified way, assigning lower barrier at the catalytic dwell position (γ = 80°), following the observation of (Watanabe et al. Reference Watanabe2012). However, to check the importance of the dwell position for torque generation and thermodynamic efficiency in the F1-ATPase system, we also considered different options of coupling the chemistry at other values of the rotational coordinate. The conformational landscape has already been determined in our previous works (Mukherjee & Warshel, Reference Mukherjee and Warshel2011, Reference Mukherjee and Warshel2015) and here we explored the effect of combining the conformational landscape with the chemical landscape at different position of the rotational coordinate.
Before moving to the actual calculations, it is useful to comment about the requirements underlying efficient torque generation in the F1-ATPase system. This point is illustrated by two schematic landscapes shown in Fig. 3, where Fig. 3a has a similar trend to the rotary–chemical coupling obtained in our previous studies that reveal a least energy path along the diagonal of the coupled surface (Mukherjee & Warshel, Reference Mukherjee and Warshel2011, Reference Mukherjee and Warshel2015). In the schematic landscape of Fig. 3a , the resultant torque that rotates the γ subunit reflects the fact that the lowest point on the coupled landscape occurs where the rotational and chemical states are at 120° and ADP +Pi, respectively. In this case, the evolution of the system toward the 120° point hydrolyzes ATP and rotates the γ subunit in a unidirectional way. On the other hand, in Fig. 3b we have no net gradient for moving in the direction that rotates the γ from 0 to 120°. In this case, although the free energy decreases along the chemical coordinate as a result of the release of the chemical free energy, we still do not have an average net torque due to loss of effective coupling between the chemical and rotational coordinates. The relationship between the shapes of the landscape and the torque will guide us in the subsequent discussion and it will also serve to remind us that the torque cannot occur without proper coupling to the chemical energy.

Fig. 3. The relationship between the shape of the landscape and the generated torque is illustrated in the schematic landscapes for chemical and rotational coordinate (omitting the chemical or conformational barrier). Two extreme situations occur when the chemical free energy is tightly coupled (a) and loosely coupled (b) to the rotational coordinate. In (a) the chemical free energy decreases more when the rotational coordinate ϕ approaches 120° than when it approaches 0°. Hence the average trajectory ultimately move from ϕ = 0° to ϕ = 120° and the system generates torque. In (b) due to loose coupling of the rotational and chemical coordinate, the system can move to different rotational angles over time so that we obtain zero torque in the ϕ direction (this figure is a reproduction of Fig. 1 from (Mukherjee & Warshel, Reference Mukherjee and Warshel2015).
In order to study the advantage that F1-ATPase might have due to the catalytic dwell at γ = 80°, we added the chemical landscape at several regions of the conformational landscape and generated the surfaces shown in Fig. 4. In particular, we considered the case where chemical energy is not coupled to the conformational surface (Fig. 4a ), where chemical barrier is similar throughout the rotational path (Fig. 4b ), where the chemical barrier is lowest at γ = 20° (Fig. 4c ), the experimentally observed case where the chemical barrier is lowest at γ = 80° (Fig. 4d ) and where the chemical barrier is lowest at γ = 100° (Fig. 4e ). With the different rotary–chemical surfaces at hand (Fig. 4a–e ), we estimated the torque generated on each of those surfaces using the FEP formulation of Eq. (2). The corresponding PMFs are shown in Fig. 4f . As seen from the figure, there is negligible torque generation for the case when chemical energy is not coupled to the conformational landscape, which reflects the obvious fact that torque cannot be generated without expending chemical energy (red curve of Fig. 4f ). On the other hand, significant torque is obtained for other cases where the chemical energy of ATP hydrolysis is coupled to the conformational surfaces. In the case of Fig. 4d we obtained a maximum free energy drop of around 7·6 kcal mol−1 between the 0° and 120° states in the direction of the rotational coordinate that can generate a maximum average torque of about 53 pN.nm on the γ rotational surface. This value lies close to the experimentally observed average torque range of 40–50 pN.nm (Noji et al. Reference Noji, Yasuda, Yoshida and Kinosita1997; Panke et al. Reference Panke, Cherepanov, Gumbiowski, Engelbrecht and Junge2001). Now, the above torque was obtained by having the lowest chemical barrier at the observed catalytic dwell at γ = 80°. However, changing the position of the lowest chemical barrier with respect to the rotational angle (for e.g. γ = 20° or 100°) did not alter the generated torque significantly. Apparently, torque is also generated for the case where the chemical barrier is similar for all values of the rotational coordinate (Fig. 4b ), implying that there is no specific advantage of having the catalytic dwell at a specific position, as long as the chemical energy is properly coupled to the rotational coordinate so that net unidirectional torque could be generated. Our finding indicates that the rotary–chemical surface is sufficiently robust so that the position of the dwell is not crucial for the overall efficiency of F1-ATPase, especially in terms of torque generation. This result is consistent with the observation (Furuike et al. Reference Furuike, Nakano, Adachi, Noji, Kinosita and Yokoyama2011) that the bacterial V-type ATPase shows different rotational stepping behavior as compared to the F1-ATPase, while still functioning efficiently.

Fig. 4. Illustrating the relationship between the chemical barriers at different γ positions with the generated torque. The surfaces represent a single 120° rotational event of F1-ATPase, where the X-axis represents the α/β conformational changes and the Y-axis represents the γ rotation. The coordinate for the chemical transformation (i.e. ATP to ADP) lies on the X-axis parallel to the catalytic subunit changes. The points (T) and (D) represent the ATP and ADP states of the chemical process, respectively, whereas (chem) represents the chemical barrier. The maps represent wild-type surfaces for the following cases: (a) without the chemical surface; (b) with similar chemical barrier for all rotational γ values; (c) with reduced chemical barrier at γ = 20°; (d) with reduced chemical barrier at γ = 80°; and (e) with reduced chemical barrier at γ = 100°. The PMF calculated from all surfaces are shown in (f) where the red, green, blue, magenta and cyan curves correspond to the surfaces of (a) to (e), respectively.
Concluding remarks
In exploring the origin of the action of F1-ATPase and related systems, it is crucial to provide a clear structure function relationship of the rotary–chemical coupling. Such a relationship should be based on well-defined physical principles that can actually reproduce the observed coupling rather than modeling it using phenomenological assumptions. Here we are fortunate to have detailed and insightful experimental observations of the rotary–chemical behavior and torque generation for the F1-ATPase system, whose reproduction provides a powerful challenge for structure-based simulation approaches. In an effort to establish the origin of the rotary–chemical coupling and to simulate the experimental observations, it is important to clarify significant misunderstandings that have arisen. In particular, it is important to clarify that the experimentally observed torque and related observations cannot be reproduced without a proper coupling of the chemical and the conformational landscapes. Trying to force the γ stalk to rotate between different states of the enzyme can only reflect the applied TMD force used to generate the rotation (e.g. Pu & Karplus, Reference Pu and Karplus2008), rather than elucidate any inherent property of the system (stronger forces would simply lead to stronger response and faster rotation). The torque can be obtained from a structure-based simplified landscape of the entire system by several approaches, such as the one adopted in this study; calculating the PMF along the rotational degree of freedom by free-energy perturbation method on the rotary–chemical surface.
Our approach of combining the chemical energy profile with the CG conformational surface to give the rotary–chemical surface provided the unique ability to explore the possible role of having the catalytic dwell at different positions of the rotational angle. Our study indicates that despite nature's elegant design of an intriguing structure/energy relationship that results in having the chemical step at the 80° rotational angle, the position of the catalytic dwell does not seem to be essential for the torque generation process.
It would be worthwhile to note that the implications of the importance of dynamical effects in the chemical step (Watanabe et al. Reference Watanabe2012) are interesting, but problematic at the conceptual level. This point has been demonstrated carefully in our work (Kamerlin & Warshel, Reference Kamerlin and Warshel2010) where it was shown that the dynamical proposal (when formulated correctly) cannot account for any significant catalytic effect since it implies transfer of the conformational energy to the chemical coordinate and such energy is dissipated long before we have a productive fluctuation that leads to the chemical event. In other words, as shown in the only study that accurately simulated this effect (Pisliakov et al. Reference Pisliakov, Cao, Kamerlin and Warshel2009), the internal friction inside the protein completely dissipates the memory of the conformational motion in the millisecond time needed to wait for a productive chemical fluctuation. Interestingly, regardless of the conceptual basis, our findings are consistent with the findings of Watanabe et al. (Reference Watanabe2012) that concluded that the torque is influenced by the binding and release of the ligands and not so much by the catalytic event.
An interesting attempt to explore the conformational/chemistry landscape was presented in Watanabe et al. (Reference Watanabe, Hayashi, Ueno and Noji2013) using phenomenological two-dimensional rotational/chemical surfaces. Although such an approach is insightful, it does not provide a unique connection to the underlying structure function correlation of the enzyme. That is, the chemical coordinate should reflect the free-energy changes along the α/β subunits conformational changes, as well as, the ATP hydrolysis of the specific bound ATP (see Fig. 2 of the present work and Fig. 3 of Mukherjee & Warshel, Reference Mukherjee and Warshel2011). Unless the conformational landscape is known through structure based calculations (for e.g. the CG approach used in Mukherjee & Warshel, Reference Mukherjee and Warshel2011), it is very hard to separate the effect of the chemical reaction from the conformational effect.
As stated in the methods section, we have studied the issue of coupling between the ATP hydrolysis and the conformation of the α/β subunits using the EVB approach in Strajbl et al. (Reference Strajbl, Shurki and Warshel2003) that also generated the catalytic free-energy surface for different ATPase configurations. The same issue was also explored by Dittrich et al. (Reference Dittrich, Hayashi and Schulten2003) using a QM/MM energy minimization study that, however, did not involve proper sampling (see discussion in Kamerlin et al. Reference Kamerlin, Sharma, Prasad and Warshel2013). The conformational dependence of the catalytic step has been explored recently by Beke-Somfai et al. (Reference Beke-Somfai, Lincoln and Norden2013), who used QM/MM calculations and phenomenological description to model the process. It was concluded that the activation barrier is reduced significantly upon T to D change of the active site.
Considering the potential insights that can be gained from all atom explicit simulations (Warshel, Reference Warshel2014), it is quite tempting to assume that an understanding of molecular motor functionality can likewise be provided with all atom explicit models. Thus, it may seem reasonable to believe that the current reported all atom simulation approaches (Czub & Grubmuller, Reference Czub and Grubmuller2014; Nam et al. Reference Nam, Pu and Karplus2014; Okazaki & Hummer, Reference Okazaki and Hummer2013; Pu & Karplus, Reference Pu and Karplus2008) will be sufficient to provide a quantitative understanding of the underlying principles that can lead to unidirectional rotation in biologically relevant timescales generating the correct torque and can also reproduce the substep features along the rotary–chemical pathway of F1-ATPase. Here one must ask oneself, what is the actual information being provided by current atomistic simulation studies with explicit models and what is really needed in order to understand the rotary–chemical coupling with the currently available computational resources. It is needless to say that atomistic studies of local structural rearrangements in catalytic sites obtained from relatively smaller time scale simulation trajectories can benefit the understanding of the chemical, ligand binding and release processes greatly (for e.g. see Czub & Grubmuller, Reference Czub and Grubmuller2014; Nam et al. Reference Nam, Pu and Karplus2014; Okazaki & Hummer, Reference Okazaki and Hummer2013; Strajbl et al. Reference Uchihashi, Iino, Ando and Noji2003). However, the situation is very different when one tries to explore the molecular basis of the torque generation, unidirectionality and substep behavior of the enzyme occurring at much larger time scales. To realize the possible problems with explicit models it may be useful to consider a recent work (Nam et al. Reference Nam, Pu and Karplus2014) that claim to provide insight into the torque generation and rotary–chemical coupling in F1-ATPase. This work tried to explore the motion between the two X-ray structures close to the catalytic dwell and ATP waiting states (effectively near the 80° and 120° states shown in our study (Mukherjee & Warshel, Reference Mukherjee and Warshel2011)), using an explicit all atom model and TMD simulations with huge external forces. The TMD simulations were done for systems with different Pi occupations in the β E subunit, from the neighborhood of the catalytic dwell structure towards the ATP waiting structure. It was found that the system without Pi reached the ATP waiting state, while the one with Pi did not do so. Although this finding was presented as an advance in understanding the rotary–chemical coupling in the enzyme, it does not provide the needed information. That is, the difficulties of pushing one state to the other, by applying strong TMD forces within a very short time, does not tell us about the least free-energy path and functional free-energy barriers for the large-scale conformational changes. Similarly, such an approach does not provide a way to evaluate the torque generated during the actual biological process. In this respect, note that our previous CG study (Mukherjee & Warshel, Reference Mukherjee and Warshel2011) has already reproduced the key features along the rotary–chemical surface before it has been suggested by Nam et al. (Reference Nam, Pu and Karplus2014), namely, relatively small barrier for the 80° rotation (i.e. the ATP waiting state to the catalytic dwell state) followed by higher barriers for the 40° rotation (i.e. the catalytic dwell state to the next ATP waiting state), where the 40° rotation is tightly coupled to the α/β conformational changes. The key difference is that, in spite of providing interesting structural details of the key states involved along the rotary–chemical process, it is unlikely that even few TMD trajectories would capture the relevant free energies that can be used to generate a unified understanding of the F1-ATPase functionality.
The above discussion should serve as an illustration of the difficulties in using explicit models in trying to learn about the molecular origin of complex biological systems. Obviously, it is very tempting to assign observed functional behavior of the system to specific structural elements and to picoseconds-nanoseconds range dynamical features. However, a theoretically well-founded structure-based approach should attempt to reveal the mechanism of the complex biological system through construction of the relevant functional free-energy landscapes.
One of the most dominant proposals from some of the theoretical and experimental studies is that the torque in F1-ATPase is generated due to vdW or steric interactions (for e.g. see (Chiwata et al. Reference Chiwata, Kohori, Kawakami, Shiroguchi, Furuike, Adachi, Sutoh, Yoshida and Kinosita2014; Martin et al. Reference Martin, Ishmukhametov, Hornung, Ahmad and Frasch2014; Nam et al. Reference Nam, Pu and Karplus2014; Pu & Karplus, Reference Pu and Karplus2008). Unfortunately, such an implication based on the TMD simulations of F1-ATPase rotation is problematic (Nam et al. Reference Nam, Pu and Karplus2014; Pu & Karplus, Reference Pu and Karplus2008) as no proper calculation has yet established that the rotation is solely due to vdW interactions. In other words, implications that the studies of (Nam et al. Reference Nam, Pu and Karplus2014; Pu & Karplus, Reference Pu and Karplus2008) support the vdW idea overlooks the fact that these studies simply induced the rotation by TMD between the initial to final X-ray structures using large external force without evaluating the free-energy landscape and hence cannot correctly estimate the contribution of the vdW forces to such a landscape. The problem with the vdW idea is highlighted when one considers the experimental observations that the inherent F1-ATPase rotary property is still retained when one deletes major portion of the γ subunit embedded within the α/β subunit cavity (Chiwata et al. Reference Chiwata, Kohori, Kawakami, Shiroguchi, Furuike, Adachi, Sutoh, Yoshida and Kinosita2014). Moreover, one can only arrive at such conclusions after considering all aspects of the problem and after studying other relevant proposals, such as the role of electrostatics with valid theoretical models capable of doing so. Here it is important to realize that our studies actually produced for the first time a reasonable functional landscape for the rotary–chemical cycle (without forcing the system to rotate) and has obtained such a landscape through proper modeling of the long-range electrostatic forces of the system (Mukherjee & Warshel, Reference Mukherjee and Warshel2011, Reference Mukherjee and Warshel2015), while omitting the vdW forces (whose contributions to the landscape was found to be minimal). Furthermore, the basic paradigm of electrostatics-driven F1-ATPase has been able to reproduce the behavior of several γ-truncated systems studied experimentally and have also provided a detailed knowledge of important and redundant areas of the γ actually involved in the rotary–chemical coupling and the substep feature of the enzyme (Mukherjee & Warshel, Reference Mukherjee and Warshel2015). In our view, our CG free-energy landscape studies has provided a major support for the electrostatics-driven mechanism, while one has yet to find any quantitative description of the proposed vdW driven mechanism of such processes.
Recent works has considered Brownian ratchet mechanism (Astumian, Reference Astumian2001; Frank & Gonzalez, Reference Frank and Gonzalez2010) as the underlying theoretical framework for many biological motors. This concept seems to stress the fact that ratcheted random thermal motions guide the action of molecular motors, but one still needs to elucidate the nature of the free-energy landscape that drives the ratcheting action. Once the free-energy surface is determined, the motor functionality and the corresponding vectorial processes are almost deterministic, since the individual rate constants follow the Boltzmann-based transition state theory. In fact, attributing functional features to fluctuations between several thermally accessible states (Frank & Gonzalez, Reference Frank and Gonzalez2010) (which might look to some as elements of a Brownian ratchet) is not so relevant to the question of how the motor operates. Having high chemical and conformational barriers is not fully compatible with the standard Brownian ratchet model and requires one to use a discrete chemical kinetics description (Kolomeisky & Fisher, Reference Kolomeisky and Fisher2007). Thus it is not useful to say that Brownian motions drive the motor (there are always thermal motions in all molecular systems), what counts are the rate limiting high barriers and the overall downhill free-energy profile, which is not related to or generated by the stochastic Brownian motions.
Overall, this study implies the fact that any special preference of the chemical event occurring at a specific rotational angle is not required for the efficient torque generation of F1-ATPase. The experimental finding of other ATPase systems where the catalytic dwell is at some other position apart from 80° (Furuike et al. Reference Furuike, Nakano, Adachi, Noji, Kinosita and Yokoyama2011) can be used as an evidence that the specific position of the stall might not be crucial for efficient energy conversion and torque generation. However, one must note that the chemical energy is absolutely needed as the driving force for the torque generation, but its actual position of coupling to the rotational coordinate might not be important and might just be a result of the structural arrangement of the multi-subunit enzyme.
Acknowledgements
This work was supported by the National Science Foundation grant MCB-1243719, National Institute of Health R01-AI055926, and National Cancer Institute grant 1U19CA105010. We also thank the University of Southern California High Performance Computing and Communication Center (HPCC) for computational resources.





 Open access
Open access