


Major depressive disorder (MDD) is a complex and sometimes devastating condition that affects millions of people across the globe every year. To date, no single, direct causal mechanism has been identified in MDD but MDD morbidity has been linked to genetic and epigenetic processes, developmental history, and unique environmental exposures. Epigenetic mechanisms, especially DNA methylation, are increasingly being recognized as factors in the pathogenesis of MDD, and several published studies from both small and large samples have shown depression-related differences in methylation of genes related to inflammation, G-protein coupled receptor protein signaling, and axon guidance among others (Byrne et al., Reference Byrne, Carrillo-Roa, Henders, Bowdler, McRae, Heath and Wray2013; Crawford et al., Reference Crawford, Craig, Mansell, White, Smith, Spaull and Murphy2018; Davies et al., Reference Davies, Krause, Bell, Gao, Ward, Wu and Wang2014; El-Sayed et al., Reference El-Sayed, Halossim, Galea and Koenen2012; Sabunciyan et al., Reference Sabunciyan, Aryee, Irizarry, Rongione, Webster, Kaufman and Consortium2012; Shimada et al., Reference Shimada, Otowa, Miyagawa, Umekage, Kawamura, Bundo and Sasaki2018; Story Jovanova et al., Reference Story Jovanova, Nedeljkovic, Spieler, Walker, Liu, Luciano and Amin2018; Uddin et al., Reference Uddin, Koenen, Aiello, Wildman, de los Santos and Galea2011; Zhao et al., Reference Zhao, Goldberg, Bremner and Vaccarino2013).
DNA methylation involves the addition of a methyl group to the fifth carbon of a cytosine base. It occurs most frequently within CpG islands (i.e., genomic regions that contain a high frequency of CpG sites) in the promoter regions of many genes, although most CpG islands are unmethylated. Unlike the DNA sequence itself, which is stable during development, DNA methylation is highly dynamic and often induced by exposure to a range of factors such as nutrition, behavior, and chemicals (Dolinoy & Jirtle, Reference Dolinoy and Jirtle2008; Dolinoy et al., Reference Dolinoy, Weidman and Jirtle2007). These factors can directly affect methylation, chromatin remodeling, and other epigenetic pathways, resulting in alterations of epigenome and subsequent gene expression, thereby contributing to disease etiology (Dolinoy & Jirtle, Reference Dolinoy and Jirtle2008; Dolinoy et al., Reference Dolinoy, Weidman and Jirtle2007; Jiang et al., Reference Jiang, Bressler and Beaudet2004; Tsankova et al., Reference Tsankova, Renthal, Kumar and Nestler2007). However, the biological pathways linking aberrant methylation to MDD remain largely unknown, either because the function of the methylated genes is not known or because the methylation results have not been compared to actual differences in gene expression or other mechanisms. This uncertainty hampers our ability to implement early diagnosis, prevention, and treatment for this debilitating disorder.
To address these challenges, we designed a twin study known as the Mood and Methylation Study (MMS). This study recruited monozygotic (MZ) and dizygotic (DZ) twin pairs discordant or concordant for a lifetime history of MDD, and collected detailed clinical phenotypes for MDD, MDD-related traits, and environmental factors for each twin participant. Biospecimens were also collected for the primary methylome and transcriptome analyses but also to promote future multi-omic studies. Discordant MZ twin pairs provide a natural experiment to study epigenetic differences in MDD, because MZ twins match exactly on genetic background, age, and sex, thus totally eliminating the effects of these important confounders (Fraga et al., Reference Fraga, Ballestar, Paz, Ropero, Setien, Ballestar and Esteller2005; Relton & Davey Smith, Reference Relton and Davey Smith2010). In addition, identical twins share developmental environment, providing further control for confounding by early life experience, which is known to have a long-lasting impact on the epigenetic plasticity of the human genome (Rutten & Mill, Reference Rutten and Mill2009). Therefore, a discordant co-twin control approach provides a powerful model for epigenomic research.
MDD is typically considered a disorder of the central nervous system, and it is known that DNA methylation is, to some extent, cell-type- and tissue-specific. But, whatever the degree of tissue specificity, it is unfeasible to obtain a large sample of brain tissues for epigenetic analyses. That said, there is increasing evidence that leukocytes may be a useful cell model to evaluate epigenetic changes, because epigenetic variation may not be limited to the affected tissue but can also be detected in peripheral blood leukocytes (Crawford et al., Reference Crawford, Craig, Mansell, White, Smith, Spaull and Murphy2018; Story Jovanova et al., Reference Story Jovanova, Nedeljkovic, Spieler, Walker, Liu, Luciano and Amin2018). In fact, the study of peripheral blood may have some advantages over the use of brain tissue in that they are likely to accumulate fewer epigenetic changes induced by disease-related external factors, which could be confounding factors in postmortem studies (Pidsley & Mill, Reference Pidsley and Mill2011). Importantly, blood samples are much easier to obtain and could be used for large-scale epidemiologic studies, tracking changes associated with depression as the disease develops (Story Jovanova et al., Reference Story Jovanova, Nedeljkovic, Spieler, Walker, Liu, Luciano and Amin2018). Although a great deal more research is needed, it appears that the patterns of methylation across CpG sites in brain and peripheral blood leukocytes are similar (Murphy et al., Reference Murphy, O'Reilly and Singh2008; Yuferov et al., Reference Yuferov, Nielsen, Levran, Randesi, Hamon, Ho and Kreek2011) and correlations of DNA methylation profiles between different human tissues are reasonably high and can be evaluated using publicly available tools such as the Blood–Brain Epigenetic Concordance (BECon) application (Bromberg et al., Reference Bromberg, Levine, Belmaker and Agam2010; Brown & Robinson, Reference Brown and Robinson2000; Edgar et al., Reference Edgar, Jones, Meaney, Turecki and Kobor2017; Hannon et al., Reference Hannon, Lunnon, Schalkwyk and Mill2015). Because there are several cell types within leukocytes, we decided to isolate one type — monocytes — for this study.
Thus, we designed our study to first conduct an epigenome-wide association study (EWAS) using monocyte DNA extracted from whole blood in twin pairs discordant for lifetime history of MDD. In an effort to replicate significant differentially methylated regions (DMRs) in the central nervous system, we obtained clinically well-characterized postmortem brain tissues from the Stanley Medical Research Institute's Depression Collection, with control samples from non-depressed brains. We also downloaded brain DNA methylation data (HumanMethylation450K BeadChip) through the publicly available Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/info/datasets.html) database GSE41826. Finally, we planned to determine the functional impact of DMRs via gene expression analyses using RNA-seq with postmortem brain gene expression data (RNA-seq) through GEO database GSE101521. With this combination of approaches, we anticipate discovering epigenetically dysregulated genomic regions for MDD, in addition to uncovering functional epigenetic alterations that may play a causative role in MDD pathogenesis.
Although results along these lines are still expected to provide novel epigenetic targets for diagnosis, prevention, and therapeutic intervention in MDD, given the advance of both theory and technology since initial submission and funding, the final methods for this study were structured to allow a study initially focused on epigenomics to become a ‘multi-omics’ study. That is, a sufficient volume of blood has been drawn, processed, and stored both to complete the proposed study and to allow for future metabolome and transcriptome studies. In addition, stool sample collection was added to the protocol to facilitate analysis of the gut microbiome and its association with both MDD and MDD-related DMRs. Because each of the omics has both genetic and environmental contributions, we also decided to recruit dizygotic twin pairs and pairs that are concordant for MDD (see Table 1 for a breakdown of the sample by zygosity and concordance). This combination of twin pairs allows for maximum flexibility in answering important questions about the heritability of individual phenotypes and the simultaneous effects of genes and environment on multiple phenotypes. Finally, the twins in this sample are community volunteers interested in participating in research and significant efforts are made by the Washington State Twin Registry to say in contact with the volunteers. It will therefore be possible to return a large percentage of them in the future for longitudinal analysis and new collection of MRI analyses to assess the connectome.
TABLE 1 Study Recruitment Goals

Methods
Washington State Twin Registry Description and Involvement
Recruitment for the present study was completed via the community-based Washington State Twin Registry (WSTR; previously known as the University of Washington Twin Registry). Complete details of the construction and current components of the WSTR are available elsewhere (Afari et al., Reference Afari, Noonan, Goldberg, Edwards, Gadepalli, Osterman and Buchwald2006; Strachan et al., Reference Strachan, Hunt, Afari, Duncan, Noonan, Schur and Buchwald2013). Briefly, recruitment into the WSTR itself relies on initial identification of twins from the Washington State Department of Licensing (DOL), which asks all driver license and state ID applicants: ‘Are you a twin (Y/N)?’ Based on a data-sharing agreement between DOL and the University, the names and contact information of all twin respondents are then sent to the WSTR. Twins are contacted with information about the Registry and an enrollment survey. After both members of the pair complete the survey, the pair is enrolled in the Registry. The Registry contacts the twins multiple times each year with birthday cards, newsletters, and study invitations in order to keep contact information current. At the time recruitment began, the average age of enrolled twins was 37, and 41% of the twins reported being married. Approximately 4% of the Registry reported being Hispanic and 86% reported being White. All other racial categories were <4%. In terms of educational achievement, 25% completed high school or GED, an additional 34% had some college/vocational school, and 33% had a bachelor's degree or higher.
Twin Sampling and Recruitment for the Mood and Methylation Study
All participants provided informed consent for the study procedures and all procedures have been reviewed and approved by the University of Washington Institutional Review Board. Enrollment into the study required at least one member of the pair to have a lifetime history of MDD as measured by expert structured interview. Non-depressed twins were required to have no lifetime history of a major depressive episode and to have Beck Depression Inventory (BDI-II) scores below 13 at prescreening (Beck et al., Reference Beck, Steer and Brown1996). In addition, both members of the pair had to be willing to come to the University of Washington Medical Center for the in-person blood draw and data collection visit. Provision of stool samples, urine, saliva, and toenail clippings was optional. The primary exclusionary conditions for twin pairs were schizophrenia, bipolar disorder I and II, current substance use disorder, cancer, uncontrolled sleep apnea, and certain autoimmune, and uncontrolled endocrine disorders known to be associated with depressive symptoms. In order to avoid problems related to population stratification, only adult, same-sex, non-Hispanic, White twin pairs who were reared together, were enrolled.
Recruitment
In order to enroll participants from the WSTR into the study, we sent an introductory letter to potentially eligible WSTR members via postal mail and/or email. Participants in the Registry have already agreed to be approached for research projects by Twin Registry staff (all WSTR members were aged 18+ at the time this study began). We then followed up with those who expressed an interest with a prescreen survey (phone or online depending on participant preference) using approved scripts to screen for interest and eligibility. Once both members of the twin pair completed the prescreening without triggering any exclusionary criteria, each member was individually interviewed via telephone by a PhD-level psychologist using the relevant sections of the Structured Clinical Interview, Research Version, for the DSM-IV (SCID-RV), which was the latest version available at the time the study began (First et al., Reference First, Spitzer, Williams and Gibbon1995). The SCID-RV is a semi-structured interview guide for making Axis I DSM-IV diagnoses. For the purposes of this study, participants completed the Mood Episodes, Psychotic and Associated Symptoms, Psychotic Disorders, Mood Disorders, Substance Use Disorders, and Anxiety Disorders modules. They did not complete the Somatoform Disorders, Eating Disorders, or Adjustment Disorders modules. Any questions or concerns about the depression phenotype or exclusionary psychiatric conditions were referred to the senior clinician for evaluation and final decision-making.
Figure 1 shows the enrollment funnel for the study and Table 1 shows the breakdown by sex and zygosity. At the prescreen stage, the primary reasons for exclusion were having an exclusionary medical condition (e.g. recent cancer, history of autoimmune disease), or that one or both members of a pair were not interested in participating. At the clinical interview stage, the primary reasons for exclusion were no history of MDD in either twin, current substance use disorder, and history of bipolar illness. In terms of overall numbers, we anticipated being able to recruit twin pairs in locations other than western Washington using a contract service to provide in-home consent documentation along with specimen and questionnaire data collection. However, with the need to isolate monocytes, the contract service proved prohibitively expensive and we were forced to adjust our recruitment targets down (Table 1).

FIGURE 1 Enrollment funnel for the Twin MMS study.
Biospecimen and Phenotype Data Collection
Table 2 shows the interviews and questionnaires administered over the course of the study and Table 3 shows the outline of the biospecimen collection procedures.
TABLE 2 Study Measures and Administration

TABLE 3 Biospecimen Collections

At-home procedures
Prior to coming in for the in-person study visit, participants received a package that contained questionnaires along with stool, saliva, and toenail sample collection kits. The questionnaires included two measures of depression symptom severity, the BDI-II mentioned above and the Quick Inventory of Depressive Symptomatology (QIDS-SR16). The BDI-II is a self-administered 21-item scale that has acceptable sensitivity and specificity with regards to DSM criteria for MDD. The QIDS is also self-administered and was designed to assess the severity of depressive symptoms (Rush et al., Reference Rush, Tirivedi, Ibrahim, Carmody, Arnow, Klein and Keller2003). The questionnaire packet also included measures of lifetime exposure to stressful events that may influence epigenetic variations leading to disease (Franklin et al., Reference Franklin, Russig, Weiss, Graff, Linder, Michalon and Mansuy2010; Waterland, Reference Waterland2009). Exposure to childhood traumatic stress was measured with the Early Trauma Inventory (Bremner et al., Reference Bremner, Vermetten and Mazure2000). This self-administered instrument assesses not only the frequency and nature of traumatic events, but also the emotional impact of the stressor on the individual immediately after and across the life span, and the impact on social and occupational functioning. We also measured hostility using the Cook–Medley Hostility Inventory (Barefoot et al., Reference Barefoot, Dodge, Peterson, Dahlstrom and Williams1989) and anger by means of the Spielberger's Anger Expression Inventory (Spielberger, Reference Spielberger1988). These factors are related to incidence of MDD (Keilp et al., Reference Keilp, Gorlyn, Oquendo, Brodsky, Ellis, Stanley and Mann2006) and might also influence epigenetic alterations (Waterland, Reference Waterland2009). Finally, we measured social and emotional support by means of the Enhancing Recovery in Coronary Heart Disease (ENRICHD) Social Support Inventory, a five-item scale assessing both social and emotional support (ENRICHD Investigators, 2001). Table 4 shows initial demographic and clinical data broken down by zygosity.
TABLE 4 Demographic and Clinical Data by Zygosity
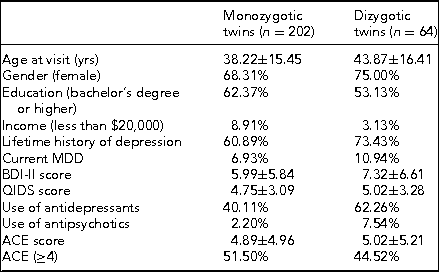
Note: BDI-II = Beck Depression Inventory-II; QIDS = Quick Inventory of Depressive Symptomatology; ACE = Adverse Childhood Experiences.
The self-collection stool sample kits consisted of a collection bowl that sits on the participant's toilet along with two tubes that have a scoop built onto the cap. The tubes have formalin fixative inside in order to stabilize the sample, along with small glass beads to aid in mixing the fixative into the sample. The participant used the cap scoop to collect a small amount of stool from the collection bowl and placed it inside the tubes. The tubes were then repeatedly inverted to distribute the fixative and stored in the participant's freezer until they attended the in-person visit. The soluble stool collection bags were flushed down the toilet.
Saliva collection was also self-administered three times within the same day. The first was upon waking in the morning, the second was between 4:30 and 6:00 pm, and the third was prior to bedtime. The kits were absorbant cotton balls that were placed in the mouth until wet, then placed into sealed bags and brought to us at the in-person visit. For toenail collection, we included clippers and a pre-labeled plastic bag for participants to cut a sample from each nail on one foot, and place them in the bag to return to us at the in-person visit.
In-person procedures
Participants came to the University of Washington Medical Center for anthropometric measurement along with blood and urine collection. The twins were permitted, but not required, to come to the visit together. Trained hospital staff completed the physical and physiological measures and drew up to 140 ml of blood into the various tubes. Participants also provided a urine sample, which was aliquoted and stored for future use. The blood samples were then processed immediately. From up to 110 ml of whole blood, we aliquoted plasma and serum. We then extracted DNA and RNA from monocytes isolated from the other peripheral blood mononuclear cells (PBMCs) with the Monocyte Isolation Kit II from Miltenyi Biotec. This isolation is achieved through negative selection. Non-monocytes are labeled with biotin-conjugated antibodies and Biotin Microbeads. The magnetically labeled cells stay in the column while the unlabeled monocytes pass through. Then the DNA and RNA were extracted with the AllPrep DNA/RNA/miRNA Universal Kit from Qiagen. This extraction started with the lysing of the monocyte cell pellet and binding of the DNA and RNA onto respective columns. The columns were then treated with specific buffers to remove unwanted proteins. The DNA/RNA were finally eluted from the column and the concentration determined using the NanoDrop Spectrophotometer.
DNA Methylation and Gene Expression Analysis
Using the collected peripheral blood monocytes, we are currently conducting an EWAS on MDD in 133 MZ and DZ pairs. DNA methylation is currently being assayed using the Infinium MethylationEPIC BeadChip. Compared to whole-genome sequencing, the BeadChip covers a small proportion of CpG sites. However, those sites are well distributed across the genome and the chip provide a cost-effective way to begin investigating methylation and disease. Gene expression—also using monocytes from the same sample of twins—will be measured by RNA-seq. Statistical analyses will be conducted to identify differentially methylated genes/regions (DMRs) associated with MDD. An initial power analysis indicated that our sample size has 80% power to detect a minimum of 9.7% methylation difference between cases and controls, assuming covariates accounting for 20% of phenotypic variability (Tsai & Bell, Reference Tsai and Bell2015). Functional importance of the identified DMR genes will be validated by linking to gene expression and further confirmed in postmortem brain tissue using available GTEx datasets. Sophisticated bioinformatics analyses will also be performed to investigate the potential functional role of the identified genes in MDD pathology. Also, given the collected specimens (e.g., plasma, stool samples), we plan to generate multi-omics datasets for all MMS twins, and conduct integrated omics analyses to identify novel biomarkers and biological pathways for MDD using a systems biology approach. DNA sample and the omics datasets will be publicly accessible through the NIMH biorepository or NIH dbGaP.
Future Directions
The methods described above are primarily dedicated to addressing the scientific aims of our original grant proposal. However, this sample of twins provides tremendous opportunity to evaluate MDD from a multi-omics perspective, and investigators interested in collaboration are encouraged to contact us. Our group has begun a pilot analysis of the gut microbiome from the MZ discordant pairs based on 16s RNA identification of gut microbiome diversity. Additional proposed research seeks to characterize MDD-associated changes in the plasma metabolome associated with microbial metabolism of neurotransmitters, neuroactive compounds, and neurometabolites and to measure differences in bacterial endotoxin exposure, measured as lipopolysaccharide binding protein, associated with leaky gut in MDD.
We anticipate that this twin sample will reveal promising directions for research on MDD treatment and prevention, but we also recognize that any significant results will require extensive replication and validation in samples different from the one described here. Greater racial diversity will be critical, along with additional focus on age-related effects and differences in early childhood exposures.
Acknowledgment
This research was funded by the National Institute of Mental Health (R01 MH097018: Zhao, Strachan).
Conflicts of Interest
None.





