


Introduction
The synthesis of large DNA constructs for applications in Synthetic Biology is routinely carried out by multiple cycles of polymerase chain reaction (PCR) amplification of oligonucleotide pools, mismatch repair, cloning, sequencing and selection. Various approaches based on these principles have been used to assemble entire bacterial and mitochondrial genomes and regions of eukaryotic chromosomes of 1 megabase in length (Dymond et al. Reference Dymond, Richardson, Coombes, Babatz, Muller, Annaluru, Blake, Schwerzmann, Dai, Lindstrom, Boeke, Gottschling, Chandrasegaran, Bader and Boeke2011; Gibson et al. Reference Gibson, Glass, Lartigue, Noskov, Chuang, Algire, Benders, Montague, Ma, Moodie, Merryman, Vashee, Krishnakumar, Assad-Garcia, Andrews-Pfannkoch, Denisova, Young, Qi, Segall-Shapiro, Calvey, Parmar, Hutchison, Smith and Venter2010a, Reference Gibson, Smith, Hutchison, Venter and Merrymanb). In efforts to minimize errors in the sequence of the synthesized DNA, high-fidelity proofreading DNA polymerases are employed in the DNA amplification steps, and enzymatic mismatch repair is incorporated into the workflow (Kunkel, Reference Kunkel2004). Using such protocols mutations are kept to a low level, in the best cases to around 1 error in 106. This is a remarkable achievement considering that solid-phase oligonucleotide synthesis (Caruthers, Reference Caruthers1991), which provides the oligonucleotide-building blocks for gene synthesis, produces much higher error rates. These include nucleotide deletions due to inefficient coupling and nucleobase modifications arising from various side reactions. The time-consuming mismatch repair, cloning and sequencing steps used commonly in the synthesis of large DNA constructs are essential to edit out these errors, adding significantly to the cost of synthesis. This complex protocol presents a significant bottleneck in the supply of long DNA strands, for which demand is escalating. Further improvements in oligonucleotide synthesis chemistry could have a major impact in this area.
A more demanding factor that should be considered into future DNA synthesis strategies is the need for chemically modified DNA. It has been established that epigenetic modifications to cytosine in eukaryotic DNA control levels of gene expression through mechanisms that are only partly understood. The major epigenetic modifications are 5-methylcytosine (mC) and 5-hydroxymethylcytosine (hmC) (Bachman et al. Reference Bachman, Uribe-Lewis, Yang, Williams, Murrell and Balasubramanian2014; Branco et al. Reference Branco, Ficz and Reik2012), but their chemically related ‘oxidation products’ 5-formylcytosine (fC) and 5-carboxylcytosine (caC) are also present at lower levels (Pfaffeneder et al. Reference Pfaffeneder, Hackner, Truss, Munzel, Muller, Deiml, Hagemeier and Carell2011). Our current understanding is that these modified cytosine bases, along with A, G, C and T are the eight bases that constitute the human epigenome. Synthesizing DNA constructs (including entire genomes) containing modified cytosine nucleobases should enable gene expression to be precisely tuned, leading to more efficient engineered organisms and increased yields of gene products. This has implications for the production of economically important biomolecules, such as proteins and small molecule drugs. We should also not overlook the possibility that other hitherto undiscovered DNA modifications might exist, and could exert an additional level of control over the genome. In addition, the synthesis of DNA containing chemically modified bases or sugars has many potential applications in biology and nanotechnology. This makes the synthesis of chemically modified DNA even more important.
The PCR-based strategies described above are not viable for the synthesis of epigenomes. This is because they do not permit the site-specific incorporation of cytosine derivatives. All four modified cytosine bases form stable base pairs with guanine, so there is no obvious way to control their relative incorporation during PCR amplification. For example, mixtures of dCTP and dmCTP will produce PCR products with random mixtures of C and mC opposite the G bases in the template. To address this problem, a combination of PCR amplification followed by controlled insertion of modified oligonucleotides can be used for locus-specific insertion of modified cytosines, but it would be extremely tedious and inefficient to make large DNA constructs by this strategy. Templated ligation using ligase enzymes to assemble DNA entirely from synthetic oligonucleotides is another option. This requires the availability of highly pure oligonucleotides, precise control of stoichiometry between the constituent oligonucleotides and highly efficient and robust ligase enzymes. It is an area that is rife for development.
Given that every nucleotide in a DNA construct synthesized by ligation of oligonucleotides would originate from chemical synthesis, and the only remaining enzymatic steps would involve joining 5′-phosphate to 3′-hydroxyl groups of adjacent DNA strands, a simple question arises. Can the entire DNA synthesis process, including DNA strand ligation, be carried out chemically without the need for enzymes? This would be challenging because although it is possible to synthesize oligonucleotides up to 200 nucleotides in length for ligation, the aforementioned limitations of solid-phase synthesis make it impossible to make them in a pure state. This is in part due to the physical properties of the solid supports on which the synthesis is conducted, but it is also a consequence of the limitations of the chemistry, namely imperfect coupling and side-reactions that create mutagenic modifications to the nucleobases. For long oligonucleotides such modifications cannot be removed by purification, and error rates as high as 10−2 have been reported (Hecker & Rill, Reference Hecker and Rill1998). Circumventing the problem by assembling DNA from purified short oligonucleotides will only serve to increase the number of ligation steps that are necessary. This is a concern, because the phosphodiester bonds between oligonucleotide strands have proved to be difficult to create chemically in high yield, although some progress has been made using cyanogen bromide as a coupling agent (Luebke & Dervan, Reference Luebke and Dervan1991; Sokolova et al. Reference Sokolova, Ashirbekova, Dolinnaya and Shabarova1987). An alternative strategy is to design an artificial chemical linkage that mimics the natural phosphodiester bond and which can be formed in high yield in aqueous media from functional groups that are orthogonal to those present in DNA. This greatly broadens the chemical space that can be explored. The templated reaction between 3′-phosphorothioate and 5′-tosylate (or iodide) modified oligonucleotides has been studied (Herrlein et al. Reference Herrlein, Nelson and Letsinger1995; Xu & Kool, Reference Xu and Kool1998), but this chemistry does not satisfy all of the key requirements for robust chemical DNA synthesis which are: (i) the use of functional groups that are stable in aqueous media; (ii) the ability to synthesize bifunctional oligonucleotides with reactive groups at each end for use in multiple ligation reactions; (iii) a trigger to initiate the ligation reaction when participating oligonucleotides have been carefully hybridized to complementary splints under thermodynamic control (to arrange the DNA strands in the desired order by templated pre-assembly); and (iv) the creation of a stable backbone linkage.
An efficient chemical ligation method would increase the maximum achievable size of chemically synthesized oligonucleotides, and could in principle be optimized for the total chemical synthesis of large DNA constructs. It could be of value in nanotechnology, but its potential in biology will only be fully realized if DNA strands containing the artificial linkages can act as templates in replication and transcription. However, after billions of years of evolution the phosphodiester linkage has become ubiquitous in the DNA and RNA of living systems, and there is no reason a priori to expect DNA polymerases to tolerate an artificial backbone. Moreover, any change to the structure of DNA caused by the presence of a modified backbone is likely to alter the biophysical properties of the duplex, including its thermodynamic stability and the shape of the helix. Despite these reservations there is reason to believe that alterations to the chemical structure of the DNA backbone can be tolerated in biology; some DNA polymerases have the capacity to read through damaged DNA, and engineered enzymes can perform this feat with remarkable efficiency (d'Abbadie et al. Reference d'Abbadie, Hofreiter, Vaisman, Loakes, Gasparutto, Cadet, Woodgate, Paabo and Holliger2007). This provided us with further impetus for the design of a biocompatible DNA backbone linkage.
Design and evolution of a biocompatible modified DNA linkage
In order to produce a biocompatible chemical linkage we turned to the CuAAC reaction, which is the copper(I)-catalysed version of Huisgen's [3 + 2] azide-alkyne cycloaddition reaction (Huisgen, Reference Huisgen1963). The CuAAC reaction was discovered independently by Sharpless and Meldal in 2002 (Rostovtsev et al. Reference Rostovtsev, Green, Fokin and Sharpless2002; Tornoe et al. Reference Tornoe, Christensen and Meldal2002) and is the archetypal ‘click’ reaction. Click chemistry is a concept that was developed to join together organic molecules under mild conditions in the presence of a diverse range of functional groups (Kolb et al. Reference Kolb, Finn and Sharpless2001). We chose this highly selective and efficient reaction for nucleic acid ligation for several reasons: (i) alkynes and azides can readily be attached to nucleic acids; (ii) the reaction proceeds efficiently in aqueous media; (iii) it can be triggered by addition of CuI; and (iv) the resultant triazole ring is chemically robust and generally considered to be non-toxic. When we started this project ourselves and others had already shown that this reaction to be compatible with DNA (Gartner et al. Reference Gartner, Grubina, Calderone and Liu2003; Kanan et al. Reference Kanan, Rozenman, Sakurai, Snyder and Liu2004; Kumar et al. Reference Kumar, El-Sagheer, Tumpane, Lincoln, Wilhelmsson and Brown2007). The new challenge was to design a biocompatible triazole linkage that can be tolerated by DNA and RNA polymerase enzymes in vitro and in vivo.
When designing a triazole linkage certain constraints are imposed by synthetic chemistry. The chosen linkage must be compatible with phosphoramidite oligonucleotide synthesis and involve chemical moieties that are accessible without a disproportionately large synthetic effort. With this in mind initial investigations involved the synthesis of DNA containing triazole linkage A (Fig. 1d ). This is formed by the reaction between oligonucleotides functionalized with 3′-azido-dT and 5′-propargylamido-dT in the presence of a complementary template oligonucleotide (splint) which holds the reactants in close proximity. The click reaction occurs in the absence of the splint, but it is several orders of magnitude slower and requires much higher oligonucleotide concentrations. Splints confer another equally important advantage; they allow the assembly of multiple alkyne/azide oligonucleotides in the correct order prior to CuI-catalysis. This is due to the complementarity of Watson–Crick base pairs which allow thermodynamic control of the DNA assembly process. In our initial biochemical studies a click-ligated DNA strand containing triazole linkage A was used as a template in PCR (Fig. 1a ). Amplification was successful but DNA sequencing of the progeny strands revealed only a single thymidine at the ligation site instead of the two adjacent thymidines that were present in the original template (El-Sagheer & Brown, Reference El-Sagheer and Brown2009). Although this was disappointing it showed for the first time that DNA strands can be ligated to produce an artificial linkage that is tolerated during PCR amplification. This gave us encouragement, suggesting that it might be possible to design a truly biocompatible DNA linkage if the origin of the limitations of this first-generation triazole linkage could be rationalized. A possibly undesirable structural feature of triazole linkage A is the presence of the rigid amide bond which will prefer to adopt the planar trans-configuration (Fig. 1b ). This could cause the thymine base adjacent to the triazole to turn away from the incoming deoxyadenosine triphosphate (dATP) during replication (Fig. 1c ) resulting in a single-base deletion mutation. The lack of binding sites provided by the 3′-oxygen and 5′-methylene of normal DNA might also have caused the polymerase to skip over this region of the DNA template.
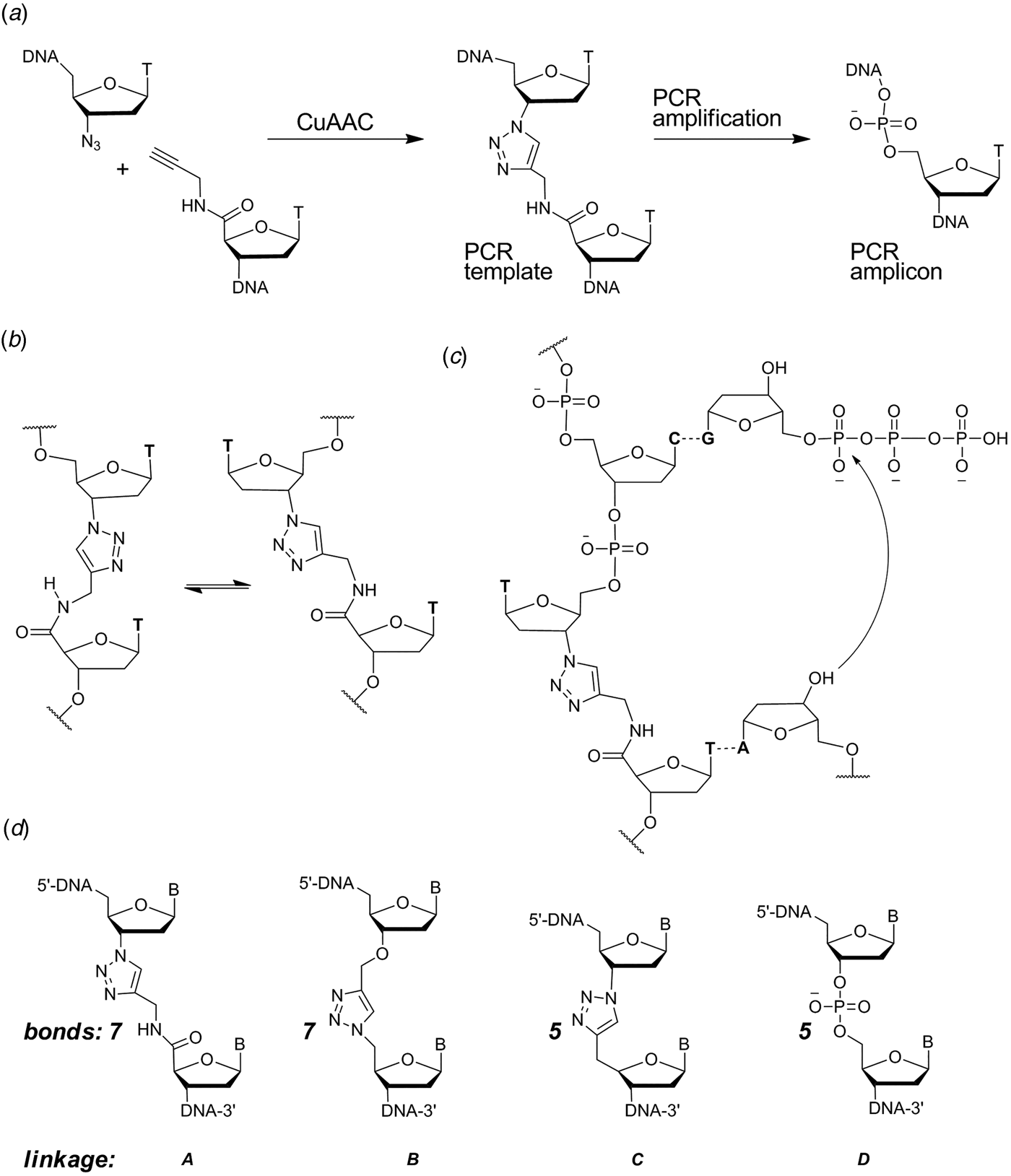
Fig. 1. (a) Synthesis and PCR amplification from the first generation triazole DNA backbone linkage. (b) Isomerization at the amide bond. (c) Replication bypasses one of the thymine bases around the triazole linkage. (d) First-generation triazole linkage A, second generation triazole linkage B, triazole linkage C in TL-DNA, canonical DNA linkage D. The number of bonds between the C3′ and C4′ atoms of adjacent sugar rings is indicated. From El-Sagheer & Brown (Reference El-Sagheer and Brown2012).
With these considerations in mind, a more flexible second-generation triazole linkage with a 5′-methylene, 3′-oxygen and greater conformational flexibility was synthesized (linkage B, Fig. 1d ) (El-Sagheer et al. Reference El-Sagheer, Sanzone, Gao, Tavassoli and Brown2011). The DNA strands required to create this linkage by click ligation (3′-alkyne oligonucleotide, 5′-azide oligonucleotide and bifunctional 3′-alkyne-5′-azide oligonucleotide) can all be made by the solid-phase phosphoramidite method with minimal changes to standard protocols. Several 81-mer DNA templates, each containing a single triazole, were made to test the biocompatibility of the artificial linkage, and in all cases the PCR reaction produced amplicons in which the entire sequence, including the bases around the triazole, were read through accurately. In a more rigorous experiment mimicking replication, successful linear copying of one of the 81-mers was then carried out to prove that PCR amplification of the chemically modified DNA did not have its origin in a rare event (El-Sagheer et al. Reference El-Sagheer, Sanzone, Gao, Tavassoli and Brown2011). We saw this as a major breakthrough, and to demonstrate that the CuAAC reaction could be used for the assembly of large DNA molecules with multiple click ligation sites, three 100-mer oligonucleotides were ligated to give a 300-mer DNA construct with two triazole linkages. PCR amplification and DNA sequencing indicated that the amplicon was a faithful copy of the original template. Next a 100-mer oligonucleotide with the required 5′-azide and 3′-alkyne functionalities was cyclized to produce a DNA construct containing a single triazole linkage to evaluate whether DNA polymerase could read through the triazole repeatedly. Rolling circle amplification (RCA) (Dean et al. Reference Dean, Nelson, Giesler and Lasken2001; Tsaftaris et al. Reference Tsaftaris, Pasentzis and Argiriou2010) was successfully carried out to produce a high molecular weight linear product containing multiple natural copies of the original cyclic template (El-Sagheer et al. Reference El-Sagheer, Sanzone, Gao, Tavassoli and Brown2011). It should be noted that PCR, RCA and linear copying were used to demonstrate that the triazole linkage is compatible with DNA polymerases. It was not our intention to use them in a long-term strategy for the synthesis of large chemically modified DNA constructs for the reasons discussed above.
Transcription through the triazole linkage
To evaluate the compatibility of the triazole linkage for RNA synthesis, we carried out in vitro transcription experiments using T7 RNA polymerase (T7-RNAP), an enzyme which is commonly used in the synthesis of small RNAs and proteins. The RNA transcript selected for this study contained the 54-mer sequence which inhibits the growth of Escherichia coli (Faubladier, Cam & Bouche, Reference Faubladier, Cam and Bouche1990). Two triazole-containing DNA template strands were studied, one with the triazole linkage inside the coding sequence and a second with the modification within the essential T7 RNA promoter region. When the triazole linkage was placed in the coding region the transcription reaction produced ~80% of the quantity of RNA obtained from the native control DNA template, and mass spectrometry showed that the two transcripts were identical (El-Sagheer & Brown, Reference El-Sagheer and Brown2011). This demonstrates that T7-RNAP can accurately transcribe through triazole-modified DNA to synthesize fully complementary RNA in good yield. This was the first example of transcription through a purely synthetic analogue of a DNA backbone, and it suggests that DNA constructs made by solid-phase synthesis and click ligation can be used directly in protein synthesis. When the transcription reactions were repeated with the triazole linkage in the promoter region of the DNA template there was no RNA product, presumably due to a lack of binding between the transcription initiation complex and the triazole DNA.
In vivo biocompatibility of the triazole linkage
Following on from the above successful in vitro experiments we investigated the properties of triazole-modified DNA in living organisms to determine whether native DNA and RNA polymerases can tolerate the triazole linkages. Initial experiments were carried out by transfecting E. coli. with a plasmid containing a triazole in each strand of its antibiotic marker gene (Fig. 2) (El-Sagheer et al. Reference El-Sagheer, Sanzone, Gao, Tavassoli and Brown2011). The number of E. coli colonies that we obtained was similar to the control in which a native plasmid was used. DNA sequencing of plasmids recovered from the triazole colonies confirmed that the nucleobases around the artificial linkages were copied correctly. We repeated the experiment in a strain of E. coli deficient in DNA repair (Baba et al. Reference Baba, Ara, Hasegawa, Takai, Okumura, Baba, Datsenko, Tomita, Wanner and Mori2006; Truglio et al. Reference Truglio, Croteau, Van Houten and Kisker2006) and this gave 93% of the number of colonies from the native plasmid. As previously, DNA sequencing confirmed that the region around the triazole was replicated accurately. Thus it appears that DNA repair in E. coli does not make a significant contribution to the biocompatibility of the artificial DNA backbone, and accurate replication occurs in the presence of the triazole linkages. However, there possibility that the success of these experiments was a consequence of selective pressure on the essential antibiotic resistant click-containing gene. To resolve this we repeated the work with a plasmid containing click DNA backbone linkages in the gene encoding the fluorescent protein mCherry which is clearly not required for survival of the bacteria (Sanzone et al. Reference Sanzone, El-Sagheer, Brown and Tavassoli2012). The effect of the proximity of the click linkers on biocompatibility was also probed by placing two click DNA linkers 4-bp apart in the region encoding the mCherry fluorophore. The click-containing plasmid was found to encode mCherry in E. coli at a similar level to the canonical equivalent. The ability of the cellular machinery to read through click-linked DNA was further probed using the click-linked plasmid to express mCherry in an in vitro transcription/translation system. As in the equivalent bacterial experiment, the yield and fluorescence of recombinant mCherry expressed from the click-linked plasmid was found to be the same as from the entirely natural plasmid. To determine whether biocompatibility could be achieved in eukaryotic cells which have more complex biochemistry we microinjected a plasmid containing triazole-linked DNA encoding mCherry into human MCF-7 breast cancer cells. Single-cell analysis of mCherry expression from the triazole-linked DNA indicated that the gene had been correctly expressed (Birts et al. Reference Birts, Sanzone, El-Sagheer, Blaydes, Brown and Tavassoli2014).

Fig. 2. Insertion of triazole linkage B into the BLA gene (blue) of plasmid DNA followed by transformation and growth of E. coli. The insert is yellow and the triazole linkages are purple.
Biophysical properties of triazole DNA
At first glance the triazole linkage does not appear to be a close structural analogue of a phosphodiester group in DNA, so how can we rationalize the fact that polymerases can copy a DNA template containing this unnatural backbone linkage? Clues come from the X-ray structure of the complex between Taq polymerase and DNA (Li et al. Reference Li, Korolev and Waksman1998). Around 12 nucleotides in the DNA template are bound to the enzyme, and if the click-ligated DNA template is substituted for the natural one, just one of the corresponding inter-nucleotide linkages will be unnatural. Hence, only one or two of many hydrogen-bonding interactions between the DNA backbone and the enzyme can be lost by substitution of triazole for phosphodiester (El-Sagheer et al. Reference El-Sagheer, Sanzone, Gao, Tavassoli and Brown2011). This might explain why triazole linkage B is read through correctly during PCR amplification. Further structural evidence comes from our nuclear magnetic resonance (NMR) study of a model DNA duplex containing a single triazole linkage in comparison with its unmodified counterpart (Dallmann et al. Reference Dallmann, El-Sagheer, Dehmel, Mügge, Griesinger, Ernsting and Brown2011). The triazole induces local structural changes but leaves the B-DNA duplex and Watson–Crick base pairs intact. Moreover, the location of the N(3)-atom of the triazole ring permits it to mimic the hydrogen bond acceptor properties of the DNA phosphodiester group, as it lies close to the position of a phosphate-branching oxygen (Fig. 3). This triazole nitrogen atom is known to have a large dipole moment and proven hydrogen bond acceptor capacity (Angell & Burgess, Reference Angell and Burgess2007), so it is likely that the triazole linkage can form H-bonds with DNA polymerase in a similar manner to a normal phosphodiester.

Fig. 3. Structure of triazole linkage B from Fig. 1 in DNA as determined by NMR spectroscopy. The backbone of the triazole DNA strand is distorted to accommodate its extra length and to allow efficient base stacking. This requires the 5′-carbon to point down, shifted by 3.2 Å relative to its position in the native duplex. The N3 nitrogen of the triazole duplex and phosphate oxygen of the canonical duplex are only 1.3 Å apart from El-Sagheer & Brown (Reference El-Sagheer and Brown2012).
The effect of the triazole linkage on the kinetics and thermodynamics of duplex formation has been explored by melting studies on a model 13-mer DNA duplex containing a single triazole linkage (Dallmann et al. Reference Dallmann, El-Sagheer, Dehmel, Mügge, Griesinger, Ernsting and Brown2011). Both triazole and unmodified control duplexes gave cooperative melting transitions (at 55 and 63 °C, respectively) with similar hyperchromicity, indicating that the triazole duplex is fully base paired. The effect of placing a mismatched base pair at the triazole site is particularly interesting. It lowers the duplex melting temperature by 11 °C on average, similar to the effect of a mismatch in the native duplex. The average decrease in hyperchromicity caused by a mismatch was 18.8% for the triazole duplex but only 7.5% for the native duplex, indicating that the combination of a triazole linkage and mismatched base pair causes a greater loss of base stacking than is observed in native mismatched DNA.
Our aforementioned NMR study indicated that the opening rates of the base pairs on either side of the triazole are increased relative to a normal duplex, causing destabilization of the four base pairs surrounding the triazole linkage (Dallmann et al. Reference Dallmann, El-Sagheer, Dehmel, Mügge, Griesinger, Ernsting and Brown2011). The free-energy required to dissociate each of the two base pairs on either side of the modified backbone is reduced by 11 kJ mol−1 compared with the native duplex, and the base pair on the 5′-side of the triazole modification is perturbed rather more than that on the 3′-side (7.2 and 3.4 kJ mol−1, respectively). Despite this destabilization, the duplex accommodates the unnatural linkage in a normal B-DNA helical structure with little distortion of the major and minor grooves.
The above biophysical data suggests a model of replication through the triazole linkage. When the correct dNTP is selected by the template-polymerase complex, a normal Watson–Crick base pair forms as the incoming nucleotide is incorporated. In contrast, on the rare occasion that a mismatched dNTP is inserted opposite the template base the duplex is strongly destabilized, and local melting triggers removal of the rogue nucleotide by the 3′-exonuclease domain of the polymerase enzyme. The double destabilization caused by the triazole linkage and mismatched base pair will be an easy target for the proofreading functionality of DNA polymerase.
Although the relative instability of DNA duplexes containing the triazole linkage does not appear to prevent correct read-through by DNA polymerases, with antisense applications in mind (Kurreck, Reference Kurreck2003) we were keen to design a more stable triazole linkage in DNA. Our approach was to strengthen the base pairs adjacent to the triazole linkage to compensate for the destabilization arising from the unnatural backbone. To achieve this we synthesized oligonucleotides containing a triazole linkage with an aminoethylphenoxazine nucleobase (G-clamp) on its 3′-side. G-clamp is an analogue of cytosine that forms an additional hydrogen bond with guanine and has enhanced base stacking (Fig. 4). We carried out ultraviolet melting studies on a series of DNA duplexes and DNA:RNA hybrids, comparing the stability of triazole G-clamp DNA with triazole DNA without the G-clamp, and with completely unmodified DNA. Duplex stability was greatly increased by the presence of the extra inter-base hydrogen bond of G-clamp, to the point where the triazole G-clamp duplex was more stable than the unmodified control. A single base pair mismatch located at or adjacent to the modifications was strongly destabilized, thus the triazole G-clamp combination is a potent mismatch sensor. A DNA strand containing the triazole G-clamp nucleotide surrogate was successfully amplified by PCR to produce unmodified copies of the original template, with deoxyguanosine inserted opposite to the G-clamp-triazole nucleotide analogue. This study showed for the first time that a DNA polymerase enzyme can correctly read through a combined backbone and nucleobase modification (El-Sagheer & Brown, Reference El-Sagheer and Brown2014).

Fig. 4. The triazole G-clamp nucleotide analogue base paired with guanine in complementary DNA. The additional steric bulk of the G-clamp nucleobase relative to cytosine is apparent.
Our research shows that a phosphodiester linker is not essential for joining together oligonucleotides to produce DNA for use in biological applications, enabling the possibility of replacing enzymatic ligation with efficient and selective chemical reactions. We have also carried out similar work on RNA (El-Sagheer & Brown, Reference El-Sagheer and Brown2010), and this is a fruitful area for future research. The chemical ligation approach is not necessarily limited to the linker reported here; alternative chemical reactions and backbone linkages might also be suitable for this purpose. However, this suggests an obvious question: is the precise structure of triazole linkage B in any way special, or could a wide variety of triazole linkages (or other heterocycles) also be biocompatible? There are five bonds between the C3′ and C4′ atoms of adjacent sugar rings in a normal DNA backbone. In contrast, triazole linkage B has seven bonds and greater conformational flexibility, possibly explaining why it destabilizes the DNA duplex. Paradoxically this flexibility might be necessary for its interactions with DNA polymerase, because triazole linkage A which contains a rigid amide bond is not read through properly by DNA polymerases. Triazole linkage C (Fig. 1) with 5 bonds and increased rigidity has been used to construct an oligo dT analogue in which every phosphate linkage is replaced by a triazole. This forms a stable duplex with oligo dA (Fujino et al. Reference Fujino, Yamazaki and Isobe2009, Reference Fujino, Miyauchi, Tsunaka, Okada and Isobe2013; Isobe et al. Reference Isobe, Fujino, Yamazaki, Guillot-Nieckowski and Nakamura2008). This attracted our attention, and we are currently studying oligonucleotides containing isolated triazole linkages of type C. Initial results indicate that this triazole linkage destabilizes duplexes in a similar manner to triazole linkage B. In addition, we have found that DNA polymerases do not read through linkage C in a clean manner, but give rise to a mixture of deletion mutations. Thus from a limited number of examples it seems that polymerase enzymes impose certain limitations on the design of functional artificial DNA backbones.
Outlook
As the biological significance and utility of DNA nucleobase modifications becomes more apparent, the need for a purely chemical approach to the synthesis of modified genes and genomes increases. In this context we have designed a triazole linkage that can be created by templated chemical ligation, is treated as normal by DNA and RNA polymerases, and is functional in bacterial and human cells. Click ligation is currently being investigated for the assembly of synthetic genes and for site-specific incorporation of synthetic fragments containing nucleobase and sugar modifications, fluorescent markers and other labels into DNA and RNA.
Acknowledgements
The authors wish to thank the European Community for the 7th Framework Programme grant ‘READNA’ and the UK BBSRC for the sLOLA grant BB/J001694/1: ‘Extending the Boundaries of Nucleic Acid Chemistry.’





 Open access
Open access