Aging is a natural process of every life form and it intrigues researchers to investigate its driving force. In the 1960s it was suggested that aging is the outcome of accelerated accumulation of somatic mutations at DNA level (Curtis, Reference Curtis1966), hence introducing errors in the primary structure of proteins (Orgel, Reference Orgel1963, Reference Orgel1973). These theories predict that somatic mutations appear stochastically from the beginning of life onwards, and the accumulation of erroneous proteins and somatic mutations eventually affect key functions of somatic maintenance (synthesis, degradation, repair), leading to a cascade of detrimental consequences (error catastrophe). The lack of high throughput detection techniques, however, hampered any firm conclusions on these theories in humans (Kirkwood, Reference Kirkwood1977). The question here is whether current high throughput next-generation sequence technology can provide some of the necessary insights, by using a study design in which the whole-genome sequence is compared between co-twins from monozygotic (MZ) twin pairs.
MZ twins generally show remarkable similarity in diverse aspects of behavior, health, and disease (e.g., Martin et al., Reference Martin, Boomsma and Machin1997; van Dongen et al., Reference van Dongen, Slagboom, Draisma, Martin and Boomsma2012). However, even for highly heritable traits, discordance within MZ pairs also has been documented (e.g., Zwijnenburg et al., Reference Zwijnenburg, Meijers-Heijboer and Boomsma2010). A remarkable pair of MZ twins discordant for multiple sclerosis (MS) has been investigated, aiming at discordances at genome, epigenome, and transcriptome levels (Baranzini et al., Reference Baranzini, Mudge, van Velkinburgh, Khankhanian, Khrebtukova, Miller and Kingsmore2010) by the first whole-genome sequencing study of CD4+ T cells. Potentially discordant variants between discordant MZ twins were detected but none of these were reproducible between methods. The result also illustrates the challenges of identification of low rate de novo mutations in next generation sequencing (NGS) data, which contain high rates of sequencing errors.
To examine the level of detectable somatic variants that may occur in a lifetime, whole-genome NGS was done on two randomly chosen MZ twin pairs of 40 and 100 years. The 40-year-old MZ twin pair is participating in the Netherlands Twin Registry (NTR co-twins) and the centenarian MZ twin pair in the Leiden Longevity Study (LLS co-twins). NGS data for these four individuals were generated using two different whole-genome sequencing platforms; by comparing within pair differences across the two platforms we aim to reduce the number of false positive hits, created, for example, by sequencing errors. To clarify whether an accumulation of somatic mutations with age can be detected in human blood, the NGS data were compared for within co-twin DNA sequencing differences in the younger and centenarian co-twins.
Materials and Methods
Study Samples
The twins of 40 years of age were randomly selected from the Netherlands Twin Registry (Willemsen et al., Reference Willemsen, Vink, Abdellaoui, den Braber, van Beek, Draisma and Boomsma2013), and the twins of 100 years of age from the Leiden Longevity Study (Westendorp et al., Reference Westendorp, van Heemst, Rozing, Frolich, Mooijaart, Blauw and Slagboom2009). Genomic DNA was isolated according to standard procedures from blood (Willemsen et al., Reference Willemsen, de Geus, Bartels, van Beijsterveldt, Brooks, Burk and Boomsma2010). The study was approved by Leiden University Medical Center and the Medical Ethics Committee of the VU Medical Centre, Amsterdam. Informed, written consent was obtained from the twins participating in the study.
Illumina Platform
Illumina Infinium 1M SNP-array and GWAS data indicated that the twins were monozygotic. Paired-end and 100 bp long reads (Bentley et al., Reference Bentley, Balasubramanian, Swerdlow, Smith, Milton, Brown and Smith2008) were generated using Illumina GAIIx instruments. Illumina sequences were aligned to the NCBI human reference genome build 37, with Burrows-Wheeler Aligner (BWA; Li & Durbin, Reference Li and Durbin2009). Short indels and structural variants were analyzed with Pindel (Ye et al., Reference Ye, Schulz, Long, Apweiler and Ning2009). The four nucleotide base counts per genomic location were directly compared between co-twins to first yield a list of potential discordant base substitutions, as in CaVEMan pipeline developed by the Wellcome Trust Sanger Institute (see Supplementary Material available online).
Complete Genomics Platform
Genomic DNA from the twins was sent to Complete Genomics (Drmanac et al., Reference Drmanac, Sparks, Callow, Halpern, Burns, Kermani and Reid2010) for whole-genome sequencing. Regions of the genome that do not have sufficient evidence (as determined by the log-likelihood scores assigned to each region) to make a reference or a variant call are made no-call. Discordant variant calling within twins was determined using the Complete Genomics tumor variant calling tool (http://www.completegenomics.com/sequence-data/cgatools/). In order to look for concordance of variant calls between co-twins, the callDiff program from cgatools was used, which groups variants into ‘super loci’ and looks for consistency. There were 3.94 million super locus variant sites across the two samples; 86.7% of the super loci were identical between the twins, leading to 525,105 loci that have mismatched calls (either called in both samples and discordant, or called in one-sample).
Comparison of Illumina and Complete Genomics Twin Discordances
To identify potential discordant single base substitutions we intersected the within-pair differences detected by Illumina sequencing and those detected by Complete Genomics sequencing. This resulted in a list of potential discordant single base substitutions for each twin pair.
Validation of Potential Discordant Single Base Substitutions
The potential discordant single base substitutions were examined using Sanger, 454 (Margulies et al., Reference Margulies, Egholm, Altman, Attiya, Bader, Bemben and Rothberg2005), and/or Ion Torrent sequencing, which are documented in the online Supplementary Material.
Results
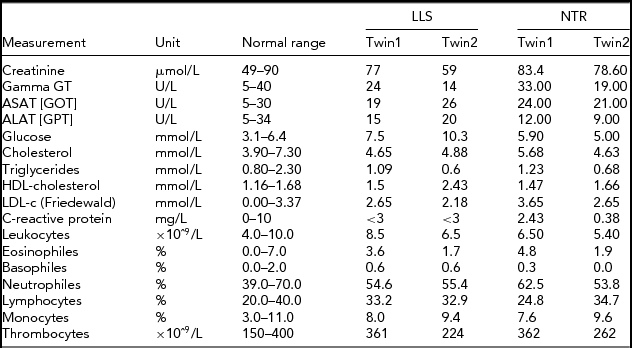
In order to obtain a first indication, using NGS technology, of whether an accumulation of somatic mutations can be detected with age in humans, we studied NGS whole-genome sequence differences in younger (40-year-old) and centenarian female co-twins. The 40-year-old pair participates in the Netherlands Twin Registry and is indicated as NTR (NTR twins), the second pair as LLS (Leiden Longevity Study). Phenotypic characteristics of the NTR and LLS twins are summarized in Table 1.
TABLE 1 Phenotypic Characteristics of the LLS and NTR MZ Co-Twins

LLS = Leiden Longevity Study (100-year-old co-twins); NTR = Netherlands Twin Research (40-year-old co-twins).
Sequencing and Germline Variant Calling
Genomic DNA was extracted from whole blood of the two pairs of twins and independently sequenced by Illumina and Complete Genomics. In terms of the number of reads, coverage of the genome, and mapping characteristics, the data from the same platform are comparable among the four samples (see Tables S1 and S2 available online). In addition, all four samples were genotyped for the Illumina 1M array and the concordance rate between these SNP array genotype calls and the calls from the Illumina whole-genome sequence was 99.98%, indicating the high quality of the data and data-processing procedure. On average, 3.2 million SNPs per genome were detected in each genome of the four samples. Using Pindel (Ye et al., Reference Ye, Schulz, Long, Apweiler and Ning2009) we detected about 0.8 million short insertions and deletions (indels) and 13,000 structural variants per individual.
The mean coverage of the Complete Genomics dataset was 20× for 92.5% of the genome and 40× for 70% of the genome (see Table S2 available online). In comparison to the reference genome, approximately 3.6 million SNPs, 0.4 million short indels, and 5,000 structural variants were observed in the two MZ twin pairs.
Somatic Variant Calling
Next, in a within-pair analysis, we compared the whole-genome sequences of the co-twins from each pair for each platform separately. After mapping Illumina sequence data using BWA (Li & Durbin, Reference Li and Durbin2009), the four nucleotide base counts per genomic location were directly compared between co-twins to first yield a list of 27,529 potentially discordant base substitutions between LLS co-twins and 31,877 between NTR co-twins, using CaVEMan developed by Welcome Trust Sanger Cancer group for somatic substitution calling from cancer samples. Using Complete Genomics tumor variant calling tool, 11,959 between LLS and 16,007 between NTR co-twins potentially discordant single base substitutions were computed from the variant lists.
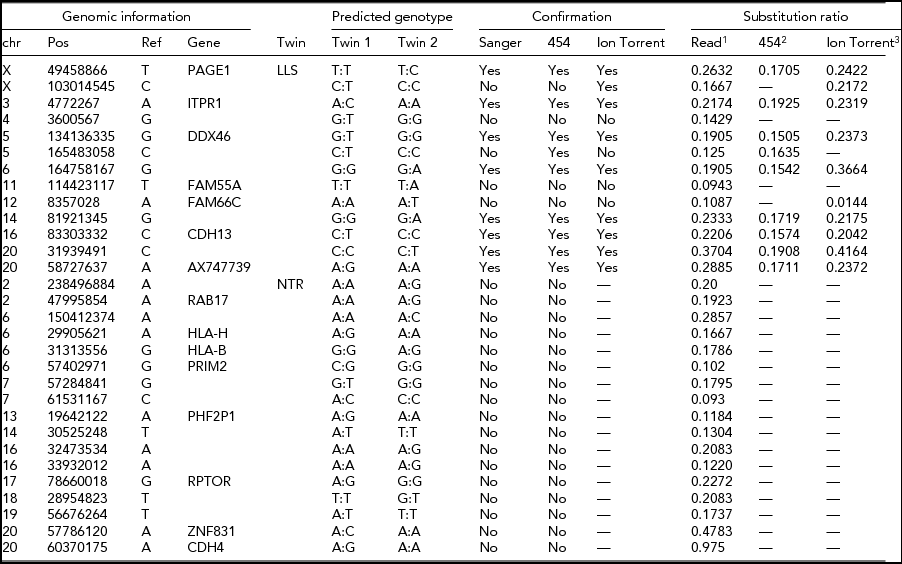
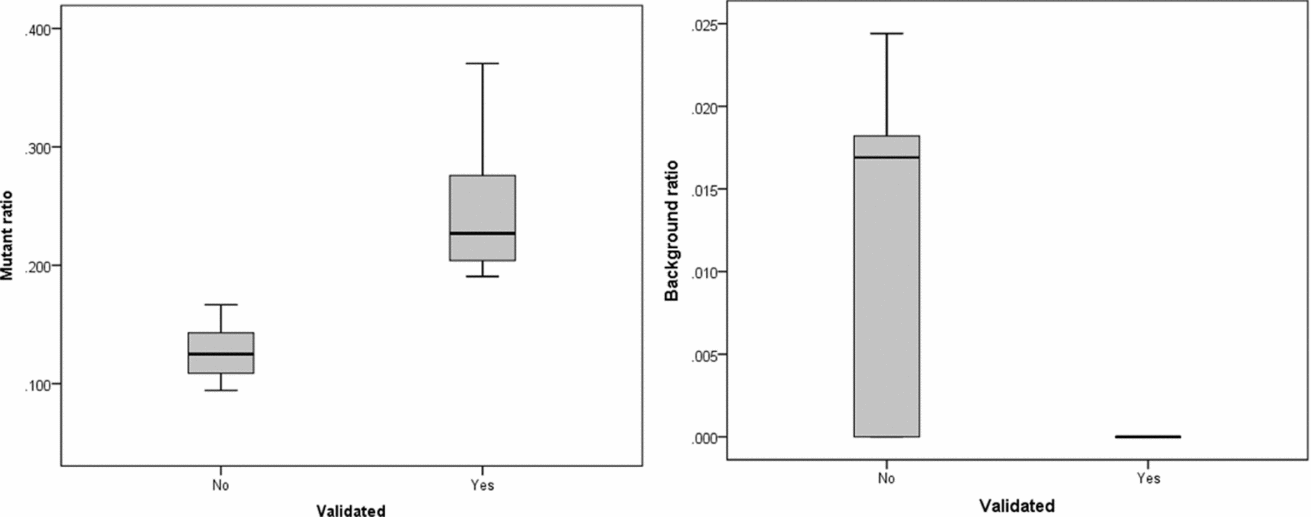
The intersection of potential single base substitutions between co-twins supported both by the Illumina and Complete Genomics platform revealed 13 and 17 potential somatic single base substitutions for LLS and NTR co-twins, respectively (Table 2), which did not cluster to particular locations of the genome. After Sanger sequencing these 30 potential single base substitutions, eight were confirmed for both strands. The substitutions identified were present in only a fraction of the reads (substitution ratio Table 2), indicating that these are somatic variations present only in a fraction of the blood cells, as shown in one example Sanger trace for position chr3:4,772,267 (Figures 1 and 2). The eight validated substitutions showed a lower ‘background ratio’ and contain a higher percentage of reads supporting substituted bases, with an average of 24.7% compared to 12.8% in the five invalidated substitutions (Figure 3).
TABLE 2 Potential Single-Base Substitutions Supported by Both Illumina and Complete Genomics Data and Subsequent Validation by Sanger Sequencing, Roche 454, and Ion Torrent Sequencing (Only for LLS Twins)
LLS = Leiden Longevity Study (100-year-old co-twins); NTR = Netherlands Twin Research (40-year-old co-twins).
1The ratio between the number of sequence reads with a substitution variant and the total coverage in the sample with somatic substitution.
2The ratio between 454 reads supporting a substitution and the total number of 454 reads covering the position.
3The ratio between the number of k-mers in Ion Torrent reads with a substitution variant and the total number of k-mers with or without the substitution variant in the sample with the predicted somatic substitution.
FIGURE 1 Sanger trace around position chr3:4,772,267. Note: The complete list of eight substitution sites are depicted in Figure 2.
FIGURE 2 Confirmed somatic mutations supported by both Illumina and Complete Genomics platforms in LLS MZ twins.
FIGURE 3 Substitution ratio in Sanger sequencing-confirmed and not confirmed single base substitutions. Left: p value for differences between validated and non-validated is .002; Right: p value for differences between validated and non-validated is .077.
The intersection may have left much discordance undetected because of platform differences. We therefore examined an additional top 40 potential somatic substitutions for each twin pair from each platform (either Illumina or Complete Genomics) using Roche 454 sequencing. However, none of those 160 (i.e., 4 × 40) sites were confirmed.
Because Illumina and Complete Genomics use very different techniques, we expect that generally the intersection represents true positive discordances among many sequencing artifacts, which are assumed to randomly distribute over the genome and largely not overlapping between the platforms. Therefore, the results present a reliable lower bound of somatic single base substitution rate between co-twins.
We detected putative discordant short indels, structural variations, or copy number variations between the co-twins but none of them were supported by both platforms. As for potential heteroplasmy sites of mitchondrial DNA among these four samples, we first computed the variant-reference read count ratio at all heteroplasmy sites discovered in any of the four individuals. The correlation of mutation ratio between co-twins is high (>0.994), indicating no somatic mutation between them (see Table S3 available online). However, there is no correlation (~0) between unrelated individuals.
Discussion
Eight single base substitutions between co-twins were supported by both platforms and eventually validated as somatic variants. More variants in the potential list could be true but as stated above, technical differences between the NGS platforms probably hamper replication. The number of somatic variants may be substantially larger but those present in smaller fractions of cells go undetected. Consistent, detectable somatic variation likely includes somatic mosaicism in blood generated during development or clonal expansion of mutations generated at any point during the lifetime. The frequency of these variants is limited in blood even after 100 years of life. The percentage of confirmed somatic substitutions overlapping with introns is surprisingly high (five out of eight). We need additional samples to investigate whether somatic protein-altering variants are under stronger selection. Our observation of low numbers of somatic variants using two NGS platforms may provide a framework for detecting disease-related somatic variants in phenotypically discordant MZ twins. This will also facilitate the study of diseases caused by somatic mutations occurring in tissues.
Two strategies for the identification and validation of extremely rare somatic variants from error-prone short-read sequencing have been reported. Reumers et al. (Reference Reumers, Rijk, Zhao, Liekens, Smeets, Cleary and Del-Favero2012) investigated filtering strategies for Complete Genomics data of a MZ twin pair discordant for schizophrenia and identified a list of 846 putative discordances. Subsequent validation by Sanger sequencing showed that only 2 out of 814 investigated discordances were validated as somatic variants (Lam et al., Reference Lam, Clark, Chen, Natsoulis, O'Huallachain, Dewey and Snyder2012; Reumers et al., Reference Reumers, Rijk, Zhao, Liekens, Smeets, Cleary and Del-Favero2012). The alternative strategy was demonstrated by Lam et al. (Reference Lam, Clark, Chen, Natsoulis, O'Huallachain, Dewey and Snyder2012) by a performance comparison of Illumina (Bentley et al., Reference Bentley, Balasubramanian, Swerdlow, Smith, Milton, Brown and Smith2008) and Complete Genomics (Drmanac et al., Reference Drmanac, Sparks, Callow, Halpern, Burns, Kermani and Reid2010) whole-genome sequence showing that a significantly higher validation rate of SNPs could be obtained among variants detected by both platforms than among the platform-specific ones (Lam et al., Reference Lam, Clark, Chen, Natsoulis, O'Huallachain, Dewey and Snyder2012). The same was illustrated in our data since 8 out of 30 potentially discordant variants detected in the overlap of Illumina and CG platforms were validated against 0 of the 40 variants that did not overlap. These two independent platforms have become particularly popular in patient- and population-based studies and their difference with respect to data generation, read structure, and variant detection was investigated in a performance comparison study by Lam et al. (Reference Lam, Clark, Chen, Natsoulis, O'Huallachain, Dewey and Snyder2012).
In summary, this study shows that the number of detectable somatic variants in blood by using NGS is very low and that accumulation of somatic mutations is not necessarily a consequence of a century of life. Stochastic somatic variation occurring in less than 20% of cells will go undetected, however. The low background somatic variation in MZ co-twins illustrated by our study will be an advantage for identification of disease-related mutations in discordant twins. Our systematic approach shows that such mutations may be detected through comparison of whole-genome next generation sequences generated by at least two independent platforms.
Supplementary Material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/thg.2013.73
Acknowledgments
We dedicate this work to the memory of the late David R. Cox, an enthusiastic collaborator in longevity genetics for many years. The research leading to these results has received funding from the European Union's Seventh Framework Program (FP7/2007-2011) under grant agreement no. 259679; the National Institute for Healthy Ageing (Grant 05060810); the European Research Council (ERC 230374); the Netherlands Genomics Initiative (NGI); Biobanking and Biomolecular Resources Research Infrastructure Netherlands (BBRMI-NL); and the Netherlands Organization for Scientific Research (NWO). This research was also partly funded by Netherlands Organization for Scientific Research VENI grant 639.021.125.