


Over the past 15 years various second-generation antipsychotic medications have become available to treat individuals with schizophrenia. Earlier publications of randomised trial results, primarily sponsored by the pharmaceutical industry, suggested that second-generation antipsychotics were superior to first-generation antipsychotics in the treatment of schizophrenia – reducing both symptoms and extrapyramidal side-effects (EPS) such as parkinsonism and tardive dyskinesia. Reference Arvanitis and Miller1–Reference Tollefson and Sanger6 A subsequent meta-analysis of short-term studies cast doubt on these conclusions, suggesting that the advantages of second-generation antipsychotics may have been exaggerated as a consequence of high doses of the comparator first-generation antipsychotic. Reference Geddes, Freemantle, Harrison and Bebbington7 This assertion was rebutted in a later meta-analysis of a larger database. Reference Davis, Chen and Glick8 More recent studies, conducted by independent investigators, and a third meta-analysis have found no substantial differences in EPS between these two classes of drugs. Reference Jones, Barnes, Davies, Dunn, Lloyd, Hayhurst, Murray, Markwick and Lewis9–Reference Leucht, Wahlbeck, Hamann and Kissling11 It has been suggested that the differences between the earlier studies and more recent studies may be related to the earlier studies commonly utilising haloperidol as the comparator – a high-potency antipsychotic often used at high doses Reference Hugenholtz, Heerdink, Stolker, Meijer, Egberts and Nolen12 – without prophylactic anticholinergics. Reference Rosenheck, Stroup, Keefe, McEvoy, Swartz, Perkins, Hsiao, Shumway and Lieberman13 Concerning tardive dyskinesia, Correll et al Reference Correll, Leucht and Kane14 reported an estimated annualised incidence of tardive dyskinesia with haloperidol of 5.4% compared with an estimated annual tardive dyskinesia risk with second-generation antipsychotics of 0.8% in randomised clinical trials, yielding a 4.6% greater attributable risk of tardive dyskinesia with haloperidol. This is consistent with older first-generation antipsychotic studies. Reference Chouinard, Annable, Mercier and Ross-Chouinard15–Reference Kane, Woerner, Weinhold, Wegner, Kinon and Borenstein18 This was supported by an analysis of the baseline CATIE Schizophrenia Trial data which found that patients with tardive dyskinesia were more likely to be receiving a first-generation antipsychotic compared with those without tardive dyskinesia. Reference Miller, McEvoy, Davis, Caroff, Saltz, Chakos, Swartz, Keefe, Rosenheck, Stroup and Lieberman19
In the initial analysis of the CATIE Schizophrenia Trial, Lieberman Reference Lieberman, Stroup, McEvoy, Swartz, Rosenheck, Perkins, Keefe, Davis, Davis, Lebowitz, Severe and Hsiao20 reported no significant difference in the percentage of patients who developed parkinsonism (mean Simpson–Angus Scale Reference Simpson and Angus21 (SAS) score ⩾1), akathisia (Barnes Akathisia Rating Scale Reference Barnes22 (BARS) global score ⩾3), or abnormal movements (Abnormal Involuntary Movement Scale Reference Guy23 (AIMS) global severity score ⩾2) in patients receiving olanzapine, quetiapine, risperidone, perphenazine or ziprasidone. However, significantly more patients receiving perphenazine discontinued treatment because of EPS than with the other antipsychotics. In a subsequent analysis of people receiving perphenazine who discontinued treatment, Stroup Reference Stroup, Lieberman, McEvoy, Swartz, Davis, Capuano, Rosenheck, Keefe, Miller, Belz and Hsiao24 found that these individuals did significantly better on quetiapine and olanzapine than on risperidone. These findings suggest that there is a subgroup of patients with schizophrenia who have a greater sensitivity to EPS.
The current analysis of the CATIE Schizophrenia Trial data was designed to more rigorously assess and compare the incidence of treatment-emergent parkinsonism, dystonia, akathisia and tardive dyskinesia associated with second-generation antipsychotics and perphenazine. In this analysis, onset of each side-effect was evaluated utilising multiple different approaches. We employed rating scale criteria as well as data on administration of concomitant medications, adverse event reports and treatment discontinuation related to such side-effects. We analysed the incidence of treatment-emergent side-effects as defined by meeting a priori criteria for each side-effect, discontinuing treatment for each side-effect and having medications added to treat the side-effect. We also applied both survival analysis (comparing time to first onset of each side-effect for individuals without the side-effect at baseline) and mixed models (comparing differences in change from baseline for all treated participants) to each type of side-effect.
Method
Study setting and design
The CATIE study was conducted between January 2001 and December 2004 at 57 US sites and included a series of treatment phases. Patients were initially assigned to olanzapine, perphenazine, quetiapine, risperidone or ziprasidone under double-blind conditions (Phase 1). Patients with tardive dyskinesia at baseline (n=231, 15% of the sample) were excluded from randomisation to perphenazine and were assigned to one of the four second-generation antipsychotics (Phase 1a). Ziprasidone was added to the trial after 40% of the patients had been enrolled. Randomisation thus took place under four separate strata within which patients had an equal chance of being randomly assigned to the treatments: including patients with tardive dyskinesia pre-ziprasidone (randomised equally to olanzapine, quetiapine or risperidone), excluding patients with tardive dyskinesia preziprasidone (randomised equally to olanzapine, quetiapine, risperidone or perphenazine), and these same two groups in the cohort of patients after ziprasidone was added.
Patients who discontinued their first treatment were invited to participate in subsequent phases of the trial. The data presented in the current analyses only deal with the time from initial randomisation until the first medication was discontinued for patients with (Phase 1a) or without (Phase 1) tardive dyskinesia.
Participants
The study was approved by an institutional review board at each site. Eligible patients were 18–65 years old with a diagnosis of schizophrenia, as determined on the basis of the Structured Clinical Interview of the DSM–IV, and were able to take oral antipsychotic medication. Patients or their guardians provided written informed consent. Patients were excluded if they had: a diagnosis of schizoaffective disorder; DSM–IV mental retardation or other cognitive disorders; an unstable serious medical condition; past adverse reactions to a proposed treatment; treatment-resistant schizophrenia; or if they were in their first episode of schizophrenia, pregnant or breast-feeding.
Interventions
Identical capsules contained olanzapine (7.5 mg), quetiapine (200 mg), risperidone (1.5 mg), perphenazine (8 mg) or ziprasidone (40 mg). Ziprasidone was approved for use by the US Food and Drug Administration (FDA) during the trial and was added in January 2002. Medications were flexibly dosed with one to four capsules daily, as judged by the study doctor. Concomitant medications were permitted, except for additional antipsychotic agents. Further details about masking, later phases of treatment, and modal dosing have been presented elsewhere. Reference Lieberman, Stroup, McEvoy, Swartz, Rosenheck, Perkins, Keefe, Davis, Davis, Lebowitz, Severe and Hsiao20,Reference Stroup, McEvoy, Swartz, Byerly, Glick, Canive, McGee, Simpson, Stevens and Lieberman25
Measures
Basic socio-demographic data documented age, race, ethnicity, gender, education, marital status and whether each patient had a recent exacerbation of their illness (i.e. hospitalisation or other increased service use). Sites were classified into six types: private non-profit, private practice, state mental health agency, university, Veterans Affairs, and other.
Extrapyramidal side-effects were measured using the six-item SAS (each scored 0–4), Reference Simpson and Angus21 the global clinical assessment item of the BARS Reference Barnes22 and the first seven items from the AIMS measure of tardive dyskinesia. Reference Guy23 These measures were collected at baseline, months 1 and 3, and quarterly thereafter until 18 months or time of treatment discontinuation. Data on concomitant medications, reasons for treatment discontinuation and reported adverse events were also used to identify onset, and to identify the occurrence of any acute dystonic reactions. Two of the authors (D.M. and S.C.) conducted a masked adjudication of physician reports to classify cases in which treatment was discontinued or concomitant medications were added for each of the four side-effect syndromes.
Criteria for parkinsonism, akathisia and tardive dyskinesia were determined a priori, preceding the analysis of the data. Patients were considered to have met criteria for parkinsonism if they scored 1 (mild) on at least two of the six SAS items or 2 (moderate) on one of the items. (This method of parkinsonism case identification is based on the method developed by Schooler & Kane Reference Schooler and Kane26 for identifying ‘cases’ of tardive dyskinesia using the AIMS, discussed below.) Because signs of parkinsonism are far more common than those of tardive dyskinesia, this highly sensitive threshold was expected to generate high rates of caseness and to help determine whether there were more subtle differences between the medications. Cases of parkinsonism were further identified if patients were started on an antiparkinsonian medication or were identified by their doctor as having discontinued their antipsychotic medication owing to parkinsonism. The summary score of all six SAS items was also used as a continuous measure.
Patients were considered to have met criteria for akathisia if they scored at least 2 (mild) on the BARS global item, if akathisia was specifically given as the reason for starting any medication, or if they were identified by their doctor as having discontinued their antipsychotic medication owing to akathisia. The summary score of the BARS global item was also used as a continuous measure.
Patients were considered to have met criteria for tardive dyskinesia if they met Schooler–Kane criteria, Reference Schooler and Kane26 i.e. if they scored 2 (mild) on at least two AIMS items or 3 (moderate) on one of the items at two or more successive assessments. Patients not completing at least two post-baseline assessments were excluded from this analysis. Analyses were also conducted using modified Schooler–Kane criteria such that meeting the AIMS criteria on only one assessment was required (i.e. ‘probable’ tardive dyskinesia). The summary score of all seven AIMS items was also used as a continuous measure.
Statistical methods
The primary outcome measure was the proportion of patients who did not meet the criteria for a particular EPS syndrome at baseline but who met at least one of the three criteria for that symptom during the subsequent follow-up period. For tardive dyskinesia, patients were excluded from the primary analysis if they met modified Schooler–Kane criteria at baseline, or were identified as having borderline tardive dyskinesia, which was defined by not meeting the full modified Schooler–Kane criteria but having at least one AIMS item score of mild, were taking medications for tardive dyskinesia, or being placed in Phase 1a, the phase which did not allow randomisation to perphenazine. A supportive analysis was repeated in which all patients with borderline tardive dyskinesia were included.
A second set of analyses involved repeated measures analysis of continuous measures representing change in severity of the three syndromes from baseline. Patients meeting criteria for each syndrome at baseline were not excluded from these analyses and baseline scores of the dependent measure were included as covariates in each analysis.
An analysis of incidence of side-effects was conducted first without adjustment for potential baseline predictors of each syndrome and then in models that included socio-demographic and other baseline measures that were significantly associated with the dependent measure.
The statistical plan used for treatment group comparisons followed the same methods as in the original publication from CATIE. Reference Lieberman, Stroup, McEvoy, Swartz, Rosenheck, Perkins, Keefe, Davis, Davis, Lebowitz, Severe and Hsiao20 Analyses (using SAS version 9.1 for Windows) were conducted on four data-sets with overlapping membership based on the tardive dyskinesia and ziprasidone cohort stratification. Each data-set only included patients with an equal chance of being randomly assigned to the treatments under comparison. Patients on perphenazine, in particular, were only compared with equivalent patients who did not have tardive dyskinesia at baseline, and patients on ziprasidone were only compared with other patients who were randomised after ziprasidone was added. The primary comparison between the four treatments available at the beginning of the trial was an overall 3 degree of freedom test. This test was performed on data-set I, excluding patients with tardive dyskinesia and those randomised to ziprasidone. If the overall test was significant at P<0.05, the three second-generation antipsychotics were compared with perphenazine with a Hochberg modification to the Bonferroni adjustment for multiple comparisons Reference Hochberg27 in which the smallest P-value was compared with P=0.05/3=0.017 and the largest with P=0.05. Next, using data-set II, which excludes perphenazine and ziprasidone and includes tardive dyskinesia patients, the three second-generation antipsychotics were compared with each other via step-down testing. If the overall 2 degree of freedom test was significant at P<0.05, an alpha of P<0.05 was applied for all comparisons.
Data-sets III and IV were used to compare ziprasidone with the other four drugs among patients randomised after ziprasidone became available; patients with tardive dyskinesia were excluded from the perphenazine comparison. Hochberg adjustment Reference Hochberg27 for four pair-wise comparisons was used to compare ziprasidone with the other three second-generation antipsychotics using data-set III and with perphenazine in data-set IV. Across both data-sets, the smallest P-value was considered significant if P=0.05/4=0.013. For evaluation of tardive dyskinesia events, in which all patients with tardive dyskinesia at baseline were excluded, this strategy simplifies to data-sets I and III.
Simple percentages of patients who experienced each type of side-effect are presented without statistical testing since they do not account for differential duration of treatment exposure. We conducted a statistical comparison of treatment groups for incidence of movement disorder events using Poisson regression to fit an exponential survival model accounting for the duration of treatment for individuals without the event and the time until first occurrence for people who had the event. Owing to the large number of parkinsonism events in the first month of treatment, the model for parkinsonism was extended to a piecewise exponential, in which separate exponential parameters were estimated for the first month and for all subsequent months. Treatment effects were summarised via the estimated probability of having an extrapyramidal event within 1 year. Reference Stokes, Davis and Koch28 Analyses were conducted with adjustment for significant baseline predictors, identified from an a priori set likely to have a relationship with the outcome. Items evaluated as potential covariates included age, gender, taking a conventional antipsychotic at baseline, baseline score of the relevant instrument, and site care setting. Potential baseline covariates for tardive dyskinesia also included items found previously to be associated with tardive dyskinesia at baseline: taking an anticholinergic, substance misuse, Positive and Negative Syndrome Scale total score, duration of antipsychotic use, and Simpson–Angus mean score. Reference Miller, McEvoy, Davis, Caroff, Saltz, Chakos, Swartz, Keefe, Rosenheck, Stroup and Lieberman19 There were no significant differences in smoking rates at baseline or substance misuse during the trial (M. Swartz, personal communication, 2007) between treatment groups. For descriptive evaluation, the following additional analyses were completed for akathisia and parkinsonism: incidence of events after 1 month of treatment, incidence of adding medications and incidence of discontinuation. Supportive Cox proportional hazards regression and exact logistic regression for rare events confirmed the primary methods. Kaplan–Meier survival plots provide a graphical display of the events over time.
Change from baseline in the continuous measurements collected over the 18 months were compared across treatment groups with a mixed model, including terms representing the baseline value of the dependent variable, time (treated as a classification variable), significant baseline predictors identified from an a priori set likely to have a relationship with the outcome, and baseline×time and treatment×time interactions. The baseline×time term adjusts for baseline differences in characteristics of patients who dropped out early and thus are not as well represented at later time points. A random subject effect and a spatial power covariance structure were used to adjust standard errors for the repeated measurements of observations from the same individual. When the treatment×time interaction was not significant, it was removed to obtain average treatment group differences in change from baseline over all time points. An additional analysis evaluated the maximum (most severe) change from baseline for each individual using analysis of covariance with the same fixed covariates, as well as duration of exposure to study drug.
Results
Although 1493 patients were enrolled in the study, all data from one site (33 patients) were excluded prior to analysis owing to concerns about data integrity, and 17 patients were randomised but did not start taking the study medication (for consort diagram see Lieberman et al Reference Lieberman, Stroup, McEvoy, Swartz, Rosenheck, Perkins, Keefe, Davis, Davis, Lebowitz, Severe and Hsiao20 )
Baseline comparisons
When baseline data on patients from all four randomisation strata were examined, including patients with tardive dyskinesia at baseline, there were no substantial differences between treatment groups on either continuous or dichotomous measures of parkinsonism or akathisia, with one exception: individuals randomised to olanzapine had a higher mean BARS score than those randomised to perphenazine, although there was no difference on percentage of patients meeting the criteria for akathisia (online Table DS1). When the comparison is properly limited to the patients in Phase 1 who did not have tardive dyskinesia at baseline and were included in the randomisation that included perphenazine, there were no notable baseline differences on measures of tardive dyskinesia (i.e. after excluding those in Phase 1a, as noted in the second set of columns in the four lowest rows of online Table DS1).
Acute dystonia
There were only eight cases of acute dystonia reported during the study, four of which resulted in treatment discontinuation. Of these eight patients, one was receiving olanzapine (discontinued), one was receiving perphenazine, one was receiving quetiapine (discontinued), two were receiving risperidone (one discontinued), and three were receiving ziprasidone (two discontinued).
Parkinsonism
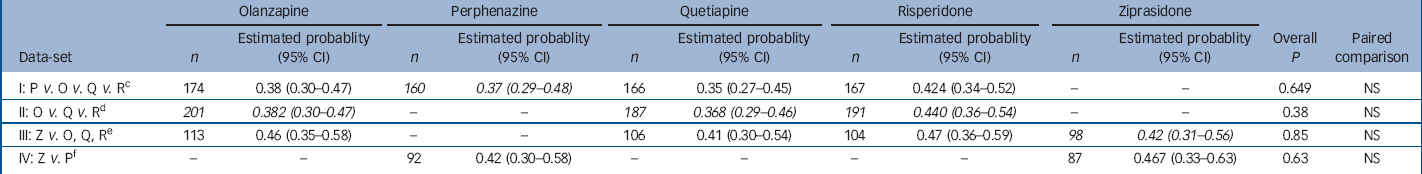
Examination of the proportion of patients showing no evidence of parkinsonism at baseline who met at least one of the three criteria for parkinsonism during the subsequent follow-up period revealed no substantial differences between treatment groups (online Table DS2). Statistical analysis, using piecewise exponential regression of the probability of having a parkinsonian event, showed no statistically significant difference between treatment groups (Table 1). Covariate-adjusted 12-month event rates were notable at 37–44% for the four second-generation antipsychotics and 37% for perphenazine (Table 1). The Kaplan–Meier survival plot graphically illustrates both the substantial incidence of parkisonian events, particularly in the first month, and the convergence of treatment groups (Fig. 1). Although there was a substantial incidence of parkinsonism events within the first month, only 2 patients discontinued treatment for EPS within the first month (data not shown). Finally, mixed model analysis of change in parkinsonian symptoms from baseline for all treated patients, as measured with the SAS, also shows no statistically significant group differences (online Fig. DS1). Analyses of maximum change in SAS score, and incidence of parkinsonism events after the first month of treatment also found no statistically significant differences (data not shown). Analysis of incidence of adding medications found an overall difference (P=0.029 for primary data-set I), with addition of parkinsonism medications most likely for patients on risperidone and least likely for patients on quetiapine. In addition, analyses of incidence of discontinuation for parkinsonism suggested there was a lower rate of discontinuation for quetiapine and ziprasidone (P<0.05 for all four data-sets, although exact logistic regression methods were statistically significant only for data-set III).

Fig. 1 Kaplan–Meier survival curve of time until parkinsonism event for people with no parkinsonism at baseline.a a. Data from all eligible patients. Perphenazine from data-set I; olanzapine, quetiapine and risperidone are from data-set II; ziprasidone from data-set III.
Table 1 Analysis of probability of having a parkinsonism event within 1 year for people with no parkinsonism at baseline a with adjustment for baseline covariates b
| Olanzapine | Perphenazine | Quetiapine | Risperidone | Ziprasidone | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Data-set | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | Overall P | Paired comparison | |||||
| I: P v. O v. Q v. R c | 174 | 0.38 (0.30–0.47) | 160 | 0.37 (0.29–0.48) | 166 | 0.35 (0.27–0.45) | 167 | 0.424 (0.34–0.52) | – | – | 0.649 | NS | |||||
| II: O v. Q v. R d | 201 | 0.382 (0.30–0.47) | – | – | 187 | 0.368 (0.29–0.46) | 191 | 0.440 (0.36–0.54) | – | – | 0.38 | NS | |||||
| III: Z v. O, Q, R e | 113 | 0.46 (0.35–0.58) | – | – | 106 | 0.41 (0.30–0.54) | 104 | 0.47 (0.36–0.59) | 98 | 0.42 (0.31–0.56) | 0.85 | NS | |||||
| IV: Z v. P f | – | – | 92 | 0.42 (0.30–0.58) | – | – | – | – | 87 | 0.467 (0.33–0.63) | 0.63 | NS | |||||
NS, not significant; O, olanzapine; P, perphenazine; Q, quetiapine; R, risperidone; Z, ziprasidone
a. Patients with no parkinsonism at baseline did not meet Simpson–Angus Scale extrapyramidal side-effects criteria and were taking no medications for parkinsonism at baseline. A parkinsonism event includes meeting Simpson–Angus Scale criteria, discontinuing because of parkinsonism, or adding medications for parkinsonism. Probability of having an event within 1 year is estimated with piecewise exponential regression adjusted for the following covariates: baseline Simpson–Angus extrapyramidal side-effects score (all normal, or one mild item), site care setting (State Mental Health, Veterans Affairs, Others), and tardive dyskinesia status at baseline (randomised into Phase 1a) where appropriate
b. Italicised values highlight treatment conditions of primary interest in each data-set
c. Excluding patients on ziprasidone and Phase 1a patients with tardive dyskinesia
d. Excluding patients on perphenazine or ziprasidone
e. Limited to ziprasidone cohort, excluding patients on perphenazine
f. Limited to ziprasidone cohort, excluding Phase 1a patients with tardive dyskinesia
Akathisia
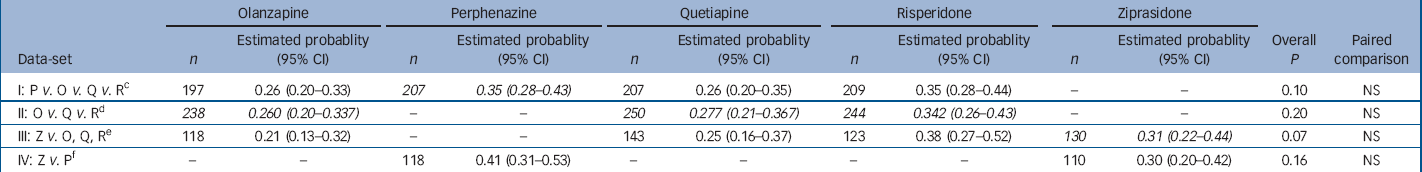
Examination of the proportion of patients who met at least one of the criteria for akathisia or who met each of the criteria, separately, among those who had no evidence of akathisia at baseline showed no substantial difference between treatment groups (online Table DS3). Poisson regression analysis of the probability of meeting any of the three criteria for akathisia revealed no significant difference between groups (Table 2). Covariate-adjusted 12-month event rates ranged from 26% to 35% for the second-generation antipsychotics, with 35% for perphenazine (Table 2). The Kaplan–Meier plot graphically shows the close grouping of survival curves across treatment groups (Fig. 2), and mixed model analysis of change from baseline on the BARS global rating similarly shows no statistically significant group differences, but does suggest a general decline in akathisia symptom levels over time (online Fig. DS2). Analysis of maximum change in BARS global rating from baseline for all treated individuals found no statistically significant differences (P=0.071, data not shown), although perphenazine had the largest estimated change (0.44) and olanzapine had the lowest (0.22). Analyses of incidence of adding medications for akathisia found no significant differences (P=0.056), although perphenazine and risperidone had numerically higher rates of medications added. No significant differences were noted for analyses of discontinuation for akathisia or akathisia events after the first month of treatment.

Fig. 2 Kaplan–Meier survival curve of time until akathisia event for people with no akathisia at baseline.a a. Data from all eligible patients. Perphenazine data from data-set I; olanzapine, quetiapine and risperidone are from data-set II; ziprasidone from data-set III.
Table 2 Analysis of probability of having an akathisia event within 1 year for people with no akathisia at baseline a with adjustment for baseline covariates b
| Olanzapine | Perphenazine | Quetiapine | Risperidone | Ziprasidone | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Data-set | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | Overall P | Paired comparison | |||||
| I: P V. O V. Q V. R c | 197 | 0.26 (0.20–0.33) | 207 | 0.35 (0.28–0.43) | 207 | 0.26 (0.20–0.35) | 209 | 0.35 (0.28–0.44) | – | – | 0.10 | NS | |||||
| II: O V. Q V. R d | 238 | 0.260 (0.20–0.337) | – | – | 250 | 0.277 (0.21–0.367) | 244 | 0.342 (0.26–0.43) | – | – | 0.20 | NS | |||||
| III: Z V. O, Q, R e | 118 | 0.21 (0.13–0.32) | – | – | 143 | 0.25 (0.16–0.37) | 123 | 0.38 (0.27–0.52) | 130 | 0.31 (0.22–0.44) | 0.07 | NS | |||||
| IV: Z V. P f | – | – | 118 | 0.41 (0.31–0.53) | – | – | – | – | 110 | 0.30 (0.20–0.42) | 0.16 | NS | |||||
BARS, Barnes Akathisia Rating Scale; NS, not significant; O, olanzapine; P, perphenazine; Q, quetiapine; R, risperidone; Z, ziprasidone
a. Patients with no akathisia at baseline had BARS global clinical assessment of less than mild (2) and were taking no medications for parkinsonism at baseline. An akathisia event includes meeting BARS global clinical assessment criteria, discontinuing because of akathisia, or adding medications for akathisia. Probability of having an event within 1 year is estimated with Poisson regression adjusted for the following covariates: baseline BARS global clinical assessment (absent or questionable), age and tardive dyskinesia status at baseline (randomised into Phase 1a) where appropriate
b. Italicised values highlight treatment conditions of primary interest in each data-set
c. Excluding patients on ziprasidone and Phase 1a patients with tardive dyskinesia
d. Excluding patients on perphenazine or ziprasidone
e. Limited to ziprasidone cohort, excluding patients on perphenazine
f. Limited to ziprasidone cohort, excluding Phase 1a patients with tardive dyskinesia
Tardive dyskinesia
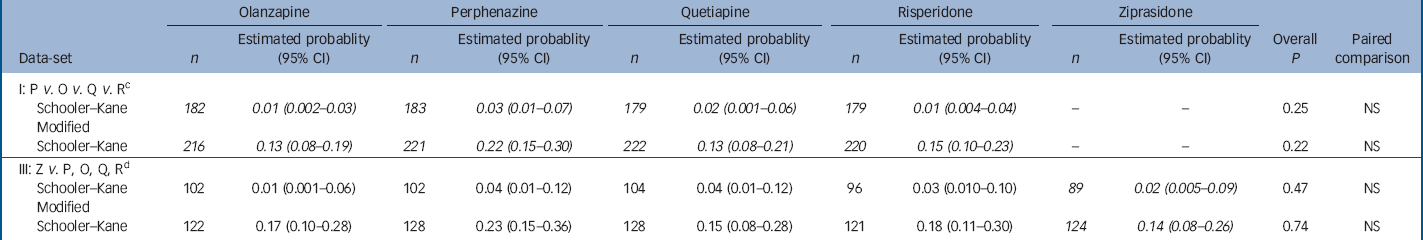
Data from patients who had no evidence of tardive dyskinesia at baseline shows that a small proportion of patients met full Schooler–Kane tardive dyskinesia criteria during Phase 1 treatment (1.1–4.5% of patients taking the second-generation antipsychotics and 3.3% of those taking perphenazine (online Table DS4). The proportion of patients who met modified Schooler–Kane criteria (i.e. at a single time-point) ranged from 8.3% to 9.6% for the second-generation antipsychotics and was 11.8% for perphenazine. The other two measures of tardive dyskinesia events (patient discontinuations and concomitant medications) were met by only 1% or fewer patients in all treatment groups. Poisson regression reveals no statistically significant difference between treatment groups on either tardive dyskinesia indicator (Table 3). Covariate-adjusted 12-month event rates for Schooler–Kane tardive dyskinesia ranged from 0.7% to 2.2% for the second-generation antipsychotics, with 2.7% for perphenazine (Table 3). Kaplan–Meier survival curves show both the infrequent incidence of tardive dyskinesia and the overlapping of treatment groups (Fig. 3), and mixed model analysis of change in tardive dyskinesia symptoms from baseline, based on the AIMS total score, also shows no statistically significant group differences (online Fig. DS3). Analyses of incidence of tardive dyskinesia events for patients with either no or borderline tardive dyskinesia at baseline, and maximum change in AIMS total score, also found no statistically significant differences between treatment groups.
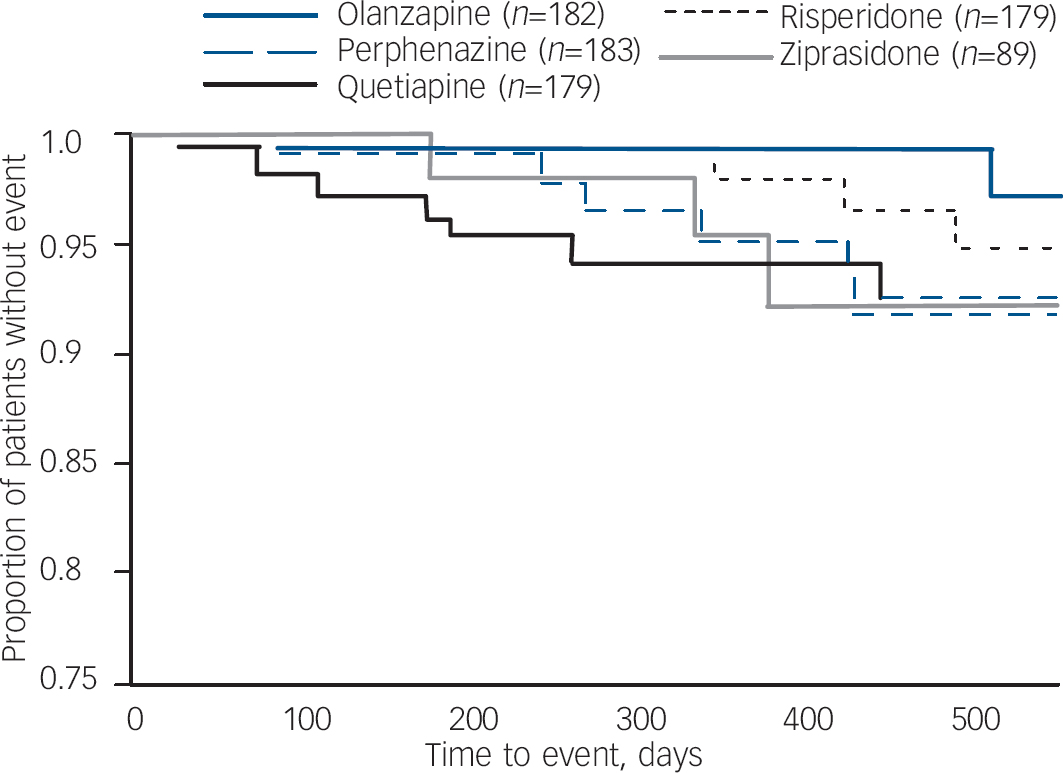
Fig. 3 Kaplan–Meier survival curve of time until Schooler–Kane tardive dyskinesia for people with no tardive dyskinesia at baseline.a a. Data from all eligible patients. Perphenazine, olanzapine, quetiapine and risperidone are from data-set I; ziprasidone from data-set II.
Table 3 Analysis of probability of having a tardive dyskinesia event within 1 year for people with no tardive dyskinesia at baseline with adjustment for baseline covariates a , b
| Olanzapine | Perphenazine | Quetiapine | Risperidone | Ziprasidone | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Data-set | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | n | Estimated probablity (95% CI) | Overall P | Paired comparison | |||||
| I: P v. O v. Q v. R c | |||||||||||||||||
| Schooler–Kane | 182 | 0.01 (0.002–0.03) | 183 | 0.03 (0.01–0.07) | 179 | 0.02 (0.001–0.06) | 179 | 0.01 (0.004–0.04) | – | – | 0.25 | NS | |||||
| Modified | |||||||||||||||||
| Schooler–Kane | 216 | 0.13 (0.08–0.19) | 221 | 0.22 (0.15–0.30) | 222 | 0.13 (0.08–0.21) | 220 | 0.15 (0.10–0.23) | – | – | 0.22 | NS | |||||
| III: Z v. P, O, Q, R d | |||||||||||||||||
| Schooler–Kane | 102 | 0.01 (0.001–0.06) | 102 | 0.04 (0.01–0.12) | 104 | 0.04 (0.01–0.12) | 96 | 0.03 (0.010–0.10) | 89 | 0.02 (0.005–0.09) | 0.47 | NS | |||||
| Modified | |||||||||||||||||
| Schooler–Kane | 122 | 0.17 (0.10–0.28) | 128 | 0.23 (0.15–0.36) | 128 | 0.15 (0.08–0.28) | 121 | 0.18 (0.11–0.30) | 124 | 0.14 (0.08–0.26) | 0.74 | NS | |||||
NS, not significant; O, olanzapine; P, perphenazine; Q, quetiapine; R, risperidone; Z, ziprasidone
a. Patients with no tardive dyskinesia at baseline met none of the criteria for modified Schooler–Kane tardive dyskinesia or borderline tardive dyskinesia. Probability of having an event within 1 year is estimated with Poisson regression adjusted for the following covariates: baseline total Positive and Negative Syndrome Scale score, years taking antipsychotic medication (0–5, 6–20, 21+), and baseline Simpson–Angus Scale extrapyramidal side-effects (EPS) score (all items =0 normal, or EPS score >0). Italicised values highlight treatment conditions of primary interest in each data-set
b. Data-sets II and IV are not applicable in this analysis since patients stratified into Phase 1a at baseline are excluded
c. Excluding patients on ziprasidone
d. Including only ziprasidone cohort patients
Discussion
Using a variety of measures of dystonia, parkinsonism, akathisia and tardive dyskinesia, the analysis of incidence rates and continuous rating scale measures from CATIE shows no consistent, substantial or statistically significant differences between any second-generation antipsychotic and perphenazine, or between any pair of second-generation antipsychotics. In the current analyses we utilised more sensitive criteria for parkinsonism and akathisia than was used in the analyses reported by Lieberman et al, Reference Lieberman, Stroup, McEvoy, Swartz, Rosenheck, Perkins, Keefe, Davis, Davis, Lebowitz, Severe and Hsiao20 and we thus found a higher incidence of these side-effects. We choose the current criteria to define ‘caseness’ of these side-effects in a similar fashion to tardive dyskinesia, and to examine any subtle differences between second- and first-generation antipsychotics.
Comparisons with previous studies examining EPS between second- and first-generation antipsychotics
Evidence from various clinical trials indicate that motor abnormalities inherent to schizophrenia-spectrum disorders Reference Chakos, Alvir, Woerner, Koreen, Geisler, Mayerhoff, Sobel, Kane, Borenstein and Lieberman29,Reference Fenton30 may be aggravated by first-generation antipsychotics, Reference Eberhard, Lindstrom and Levander31–Reference Lee, Wu, Habil, Dyachkova and Lee34 whereas some of the newer second-generation antipsychotics appear to have a lower propensity to induce motor symptoms or to improve symptoms associated with pre-existing tardive dyskinesia. Reference Tollefson and Sanger6,Reference Jeste, Okamoto, Napolitano, Kane and Martinez33,Reference Dolder and Jeste35–Reference Tarsy and Baldessarini41 In contrast, we found no advantage between any of the treatments tested in CATIE in the incidence of treatment-emergent dystonia, parkinsonism, akathisia or tardive dyskinesia in patients with chronic schizophrenia during maintenance antipsychotic treatment for up to 18 months. Although our findings are discrepant with the findings of initial clinical trials with second-generation antipsychotics, they are in close agreement with more recent trials conducted by independent investigators which have found virtually no substantial differences in EPS between these two classes of drugs. Reference Jones, Barnes, Davies, Dunn, Lloyd, Hayhurst, Murray, Markwick and Lewis9,Reference Leucht, Wahlbeck, Hamann and Kissling11 Our findings are also in accord with a large meta-analysis of 31 randomised controlled trials that included 2320 patients, which found no greater risk of EPS between low-potency first- and second-generation antipsychotics, other than clozapine. Reference Rosenheck, Perlick, Bingham, Liu-Mares, Collins, Warren, Leslie, Allan, Campbell, Caroff, Corwin, Davis, Douyon, Dunn, Evans, Frecska, Grabowski, Graeber, Herz, Kwon, Lawson, Mena, Sheikh, Smelson and Smith-Gamble10
Although the review by Correll et al Reference Correll, Leucht and Kane14 reported a lower annualised incidence of tardive dyskinesia in patients receiving second-generation antipsychotics (0.8%) compared with those receiving first-generation antipsychotics (5.4%), three of the randomised trials they reviewed used haloperidol as the comparator at relatively high mean doses (13–15 mg). The authors acknowledged that their findings may have been biased by the use of high doses of haloperidol as the comparison treatment.
In the CATIE Schizophrenia Trial, the use of perphenazine, an intermediate-potency first-generation antipsychotic given within a range of modest doses, was likely to be at least partially responsible for the lack of difference in the incidence of treatment-emergent dystonia, parkinsonism, akathisia and tardive dyskinesia seen between the first- and second-generation antipsychotic groups. In the CATIE study, the mean modal dose for the perphenazine group was 20.8 mg/day. Using the dose equivalency of 4:1 (perphenazine:haloperidol) proposed by Kane et al, Reference Kane, Leucht, Carpenter and Docherty42 this would be equivalent to a dose of 5.2 mg/day of haloperidol, which is lower than was used in the initial randomised trials of second-generation antipsychotics. At this dose, perphenazine was no less effective than olanzapine, quetiapine, risperidone or ziprasidone measured as time to discontinuation of treatment for any cause. Reference Lieberman, Stroup, McEvoy, Swartz, Rosenheck, Perkins, Keefe, Davis, Davis, Lebowitz, Severe and Hsiao20 Similarly, there were no differences between perphenazine and olanzapine, quetiapine, risperidone and ziprasidone on measures of symptoms or quality of life, Reference Rosenheck, Leslie, Sindelar, Miller, Lin, Stroup, McEvoy, Davis, Keefe, Swartz, Perkins, Hsiao and Lieberman43,Reference Swartz, Perkins, Stroup, Davis, Capuano, Rosenheck, Reimherr, McGee, Keefe, McEvoy, Hsiao and Lieberman44 or on neurocognitive functioning. Reference Keefe, Bilder, Davis, Harvey, Palmer, Gold, Meltzer, Green, Capuano, Stroup, McEvoy, Swartz, Rosenheck, Perkins, Davis, Hsiao and Lieberman45
In contrast to most previous trials, we excluded individuals who were experiencing a particular EPS syndrome at entry into the trial from the analyses for that primary outcome measure of treatment-emergent side-effects. Thus, this design is more consistent than previous studies with the basic principle of risk assessment research which states that patients who already have the outcome being studied should be excluded from the study cohort. Reference Kelsey, Whittemore, Evans and Thompson46 Such patients already are ‘cases’ and thus cannot be at any risk of becoming cases and add uninformative variance that biases results towards the null. The exclusion of patients who had EPS syndromes at baseline from our statistical analyses allowed more precise comparison of treatment-emergent incidences of these side-effects than studies that included mixed samples. Although we feel that this was the best method for comparing rates of EPS given the design of the trial, our findings may not directly relate to patients who are already experiencing an EPS on their current antipsychotic agent. However, in the analyses of change in mean rating scores over time, we included all treated patients and adjusted for treatment, baseline rating score, site care setting, Phase1a tardive dyskinesia status where applicable, classification of time, and the interaction between baseline rating score and time, and found no difference between medications for any of the side-effects.
Another substantial difference between the current analysis and previously reported randomised trials was that patients with a history of tardive dyskinesia at baseline were excluded from randomisation to perphenazine. There appear to be individuals who are particularly vulnerable (or resistant) to EPS. It has previously been reported that there is a relationship between the development of parkinsonism, akathisia and tardive dyskinesia, and various investigators have shown that antipsychotic-induced parkinsonism and/or akathisia are associated with a higher risk of developing tardive dyskinesia. Reference Crane47–Reference Umbricht and Kane50 Likewise, the baseline analysis of the CATIE data showed a significant correlation of parkinsonism and akathisia with tardive dyskinesia. Reference Miller, McEvoy, Davis, Caroff, Saltz, Chakos, Swartz, Keefe, Rosenheck, Stroup and Lieberman19 Not randomising patients with tardive dyskinesia to perphenazine systematically excluded people who had demonstrated previous vulnerability to EPS, who were likely to be at a greater risk of developing parkinsonism and akathisia having received perphenazine, which could potentially bias the results. To account for this potential bias, all comparisons of perphenazine to second-generation antipsychotics included only individuals without tardive dyskinesia at baseline. In other words, the comparisons of perphenazine with second-generation antipsychotics were based solely on those people who had not shown previous vulnerability to EPS and, thus, should not have biased the findings. To further control for this potential, we covaried for tardive dyskinesia at baseline in all of the analyses comparing perphenazine with second-generation antipsychotics and comparing second-generation antipsychotics with each other, and found no difference in the results.
Higher treatment discontinuation with perphenazine
Our results do not provide an explanation for why perphenazine was associated with the highest rate of treatment discontinuations due to EPS, as noted in the original reporting of CATIE. Reference Lieberman, Stroup, McEvoy, Swartz, Rosenheck, Perkins, Keefe, Davis, Davis, Lebowitz, Severe and Hsiao20 There are several possible methodological explanations for this. First, we examined the incidence of side-effects, and not merely the reasons for treatment discontinuation. In addition, we looked at the incidence of the different types of EPS separately, excluding from the analysis those who had a specific type of EPS at baseline. This is the appropriate method for the determining incidence of specific treatment-emergent side-effects, but does not allow us to pool the incidence of all types of EPS. Of the 22 patients on perphenazine identified as discontinued owing to EPS in the original report, Reference Lieberman, Stroup, McEvoy, Swartz, Rosenheck, Perkins, Keefe, Davis, Davis, Lebowitz, Severe and Hsiao20 8 were not included here as treatment-emergent cases because they met the criteria for EPS at baseline, 2 were deemed unclassifiable during adjudication, and 12 were included in these analyses (2 discontinued for tardive dyskinesia, 3 for parkinsonism, 6 for akathisia, and 1 for both parkinsonism and akathisia). It is also possible that the ratings of ESP in CATIE may have been relatively insensitive to milder forms of ESP. It is difficult to train raters in the measurement of mild akathisia and rigidity, and patients may have experienced mild forms of these side-effects that were not detected by the raters. This complexity could have been compounded by the difficulty patients with schizophrenia have in describing their subjective experiences of a medication.
In our secondary analyses we found that a significantly higher proportion of participants randomised to risperidone had medications added to treat parkinsonism, and significantly fewer patients randomised to quetiapine and ziprasidone discontinued treatment for parkinsonism than with the other medications. There was also a trend towards a higher proportion of individuals randomised to risperidone and perphenazine having medications added to treat akathisia. As these were secondary outcomes, they were not corrected for multiple testing.
Since most participants had received antipsychotic medications for many years (mean=14.4 years (s.d.=10.7)), the patients in this analysis may be less likely to develop antipsychotic-induced EPS than the general population of individuals with schizophrenia, including those with first-episode psychosis. In addition, it is possible that for participants who developed tardive dyskinesia during the trial, the onset could have been related to prior antipsychotic exposure. The studies that showed advantages of second-over first-generation antipsychotics were conducted on patient samples of similar age and duration of illness, so these sample characteristics may not actually account for the differences in findings. Reference Rosenheck, Stroup, Swartz, Mcevoy, Davis, Keefe, Hsiao and Lieberman51
Limitations
As with other studies comparing the incidence of tardive dyskinesia in patients treated with first- and second-generation antipsychotics, some participants may have been experiencing withdrawal dyskinesia or ‘unmasking’ of tardive dyskinesia related to switching from one antipsychotic to another. The majority of patients in the CATIE Schizophrenia Trial switched antipsychotics at baseline. It is possible that the antipsychotic prior to entry into the study or the antipsychotic that they were randomised to may have influenced the rates of withdrawal dyskinesia, although investigators were allowed to cross-titrate the previous and new antipsychotics for up to a month. We found very few cases of tardive dyskinesia within the first month of the trial and our findings did not change substantially whether we included the data from the 1-month visit or not, suggesting that withdrawal dyskinesia and ‘unmasking’ of tardive dyskinesia did not significantly affect our findings.
Another potential limitation of the study was the relatively short duration of exposure to each drug owing to high switching rates. In Phase 1, the mean duration of exposure was 9.2 months for olanzapine, 4.6 months for quetiapine, 4.8 months for risperidone, 5.6 months for perphenazine and 3.5 months for ziprasidone. None the less, the duration of exposure was similar to those in prior studies with second-generation antipsychotics. Reference Rosenheck, Stroup, Swartz, Mcevoy, Davis, Keefe, Hsiao and Lieberman51 For example, the average median duration of the trial referenced in the Correll review was 8.8 months. Reference Correll, Leucht and Kane14 Our findings were corrected for duration of exposure and it is unlikely that the results for parkinsonism and akathisia were affected by the duration of exposure as they tend to occur early in treatment. Another limitation of the study was that, as with most randomised trials of antipsychotic medications, the training for the scales used to rate EPS and tardive dyskinesia were not as rigorous as the training for ratings of psychopathology. Given that the trial was double-blind, this fact should have influenced all treatment groups equally.
Strengths
The strengths of the study are many and include the large sample size, independence from pharmaceutical industry sponsorship, and the head-to-head comparison of the four second-generation antipsychotics (that were FDA-approved at the time of the study) and a representative first-generation antipsychotic. Apart from pharmaceutical industry-sponsored studies, CATIE is the largest randomised clinical trial conducted to date. Based on the sample size and event rates for each of the three outcomes, the study had 80% power to detect with a P-value of <0.05, a 15% difference between any two treatment groups for parkinsonian events, a 14% difference for akathisia, and a 7% difference in tardive dyskinesia.
Our findings suggest that the incidence of treatment-emergent parkinsonism, akathisia and tardive dyskinesia are not significantly different between modest dosages of the intermediate-potency perphenazine and the four second-generation antipsychotics in patients with chronic schizophrenia requiring maintenance antipsychotic treatment. None the less, significantly more participants receiving risperidone had medications added to treat parkinsonism and significantly fewer participants receiving quetiapine and ziprasidone discontinued treatment because of parkinsonism. There was also a trend towards more individuals who were receiving risperidone and perphenazine to have medications added to treat akathisia. Previous reports of a relatively lower incidence of EPS with second-generation antipsychotics compared with first-generation antipsychotics were likely to be related to the use of higher dosages of the high-potency first-generation antipsychotic haloperidol. In addition, the difference in EPS between the second- and first-generation antipsychotics may have been lessened by the higher doses of second-generation antipsychotics, such as olanzapine, that were used in CATIE and are routine in clinical practice, compared with the doses used in the original studies.
Acknowledgements
This article was based on results from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project, supported by the National Institute of Mental Health (NO1 MH90001).
AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, Forest Pharmaceuticals Inc, Janssen Pharmaceutica, Eli Lilly & Co, Otsuka Pharmaceutical Co, Ltd, Pfizer Inc, and Zenith Goldline Pharmaceuticals, provided medications for the study. AstraZeneca Pharmaceuticals: D.M. (consulting and educational fees); R.A.R (research funding); J.P.McE. (research funding); B.L.S (research funds); M.H.C (research funds); M.S.S (consulting and educational fees); R.S.E.K. (research funding); J.A.L. (research funding and consulting/educational fees). Bristol-Myers Squibb: D.M. (consulting and educational fees); S.N.C. (research funds); R.A.R (research funding and consulting fees) J.P.McE. (consulting/advisory board and lecture fees); M.S.S (consulting and educational fees); R.E.S.K. (consulting/advisory board fees); T.S.S (consulting fees); J.A.L. (research funding and consulting/educational fees). Eli Lilly & Co: R.A.R (research funding and consulting fees); J.P.McE. (research funding); M.S.S (research funds and consulting/educational fees); R.S.E.K. (research funding, consulting/advisory board fees and lecture fees); T.S.S (research funding); J.A.L. (consulting and educational fees). Forest Laboratories: R.S.E.K. (consulting/advisory board fees). Forest Pharmaceutical Co: J.A.L. (consulting and educational fees). Forest Research Institute: J.P.McE. (research funding); J.A.L. (research funding and consulting/educational fees). GlaxoSmithKline: T.S.S (consulting fees); J.A.L. (research funding and consulting/educational fees). Janssen Pharmaceutica: D.M. (consulting and educational fees); R.A.R (consulting fees); J.P.McE. (research funding and lecture fees); B.L.S (lecture fees); M.H.C (research funds); R.S.E.K. (research funding, consulting/advisory board fees and lecture fees); T.S.S (consulting fees); J.A.L. (research funding and consulting/educational fees). Novartis: B.L.S (research funds and lecture fees); J.A.L. (consulting and educational fees). Organon: D.M. (consulting and educational fees); M.H.C (research funds). Ortho-McNeil Neurologics: S.N.C. (research funds). Pfizer Inc: D.M. (research funds, consulting and educational fees); S.N.C. (research funds); J.P.McE. (research funding and consulting/advisory board fees); M.H.C (research funds); M.S.S (consulting and educational fees); R.S.E.K. (consulting/advisory board fees); J.A.L. (research funding and consulting/educational fees). Solvay: M.H.C (research funds); J.A.L. (consulting and educational fees). S.M.D is an employee of Quintiles.
The CATIE Study Investigators Group includes Lawrence Adler, MD, Clinical Insights; Glen Burnie, MD; Mohammed Bari, MD, Synergy Clinical Research; Irving Belz, MD, Tri-County/MHMR; Raymond Bland, MD, Southern Illinois University School of Medicine; Thomas Blocher, MD, MHMRA of Harris County; Brent Bolyard, MD, Cox North Hospital; Alan Buffenstein, MD, The Queen's Medical Center; John Burruss, MD, Baylor College of Medicine; Matthew Byerly, MD, University of Texas Southwestern Medical Center at Dallas, Dallas; Jose Canive, MD, Albuquerque VA Medical Center; Stanley Caroff, MD, University of Pennsylvania and Philadelphia VA Medical Center; Charles Casat, MD, Behavioral Health Center; Eugenio Chavez-Rice, MD, El Paso Community MHMR Center; John Csernansky, MD, Washington University School of Medicine, St. Louis; Pedro Delgado, M., University Hospitals of Cleveland; Richard Douyon, MD, Veterans Affairs Medical Center; Cyril D'Souza, MD, Connecticut Mental Health Center; Ira Glick, MD, Stanford University School of Medicine; Donald Goff, MD, Massachusetts General Hospital; Silvia Gratz, MD, Eastern Pennsylvania Psychiatric Institute; George T. Grossberg, MD, St. Louis University School of Medicine-Wohl Institute; Mahlon Hale, MD, New Britain General Hospital; Mark Hamner, MD, Medical University of South Carolina and Veterans Affairs Medical Center; Richard Jaffe, MD, Belmont Center for Comprehensive Treatment; Dilip Jeste, MD, University of California-San Diego, Veterans Affairs Medical Center; Anita Kablinger, MD, Louisiana State University Health Sciences Center; Ahsan Khan, MD, Psychiatric Research Institute; Steven Lamberti, MD, University of Rochester Medical Center; Michael T. Levy, MD, PC, Staten Island University Hospital; Jeffrey Lieberman, MD, University of North Carolina School of Medicine; Gerald Maguire, MD, University of California Irvine; Theo Manschreck, MD, Corrigan Mental Health Center; Joseph McEvoy, MD, Duke University Medical Center; Mark McGee, MD, Appalachian Psychiatric Healthcare System; Herbert Meltzer, MD, Vanderbilt University Medical Center; Alexander Miller, MD, University of Texas Health Science Center at San Antonio; Del D. Miller, MD, University of Iowa; Henry Nasrallah, MD, University of Cincinnati Medical Center; Charles Nemeroff, MD, PhD, Emory University School of Medicine; Stephen Olson, MD, University of Minnesota Medical School; Gregory F. Oxenkrug, MD, St. Elizabeth's Medical Center; Jayendra Patel, MD, University of Mass Health Care; Frederick Reimher, MD, University of Utah Medical Center; Silvana Riggio, MD, Mount Sinai Medical Center-Bronx VA Medical Center; Samuel Risch, MD, University of California, San Francisco; Bruce Saltz, MD, Henderson Mental Health Center; Thomas Simpatico, MD, Northwestern University; George Simpson, MD, University of Southern California Medical Center; Michael Smith, MD, Harbor – UCLA Medical Center; Roger Sommi, PharmD, University of Missouri; Richard M. Steinbook, MD, University of Miami School of Medicine; Michael Stevens, MD, Valley Mental Health; Andre Tapp, MD, VA Puget Sound Health Care System; Rafael Torres, MD, University of Mississippi; Peter Weiden, MD, SUNY Downstate Medical Center; and James Wolberg, MD, Mount Sinai Medical Center.









eLetters
No eLetters have been published for this article.