


Mitochondria are energy-converting organelles that are present in the cells of virtually all eukaryotic organisms. These cellular powerhouses use oxygen to harness energy through the oxidative phosphorylation (OXPHOS) process. To achieve this, high-energy electrons derived from the oxidation of food molecules are transferred along the respiratory chain (RC) to four multisubunit protein complexes embedded in the inner mitochondrial membrane, releasing energy that is used to pump protons across this membrane. The established proton gradient and resulting mitochondrial membrane potential (Δψm) are then used by adenosine triphosphate (ATP) synthetase to generate ATP, which provides the cell with the majority of its energy requirements. This process of energy production was developed by bacteria approximately two billion years ago, before the appearance of eukaryotic cells. According to the endosymbiotic theory, mitochondria were once aerobic bacteria that united with the ancestor of the eukaryotic cell in a mutually advantageous relationship, and this led to an evolutionary explosion from which multicellular organisms evolved (Ref. Reference Lane1).
Interestingly, mitochondria exist as an intracellular network, and constantly merge through fusion and divide through fission. Fusion consists of the joining of separate mitochondria and is mainly controlled by GTPases. In mammals, mitofusins (MFN1 and MFN2) mediate the fusion of the outer membrane of two mitochondria, followed by the joining of their inner membranes mediated by OPA1 (optic atrophy 1) (Fig. 2). By contrast, DNM1L (dynamin-1 like protein; DRP1) and FIS1 (fission 1 homologue) are key components of the fission machinery, which in cooperation, lead the splitting of the mitochondrial tubule. These two opposing processes are finely balanced, thus maintaining steady state physiological conditions, and have important roles in mitochondrial function and development, as well as in programmed cell death (for a review, see Ref. Reference Chan2).
Although the majority of the estimated 1500 proteins present in the mitochondrion (Ref. Reference Taylor, Fahy and Ghosh3) are encoded by the nuclear genome, mitochondria have their own DNA within the matrix, which encodes a small set of mitochondrial proteins – 13 subunits of the RC complexes – as well as rRNAs and tRNAs. In mammalian cells, mitochondrial DNA (mtDNA) is a circular molecule that is present in several copies and is organised into bacterial nucleoid-like structures. These structures contain several proteins that are involved in mtDNA maintenance and replication, as well as in its transcription and translation (Ref. Reference Wang and Bogenhagen4). The physical proximity of the nucleoids to the RC makes mtDNA particularly vulnerable to damage by reactive oxygen species (ROS), which are unavoidable OXPHOS products. Accordingly, mitochondria have quality-control systems in place to maintain their function, and they selectively respond to oxidative stress through mechanisms acting at the molecular, organellar and cellular levels (Figs 1 and 2). At the molecular level, the first line of protection is provided by molecular chaperones and endogenous proteases, which respectively detect and eliminate abnormally folded and aggregated proteins generated by ROS. Oxidative damage to mitochondria is also counteracted at the organelle level by components of the fusion and fission machinery, which ensure the maintenance of a functional population of organelles (Ref. Reference Twig, Hyde and Shirihai5). This occurs through the alteration of mitochondrial network dynamics and leads to the segregation of defective mitochondria, which are eventually removed via autophagy (mitophagy). Significantly, a major consequence of mitochondrial damage is loss of ATP, which results in a decrease in the ratio of ATP to AMP. This, in turn, can lead to the activation of a cellular signal-transduction pathway where the AMP-activated protein kinase (AMPK) acts as a master sensor of intracellular energy status (Ref. Reference Hardie6) and reduces the activity of the mammalian target of rapamycin complex 1 (mTORC1). mTORC1 is a major positive modulator of mitochondrial metabolism and biogenesis (reviewed in Ref. Reference Laplante and Sabatini7). It is supposed that an mTORC1-dependent decrease in mitochondrial biogenesis could indirectly promote the autophagic segregation of defective mitochondria by ensuring that mitochondrial dynamics processes are used for the clearance of dysfunctional mitochondria, as opposed to their biogenesis (Fig. 2).

Figure 1. Core quality-control pathways in mitochondria. Low levels of damaged proteins in mitochondria are cleared at the molecular level by intraorganellar proteases and chaperones such as OMI/HTRA2 and TRAP1 (top left). Enhanced levels of damage probably overwhelm the capacity of the molecular quality-control machinery (middle left), leading to the proposed segregation of damaged mitochondrial components by the fusion/fission machinery. This enables the physical separation of healthy (green) and damaged (orange) daughter mitochondria. Damaged mitochondria are then recycled using the cellular autophagy pathyways. If the levels of damage exceed the capacity of both molecular and organellar quality-control pathways (top right), mitochondria can rupture, leading to the release of apoptosis-promoting factors and cell death.
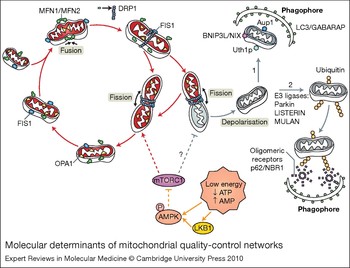
Figure 2. Molecular determinants of mitochondrial quality-control networks. Mitochondria are dynamic organelles that are continuously remodelled by fusion and fission events. Fusion requires the joining of both the inner and outer mitochondrial membranes of two mitochondria. Mitofusin 1 (MFN1) and mitofusin 2 (MFN2) carry out the initial joining of the outer membrane fusion, whereas OPA1 mediates the fusion of the inner membrane. Mitochondrial fission is mediated by DRP1 and FIS1; DRP1 oligomerizes into 8–12 subunit chains and is recruited to mitochondria via the adaptor FIS1. This recruitment allows the full oligomerisation of DRP1 into spiral chains that wrap around the mitochondria and, by constriction, causes mitochondrial fission. In this figure, fission is shown as a means to selectively eliminate damaged mitochondrial components by packing them in one of the fission products, and it has been proposed to involve the asymmetrical segregation of OPA1 (Ref. Reference Twig, Hyde and Shirihai5). In this context, fission produces metabolically different daughter units that may either maintain (red) or lose (grey) an intact membrane potential. Depolarised mitochondria are targeted to degradation by autophagy (mitophagy) through a mechanism that is still poorly defined. Here, we illustrate two possibilities: (1) the presence of proteins associated with mitochondrial membranes, such as the mammalian BNIP3L/NIX (Ref. Reference Sandoval109, Reference Schweers110), and the yeast proteins Uth1p (Ref. Reference Kissova111) and Aup1 (Ref. Reference Tal112), which are involved in the direct entrapment of mitochondria by autophagosomes; (2) the activity of E3 ligases, such as LISTERIN (Ref. Reference Chu113), MULAN (Ref. Reference Li114) and Parkin (Refs Reference Narendra28, Reference Geisler115), which might regulate the conjugation of monoubiquitin (Ub) or polyubiquitin chains to an exposed mitochondrial protein, thereby targeting the organelle to autophagic degradation. p62 and NBR1 are autophagic receptors that bind both Ub and ATG8/LC3 on the phagophore (Ref. Reference Kirkin116), which might constitute the mechanistic link between mitochondrial ubiquitylation and mitophagy. The mTOR signalling pathway acts as a major positive modulator of mitochondrial metabolism and biogenesis. We propose that the inhibition of mTORC1 upon mitochondrial damage caused by loss of ATP and consequential activation of AMPK, might be crucial to ensure an enhancement of the autophagic degradation of defective mitochondria. Abbreviations: AMPK, AMP-activated protein kinase; Aup1p, yeast mitochondrial protein phosphatase homologue; BNIP3L/NIX, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like; LC3, light chain 3 protein; LKB1, serine/threonine-protein kinase 11; mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; NBR1, next to BRCA1 gene 1 protein; OM, outer membrane; OPA1, optic atrophy 1; Uth1p, yeast outer mitochondrial membrane protein.
Ultimately, if mitochondrial damage is too severe and overwhelms the cellular networks of protective mechanisms, dysfunctional organelles break open, releasing a series of apoptotic factors that ultimately cause cell death (reviewed in Ref. Reference Youle and Strasser8). This removal of cells with severely injured mitochondria has been suggested to be another layer of protection that acts at the level of the organism (Ref. Reference Hamann, Brust and Osiewacz9). Nevertheless, the demise of postmitotic cells such as neurons can result in irreparable damage to the organism.
The chronic and selective loss of neuronal cells is indeed a typical feature of most neurodegenerative diseases and results in the progressive impairment of function in the central (CNS) and/or peripheral nervous system (PNS). Although they affect different neuronal cell populations, neurodegenerative diseases share remarkable common traits, e.g., enhanced neuronal cell death, defects in axonal transport and the accumulation of dysfunctional mitochondria.
In addition to their primary role in cellular metabolism, mitochondria are active players in Ca2+ homeostasis. Under conditions of cellular Ca2+ overload, in association with oxidative stress, mitochondrial Ca2+ uptake leads to the collapse of Δψm, which sensitises cells to apoptosis. In this regard, it is noteworthy that alterations of mitochondrial Ca2+-regulating proteins (Bcl-2 family members and uncoupling proteins) are implicated in age-related neuronal pathologies (Ref. Reference Mattson10).
The high density of mitochondria within neurons could provide a rationale for the sensitivity of the CNS to energy deficits due to mitochondrial dysfunction. Nevertheless, it is still debated whether mitochondrial impairment and oxidative damage are aetiological factors, or are solely the consequences of neurodegeneration. In this review, we provide a synopsis of the current advances in the knowledge of mitochondrial quality-control systems, with particular emphasis on how disturbances in the molecular components of such systems might contribute to neurological diseases.
Mitochondrial dysfunction and Parkinson disease
Mitochondrial dysfunction has long been associated with the onset of neurodegenerative states, including the selective loss of dopaminergic neurons in Parkinson disease (PD). PD is the most common neurodegenerative movement disorder, and it affects 1% of the population above the age of 60. The critical loss of pigmented neurons in the substantia nigra underlies the motor impairment, which is the most common clinical feature of the disease. However, neuronal loss in regions of the brain controlling autonomic functions, cognition and mood is also implicated. Neurons that degenerate in PD often contain intracellular inclusions of α-synuclein (SNCA), which is a major component of the so-called Lewy bodies (Ref. Reference Mattson, Gleichmann and Cheng11).
Despite the fact that the majority of PD cases occur sporadically as a result of unknown causes, reduced levels of mitochondrial complex I activity are generally associated with the disease (Refs Reference Schapira12, Reference Swerdlow13, Reference Navarro and Boveris14). Additionally, the toxins rotenone and MPTP cause an acute and irreversible parkinsonian syndrome that might involve the specific inhibition of complex I (Refs Reference Langston and Ballard15, Reference Gerlach16). This suggests that one of the main causes of neuron loss and motor impairment in PD is toxin-induced mitochondrial stress in dopaminergic neurons (reviewed in Ref. Reference Di Monte17).
Although classically considered to be a nongenetic disorder because of the large number of sporadic cases, genetic analyses of the small proportion of familial PD cases have led to significant insights into its pathogenesis.
Mutations in SNCA (α-synuclein) and LRRK2 (leucine-rich repeat kinase 2) genes mediate autosomal-dominant forms of PD, whereas mutations in PARK2 [Parkinson disease (autosomal recessive, juvenile) 2; parkin], PARK7 [Parkinson disease (autosomal recessive, early onset) 7; DJ-1] and PINK1 (PTEN-induced putative kinase 1) cause the disease in an autosomal-recessive manner. Mutations in HTRA2, which encodes the mitochondrial serine protease (HtrA2 serine peptidase; OMI) have also been described in families affected by PD (see Ref. Reference Bogaerts18 for a review). However, the importance of HTRA2 mutations in the development of PD has been questioned by independent studies, indicating that the role of this protease in the development of PD is still controversial.
Interestingly, the major common functional effects of the proteins encoded by these genes relate to mitochondria: PINK1 is a kinase regulated by a canonical N-terminal mitochondrial-targeting sequence (Ref. Reference Valente19); DJ-1 is a molecular chaperone that has been proposed to be localised to the mitochondria upon oxidative stress (Refs Reference Ashley20, Reference Canet-Aviles21, Reference Junn22) and stabilise complex I (Ref. Reference Hayashi23); Parkin and PINK1 seem to be involved in the regulation of mitochondrial dynamics, morphology and turnover (Refs Reference Poole24, Reference Deng25, Reference Yang26, Reference Dagda27, Reference Narendra28); and finally HTRA2/OMI and PINK1 have been shown to be components of the same mitochondrial stress-sensing pathway (Ref. Reference Plun-Favreau29). Given the fact that α-synuclein and LRRK2 are partially localised to mitochondria (Refs Reference Li, Orr and Li30, Reference Nakamura31), the majority of genes associated with PD so far implicate, either directly or indirectly, the involvement of mitochondria in the pathogenesis of this disease (Table 1). Molecular dissection of the contribution of these gene products to mitochondrial function is currently underway and is likely to be important for a better understanding of PD (reviewed in Ref. Reference Bueler32).
Table 1. Genes involved in major mitochondria-associated neurodegenerative diseases

Abbreviations: AD, Alzheimer's disease; DNM1L, dynamin-1 like protein (DRP1); HD, Huntington disease; HTRA2, HtrA2 serine peptidase (OMI); HTT Huntingtin (Htt); IM, inner membrane; IMS, Inner membrane space; LRRK2, leucine-rich repeat kinase 2; OM, outer membrane; OPA1, optic atrophy protein 1, autosomal dominant; PARK2, Parkinson disease autosomal recessive, juvenile 2 (parkin); PARK7, Parkinson disease autosomal recessive, early onset 7 (DJ-1); PARL, presenilin-associated rhomboid-like protein; PD, Parkinson's disease; PINK1, PTEN-induced putative kinase 1; PPARGC1A, peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1); PPID, peptidylprolyl isomerase D (cyclophilin D; CYPD); PREP, prolyl endopeptidase (PreP); SNCA, α-synuclein; TRAP1, TNF-receptor-associated protein 1 (Hsp75).
Any given mitochondrion is not a discrete, autonomous organelle (Ref. Reference Chan2); in fact, the entire mitochondrial population within a cell is in constant flux driven by a series of fusion and fission events. The dynamic nature of the mitochondrion population not only determines mitochondrial morphology and copy number (Ref. Reference Westermann33), but it also provides additional protection against mitochondrial damage (Fig. 2). Recently, it was proposed that PINK1 and Parkin regulate mitochondrial dynamics through interaction with the fusion–fission machinery (Refs Reference Poole24, Reference Deng25, Reference Yang26). However, it is also conceivable that these proteins affect mitochondrial dynamics indirectly by perturbing calcium and ROS homeostasis (Refs Reference Dagda27, Reference Sandebring34). Several groups reported that loss of Pink1 or parkin in Drosophila leads to significant mitochondrial enlargement, suggesting a defect in mitochondrial fission (Refs Reference Park35, Reference Clark36). Interestingly, the phenotypes of Pink1 or Parkin mutant flies are very similar, and although Pink1 is unable to rescue the defects caused by the lack of parkin, overexpression of parkin rescues the mitochondrial pathology induced by the loss of Pink1. By contrast, PINK1-deficient mammalian cells show fragmented and truncated mitochondria; nevertheless, the molecular interaction between Parkin and PINK1 is conserved in mammalian cells (Refs Reference Exner37, Reference Lutz38, Reference Weihofen39).
Parkin acts as an E3 ubiquitin ligase, and it is mostly localised to the cytosol. However, it has been shown in vitro that Parkin is recruited to the mitochondria upon loss of Δψm, leading to a selective autophagic engulfment and elimination of dysfunctional organelles (Ref. Reference Narendra28) (Fig. 3). According to Chung and colleagues, this recruitment is dependent on the direct phosphorylation of Parkin on Thr175 by PINK1 (Ref. Reference Kim40), whose kinase domain faces the cytosol (Ref. Reference Zhou and Freed41). Considering that downregulation of PINK1 causes loss of mitochondrial membrane potential in several mammalian systems (Refs Reference Valente19, Reference Exner37, Reference Abou-Sleiman42), it seems plausible to propose a role for PINK1 in flagging dysfunctional mitochondria for degradation. The mitochondrial pathology in PINK1-deficient animals might be caused by an insufficient recruitment of Parkin followed by an impairment of mitochondrial turnover (Ref. Reference McBride43). However, given that Parkin is capable of rescuing mitochondrial dysfunction caused by the loss of PINK1, it is unclear whether a direct molecular interaction between PINK1 and Parkin is essential for efficient removal of damaged mitochondria.
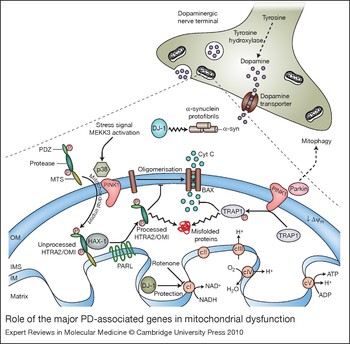
Figure 3. Role of the major PD-associated genes in mitochondrial dysfunction. Many genes associated with Parkinson disease (PD) implicate a role for mitochondria in the pathogenesis of the disease. The serine protease HTRA2–OMI is synthesised as an inactive precursor containing a mitochondrial-targeting sequence (MTS). In response to the activation of the MEKK3–p38 stress-kinase-signalling pathway, HTRA2/OMI is phosphorylated by p38 in a PINK1-dependent manner and imported into the intermembrane space (IMS), where it has been reported to bind to the Bcl-2-family-related protein Hax-1 (Ref. Reference Chao117). Despite the fact that this interaction is controversial (Ref. Reference Jeyaraju118), it has been suggested to promote the proteolytic processing of HTRA2/OMI by the mitochondrial protease PARL. Active HTRA2/OMI is thought to be involved in the degradation of misfolded proteins present in the IMS (Ref. Reference Moisoi119) and to prevent the oligomerisation of the activated form of BAX on the outer membrane (OM), thus avoiding apoptosis (Ref. Reference Chao117). Under oxidative stress conditions, PINK1 also interacts with the mitochondrial molecular chaperone TRAP1. Once phosphorylated, TRAP1 inhibits oxidative-stress-induced cytochrome c (Cyt C) release, prevents misfolding and promotes the correct assembly of mitochondrial proteins. DJ-1 is a cytosolic oxidative-stress-regulated chaperone, which redistributes from the cytosol to the mitochondria upon oxidation of specific cysteine residues. In the cytoplasm, DJ-1 prevents the aggregation and toxicity of α-synuclein (α-syn); in mitochondria, it has been suggested to protect respiratory complex I (cI) from oxidative-stress-mediated inactivation (Ref. Reference Hayashi23). Another protein involved in the mitochondrial stress response is Parkin, a cytosolic E3-ubiquitin ligase, which is selectively recruited to uncoupled or dysfunctional mitochondria, targeting them for autophagy (Ref. Reference Narendra28). A model is illustrated whereby PINK1, acting upstream of Parkin, might regulate mitochondrial function by sensing mitochondrial damage, recruiting Parkin and inducing mitophagy. Abbreviations: ADP, adenosine diphosphate; ATP, adenosine-5′-triphosphate; cII, complex II; cIII, complex III; cIV, complex IV, cV, complex V; HAX-1, HCLS1-associated protein X-1; HTRA2, high-temperature requirement A2; IM, mitochondrial inner membrane; MEKK3, mitogen-activated protein kinase kinase kinase 3; PINK1, PTEN-induced putative kinase 1; PD, Parkinson disease; PDZ domain, post-synaptic density protein (PSD95), Drosophila discs large tumour suppressor (DlgA) and zonula occludens-1 protein (zo-1) domain; TRAP1, TNF-receptor associated protein; Δψm, mitochondrial membrane potential.
PINK1 and Parkin seem to share the same genetic pathway, which probably involves the mitochondrial quality-control machinery. Additionally, two intermembrane space (IMS) proteins involved in mitochondrial quality control are substrates of PINK1: the chaperone TRAP1/ Hsp75 (Ref. Reference Pridgeon44) and the serine protease HTRA2/OMI (Ref. Reference Plun-Favreau29). Under stress conditions, these proteins interact with PINK1 (Fig. 3) and are subsequently activated to prevent the accumulation of misfolded proteins. Given the proposed role of toxins such as rotenone and MPTP in acute parkinsonian syndrome, it is attractive to argue that defects in proteins involved in mitochondrial-stress pathways could increase the susceptibility towards neurodegeneration in individuals exposed to environmental toxins.
In addition to mitochondrial dysfunction, protein aggregation also has a major role in PD pathogenesis. However, it remains to be determined whether there is a convergence between mitochondrial dysfunction and protein aggregation. α-synuclein is a fibrillar aggregation-prone protein that is a main component of Lewy bodies and is believed to contribute to PD by toxic gain-of-function effects. Although this protein is mostly cytosolic, mitochondrial abnormalities were observed in several transgenic mouse models that either lack or overexpress wild-type or mutant α-synuclein (Refs Reference Poon45, Reference Martin46), suggesting that it has a physiological role in the mitochondria (Ref. Reference Ellis47), where it might interact with mitochondrial proteins. Recent work has shown that loss of PINK1 leads to an increase of α-synuclein aggregation (Ref. Reference Liu48), whereas overexpression of PINK1 was able to suppress α-synuclein-induced phenotypes in Drosophila (Ref. Reference Todd and Staveley49). Moreover, the chaperone DJ-1 has been shown to redistribute to mitochondria upon oxidative stress (Refs Reference Ashley20, Reference Canet-Aviles21,Reference Junn22) and to prevent the aggregation and toxicity of α-synuclein in an oxidation-dependent manner (Refs Reference Zhou and Freed41, Reference Shendelman50, Reference Batelli51).
Whether or not DJ-1 recruitment is regulated in the same manner as Parkin remains to be determined. However, it has already been demonstrated that Parkin, PINK1 and DJ-1 form a functional ubiquitin E3 ligase complex that promotes ubiquitylation and degradation of unfolded or misfolded proteins (Ref. Reference Xiong52). This complex might thus be the missing link between mitochondrial function and protein aggregation.
Finally, PINK1, DJ-1 and Parkin seem to interact in vivo with LRRK2, another PD-associated protein (Refs Reference Tain53, Reference Venderova54). LRRK2 is a serine/threonine kinase responsible for the phosphorylation of actin crosslinkers, moesin (MSN), ezin (EZR) and radixin (RDX) (Ref. Reference Jaleel55), as well as the eukaryotic initiation factor 4E (eIF4E)-binding protein (4E-BP), which is a negative regulator of eIF4E-mediated protein translation and a key mediator of various stress responses. 4E-BP activity is also regulated by the TOR signalling pathway. Activated TOR hyperphosphorylates 4E-BP, inhibiting it and promoting 5’-cap-dependent translation. A switch from cap-dependent to cap-independent initiation of translation seems to be a key event in mediating the survival response to various physiological stresses (Ref. Reference Clemens56). In support of this, Whitworth and colleagues recently showed that loss of Drosophila Lrrk2 as well as treatment with rapamycin, an inhibitor of TOR, lead to the activation of 4E-BP, and this suppresses Pink1 and parkin mutant phenotypes in flies. Taken together, and with the caveat that some of these observations await confirmation, it is attractive to suggest that LRRK2 might have a role in the regulation of mitochondrial homeostasis by controlling signalling pathways involved in protein translation.
As described here, the most recent work has emphasised the central importance of mitochondrial dysfunction in PD. Nevertheless, we still need to consider several questions: is this mitochondrial pathway also relevant to sporadic forms of PD? How can we exploit our knowledge of mitochondrial involvement in PD to develop better therapies? We believe that new insights from future studies will provide us with neuroprotective therapies that could make a difference in the treatment of PD.
Mitochondrial dysfunction in Alzheimer disease
Alzheimer disease (AD) is a progressive and irreversible pathology that affects millions of people worldwide, and it represents the most common form of dementia among elderly people. AD has been attracting increasing attention from both the scientific community and governments because of its increasing social impact and costs. By the year 2050, 50% of people 85 years and older will be afflicted with AD (Ref. Reference Reddy and McWeeney57), making this disease a health priority.
AD can be classified into sporadic AD and familial AD (FAD). Only a small fraction (approximately 10%) of AD cases are familial (Ref. Reference Thinakaran58), and show autosomal-dominant transmission within affected families. Sporadic AD accounts for the majority of cases, with ageing being the most relevant known risk factor. In addition to age and family history, an increased risk of developing the disorder – both early and late-onset AD – is associated with apoEɛ4 (Ref. Reference Corder59), one of three allelic variants of the human APOE gene. More recently, variants of clusterin (CLU) and phosphatidylinositol-binding clathrin assembly protein (PICALM) proteins have also been associated with AD (Ref. Reference Harold60). A characteristic feature of all forms of AD is progressive neuronal cell death in brain regions displaying high plasticity, which is caused by the accumulation of intraneuronal neurofibrillary tangles (NFTs) and extracellular β-amyloid plaques. NFTs consist of abnormally phosphorylated tau protein, which is polymerised into paired helical filaments (PHFs); amyloid plaques are composed of β-amyloid peptide (Aβ1–42), which polymerises into insoluble fibrils with high β-sheet content.
The Aβ-aggregation process has been considered for years to be the most relevant phenomenon implicated in the aetiology of AD. However, mounting evidence is now highlighting the role of mitochondrial impairment in the pathology of AD, and the two events are reciprocally related. In particular, the finding that Aβ species accumulate intracellularly in the mitochondria (Ref. Reference Caspersen61), and the fact that this occurs before their extracellular accumulation, has changed the focus of potential therapies against this pathology. These findings provide a direct link between Aβ and the pathological dysfunctions of the mitochondria found in AD, such as deficits in tricarboxylic acid (TCA) cycle and/or complex IV. Although the molecular origin of the intramitochondrial Aβ is still unknown, the in vivo and in vitro consequences of these deposits in the cell have, in part, been elucidated. The presence of β-amyloid aggregates in the organelle has been shown to impair the enzymatic activity of cytochrome c oxidase and inhibit the activity of mitochondrial amyloid-β-binding alcohol dehydrogenase (ABAD) (Fig. 4). The selective decrease in complex IV observed in the brains of AD patients (Refs Reference Parker62, Reference Maurer, Zierz and Moller63) could be responsible for an imbalance in the electron transport chain (ETC) energy-extracting mechanism, which, in turn, would compromise the general metabolism of the neurons.

Figure 4. Mitochondrial dysfunction in AD. Degenerating neurons in brain areas affected by Alzheimer disease (AD) (e.g. hippocampus and cerebral cortex) present typical intracellular neurofibrillary tangles (NFTs) and extracellular accumulation of β-amyloid plaques. Aβ is produced from the metabolism of amyloid precursor protein (APP) through sequential cleavage by β- and γ-secretases, and it is prone to form toxic oligomeric structures both inside and outside the cell. APP and Aβ can affect mitochondrial function by different mechanisms. APP is targeted to mitochondria, where it forms complexes with the translocases of the outer and inner membranes (TOM and TIM), which drive the import of mitochondrial proteins in cooperation with molecular chaperones. However, the presence of an acidic domain within APP might be responsible for preventing its translocation into mitochondria. As a consequence, the import of mitochondrial proteins, such as respiratory chain subunits, is reduced, and this event is associated with increased free radical generation and reduced activity of the electron-transport chain. Other aspects of amyloid metabolism involve mitochondria: intramitochondrial Aβ has been shown to interact with amyloid-β-binding alcohol dehydrogenase (ABAD) and to produce reactive oxygen species (ROS). Aβ also interacts with cytochrome c oxidase, thus decreasing the activity of complex IV (cIV). Furthermore, presequence peptidase PreP and HTRA2/OMI serine protease have been shown to degrade Aβ oligomers, thus providing a mechanism to detoxify this metabolite. Intramitochondrial Aβ directly interacts with cyclophilin D (CypD), a component of the mitochondrial permeability transition pore (mPTP), which is located in the mitochondrial matrix. This interaction makes the channel more sensitive to Ca2+ and stimulates mPTP opening, thus raising the permeability of the mitochondrial inner membrane (IM) and eventually disrupting the mitochondrial outer membrane (OM). As a result, deregulation of the mPTP opening determines a functional disorder that triggers cell death. Abbreviations: AD, Alzheimer disease; ATP, adenosine-5′-triphosphate; cI, complex I; cII, complex II; cIII, complex III; cV, complex V; HTRA2, HtrA2 serine peptidase; Δψm, mitochondrial membrane potential.
The process of Aβ aggregation is accompanied by the generation of ROS and lipid peroxidation. At the mitochondrial level, such neurotoxic actions affect the components of the OXPHOS by depletion of ATP levels (Ref. Reference Casley64), and they exert a secondary effect on the mitochondrial ATPase. This, in turn, results in the altered regulation of the calcium elevations in which the ATPase is implicitly involved (Ref. Reference Boitier, Rea and Duchen65). Furthermore, the toxicity of Aβ peptides – which is associated with their oligomeric state, rather than their fibrillar insoluble state (see Ref. Reference Walsh and Selkoe66 for a review) – causes a rapid increase of intracellular calcium that eventually leads to cell death. A pivotal role in cell death commitment is played by the opening of the mitochondrial permeability transition pore (mPTP), which determines the collapse of Δψm and the subsequent release of IMS proapoptotic factors. The molecular basis for this pathogenic mechanism was provided by the work of Du and colleagues (Ref. Reference Du67) in both transgenic mouse models and human AD brains, where the Aβ peptide has been shown to interact with cyclophilin D (CYPD; CypD), a mitochondrial matrix protein involved in mPTP formation (Fig. 4).
Several cell models document the accumulation of the full-length amyloid precursor protein (APP) in the outer mitochondrial membrane as another molecular event that is responsible for mitochondrial dysfunction, owing to its translocational arrest (Ref. Reference Anandatheerthavarada68). Although several aspects of the pathogenesis of AD remain unsolved, the turnover and degradation of APP and Aβ in the mitochondrial compartment appear to be two fundamental processes whose dysregulation might lead to dramatic consequences on the functioning of the organelle, and ultimately, of the cell as a whole.
In transgenic mice, elimination of the aberrant forms of these proteins can reverse the neurological deficits without affecting the number of neurons (Refs Reference Janus69, Reference Dodart70, Reference Kotilinek71). From a therapeutic perspective, these findings suggest that some neurological defects that are associated with AD might be caused by neuronal dysfunction rather than the loss of neurons. Drugs aimed at enhancing the removal of abnormal Aβ assemblies might help to inhibit their toxic action on mitochondria and, by extension, neurons. At present, several studies are attempting to identify in vitro and in vivo modulators that influence Aβ aggregation, as well as tau in the NFTs. A potential role for HTRA2/OMI protease in the clearance of Aβ oligomers from within the mitochondria has been recently indicated (Ref. Reference Park72). A similar function in degradation of mitochondrial Aβ has been suggested for the mitochondrial matrix metallopeptidase PreP (prolyl endopeptidase, PREP), an organellar functional analogue of the human insulin-degrading enzyme (IDE) (Ref. Reference Falkevall73) (Fig. 4).
The mitochondrial dysfunction found in AD has also been related to mechanisms that are independent of Aβ. The majority of hereditary AD cases (autosomal dominant) carry mutations in the presenilin genes PSEN1 and PSEN2, the subcomponent of γ-secretase that is responsible for APP cleavage to produce Aβ peptide. Specifically, mutations in PSEN1 have been shown to destabilise mitochondrial functions through two possible mechanisms by: (1) perturbing endoplasmic reticulum (ER) calcium handling, which in turn promotes excessive mitochondrial Ca2+ uptake and apoptosis (Ref. Reference Bezprozvanny and Mattson74); and (2) impairing the axonal transport of the organelle, which affects the normal synaptic activity of neurons (Ref. Reference Verstreken75).
Altogether, these studies have attributed the causes of AD to two inter-related pathogenic events: aggregation of misfolded proteins, and mitochondrial dysfunction; however, their inter-relationship needs further investigation. Despite the broad agreement that both processes are likely to have pivotal roles in AD progression, there is insuficient evidence supporting the notion that mitochondrial alterations are a primary cause of this disease. Instead, we favour the notion that mitochondrial dysfunction is a secondary event in the disease process, which nevertheless might be a key determinant of neurodegeneration in AD.
Although mitochondrial dysfunction and aberrant protein degradation are clearly related to AD, many questions remain unanswered. A major problem inherent to AD is the absence of early diagnostic tests, which is due in part to the lack of reliable predisease biomarkers. Several new approaches designed to target the formation of β-amyloid aggregates are under investigation in clinical trials, with the aim of slowing or halting the progression of AD (refer to the Alzheimer's association page in the Further Reading section below). However, synapse loss in the neocortex and hippocampus is a well-documented structural feature of the brain lesion in AD, and it is reported to be an early event in AD (Refs Reference Scheff76, Reference Stokin77). For that reason, it would be useful to look for markers of synaptic loss, such as synaptophysin (Refs Reference Sze78, Reference Masliah79). Future studies should also investigate the molecular aspects of mitochondria in neurons of animal models of AD. It would be interesting to study the effects of specific manipulations of mitochondrial functions in these cells, to determine whether the mitochondrial damage in AD can be reversed by stimulating autophagy and/or mitochondrial biogenesis.
Huntington disease and mitochondrial dysfunction
Huntington's disease (HD) is a fatal, dominantly inherited neurodegenerative disorder characterised by chorea, involuntary movements, and cognitive impairments. Symptoms result from the selective loss of long-projection neurons known as medium spiny neurons. These neuronal cells release γ-aminobutyric acid (GABA) in the striatal brain regions that control movement, memory and emotions. HD is slowly progressive, and patients survive for about 15–20 years from disease onset (for reviews, see Refs Reference Reddy, Mao and Manczak80, Reference Mattson, Gleichmann and Cheng11). Tremendous progress has been made since the discovery of the HTT gene in 1993. HD is caused by a genetic mutation that results in an expanded polyglutamine-encoding repeat within exon 1 of the HTT gene (Ref. 81).
The HTT gene product, Huntingtin (HTT, Htt), is an extremely large protein of 350 kDa, which is ubiquitously expressed in the brain and peripheral tissues. It has been reported to act as a scaffold protein that regulates signalling pathways, and vesicle and organelle trafficking. Although Htt is mostly cytoplasmic, it is also found at lower concentrations in multiple subcellular compartments, such as the plasma membrane, nucleus, endoplasmic reticulum, Golgi and mitochondria.
A polyglutamine stretch of less than 35 in the N-terminus of Htt is normal and does not cause disease. However, an abnormal stretch of 36-39 glutamine residues results in incomplete penetrance of the disease, and 40 or more results in HD with full penetrance (see Ref. Reference Bossy-Wetzel, Petrilli and Knott82 for a review).
Strong evidence suggests that mitochondrial impairment has a key role in HD pathogenesis (Ref. Reference Lin and Beal83). Mutant Htt (mtHTT, mtHtt) might cause its neurotoxicity by evoking defects in mitochondria, which in turn lead to a bioenergetic failure, HD-linked neuronal dysfunction and cell death. Indeed, postmortem brain samples of HD patients exhibit reduced activity of mitochondrial respiratory complexes II, III and IV (Ref. Reference Gu84); and humans exposed to 3-nitropropionic acid (3-NP), a selective inhibitor of succinate dehydrogenase and complex II, exhibit motor dysfunction similar to that observed in HD patients (Ref. Reference Rubinsztein85).
Moreover, several harmful effects of mtHtt on mitochondria have been reported, including: reduced ATP levels in synaptic terminals (Ref. Reference Orr86); mitochondria depolarisation at lower Ca2+ loads (Ref. Reference Panov87); increased sensitivity to Ca2+ overload and N-methyl-D-aspartic acid (NMDA) receptor-mediated neuronal apoptosis (Ref. Reference Fernandes88); and a reduced threshold for mPTP opening and cytochrome c release (Ref. Reference Choo89). Although the bioenergetic deficits in HD are well known, mtHtt can also adversely affect mitochondria by modifying gene transcription. For instance, mtHtt binds to the tumour suppressor p53 and increases its levels, which, in turn, results in transcriptional activation of its proapoptotic mitochondrial targets Bax and Puma, causing mitochondrial damage. Since the loss of p53 prevents mtHtt-mediated neurodegeneration in Drosophila (Ref. Reference Bae90), we can easily assume that altered p53 transcriptional activity results in mitochondrial dysfunction and neuronal loss.
PPARGC1A [PGC-1α, peroxisome proliferator-activated receptor (PPAR)-γ coactivator 1α] is another gene whose expression seems to be regulated by mtHtt. By interacting with the promoter and interfering with CREB-dependent PPARGC1A gene expression, mtHtt represses the transcription of this gene (Ref. Reference Cui91). PGC-1α is a nuclear co-activator that has a major role in mitochondrial biogenesis; therefore, inhibition of PGC-1α expression limits the ability of the vulnerable neurons to adequately respond to energy demands in HD.
Although the transcriptional deregulation of mtHtt is of great relevance, it cannot fully explain all the mitochondrial defects observed in HD. Recent studies indicate that impaired mitochondrial trafficking along axons and dendrites might also have an important role in the disease pathology (see Ref. Reference Li, Orr and Li30 for a review). Mitochondria are dynamically transported along lengthy neuronal processes to provide energy to nerve terminals and maintain the normal neuronal function. ATP-dependent motor proteins regulate such mitochondrial movement: kinesins mediate anterograde transport (away from the cell body), and dynein-dynactin regulates retrograde transport (toward the cell body) (Ref. Reference Hollenbeck and Saxton92). Wild-type Htt seems to regulate the trafficking of endocytic vesicles by binding to Htt-associated protein 1 (HAP1) (see Ref. Reference Li, Orr and Li30 for a review). This protein complex might act as a docking platform that interacts with the molecular motor dynein–dynactin and kinesin, and it is known to regulate microtubule-mediated BDNF (brain-derived neurotrophic factor) vesicle and mitochondrial transport (Refs Reference Gauthier93, Reference McGuire94). Furthermore, phosphorylation of Htt seems to act as a molecular switch for bidirectional transport in neurons (Ref. Reference Colin95) (Fig. 5).

Figure 5. Role of Htt protein in mitochondrial trafficking and dynamics. Mitochondrial movement in neurons is highly diverse and complex. Normal Htt protein regulates anterograde (away from the cell body) and retrograde (towards the cell body) transport of mitochondria by interacting with several trafficking mediators. Htt stimulates trafficking by binding to HAP1, which in turn, leads to interaction with the motor proteins dynein–dynactin and kinesin. Phosphorylation of Htt acts as a molecular switch for anterograde versus retrograde mitochondrial transport. When Htt is phosphorylated, kinesin-1 is recruited and promotes anterograde transport; conversely, when Htt is unphosphorylated, kinesin-1 detaches from the motor complex and induces a switch to retrograde transport (Ref. Reference Colin95). In addition to migration and movement, mitochondria undergo cycles of fusion and fission. The key mitochondrial fission regulator is dynamin-related protein 1 (DRP1). Similarly to dynamin, DRP1 seems to act as a mechano-enzyme to constrict and divide mitochondria. Given that Htt interacts with dynamin, one can speculate that Htt might regulate fission by interacting with DRP1. Abbreviations: HAP1, Htt-associated protein 1; Htt, Huntingtin protein; P, phosphate.
Whereas wild-type Htt promotes axonal BDNF vesicle trafficking, mtHtt disrupts the formation of trafficking complexes and impairs vesicle transport (Ref. Reference Gauthier93). Abnormal interaction of mtHtt with motor proteins seems to be the main cause of the trafficking defects; however, mtHtt aggregates might also contribute by acting as a physical roadblock (Refs Reference Chan2, Reference Orr86), or by sequestering wild-type Htt and components of the trafficking machinery (Ref. Reference Trushina96).
Neuronal function is particularly dependent on the intracellular trafficking of organelles and molecules. Defective mitochondrial transport ultimately impairs neuronal transmission and results in synaptic damage and selective neuronal loss. In addition to blocking mitochondrial movement, it has been reported that mtHtt induces an imbalance in mitochondrial fission and fusion (Fig. 2). According to Monteiro and colleagues (Ref. Reference Wang97), the increase in cytotoxicity induced by mtHtt is probably mediated by an alteration in mitochondrial dynamics, which results in increased mitochondrial fragmentation. Given that mtHtt interacts with dynamin (Ref. Reference Kaltenbach98) and its distribution pattern in the mitochondria has striking similarities to DRP1 (Ref. Reference Panov87), it is possible that DRP1 could also form a complex with Htt. Thus, one can hypothesise that normal Htt might regulate fission events by interacting with DRP1, whereas mtHtt might alter the assembly and function of these complexes, which ultimately leads to imbalances in mitochondrial dynamics. Naturally, further investigation is needed to test this idea.
As described earlier in this review, mitochondrial fragmentation is a common stress response that allows the segregation and elimination of dysfunctional mitochondria by autophagy. Interestingly, autophagy is a major degradation route for mtHtt, and the pharmacological induction of autophagy seems to be of therapeutic value to neurodegenerative disease caused by aggregate-prone proteins such as HD (Refs Reference Rubinsztein85, Reference Sarkar and Rubinsztein99). Based on this, one might speculate that the induction of autophagy is responsible not only for the clearance of the mutant protein, but also for removal of the dysfunctional mitochondria present in the HD-associated neurons.
Clinical implications
Ageing is the most important risk factor for common neurodegenerative disorders. In the CNS, the physiological process of ageing has been associated with an elevated mutation load in mtDNA, defects in mitochondrial respiration and increased oxidative damage. Indeed, this mitochondrial loss of function seems to be a consequence of the cellular deterioration that occurs with age, which compromises mitochondrial biogenesis and turnover. Interestingly, it has been suggested that induction of mitochondrial biogenesis through pharmacological (bezafibrate) or metabolic modulation of the PPAR–PGC-1α pathway could represent an effective therapeutical approach for mitochondrial disorders (Ref. Reference Wenz100).
Therapeutic options currently available for patients affected by neurodegenerative diseases are extremely limited. Although characterised by specific attributes and pathological hallmarks, most neurodegenerative diseases have features of mitochondrial dysfunction, which converge in metabolic alterations that ultimately lead to neuronal cell death and affect the brain physiology. Consequently, possible therapies could be aimed at restoring the normal function of these organelles. With regards to AD, recent data suggest that cognitive decline is correlated with selective abnormalities in TCA cycle enzymes of mitochondria (Ref. Reference Bubber101). So far, treatments designed to overcome these defects, by administration of glucose and insulin, have improved memory in AD patients (Ref. Reference Craft102); however, the benefits are transient. Interesting new data are emerging for long-term benefits to AD patients from studies on Dimebon, an antihistamine drug. In contrast to the conventional drugs for AD therapy, which operate through cholinesterase inhibition or NMDA-receptor antagonism, Dimebon is a compound that seems to work through a novel mechanism by improving mitochondrial function in the brain. Moreover, Dimebon has been shown to be a promising therapeutic candidate in inhibiting brain cell death in both AD and HD preclinical models (Ref. Reference Moreira103).
Enhancing cellular defence mechanisms against different kinds of stress could be an attractive therapeutic strategy for neurodegenerative diseases. In particular, induction of expression of molecular chaperones might reduce the formation of misfolded proteins and toxic aggregates. Geldanamycin, a natural substance that modulates Hsp90 function, was previously shown to induce a heat-shock response through the activation of heat shock factor 1 (HSF1); it also reduces polyQ aggregation in mammalian cells (Ref. Reference Sittler104) and suppresses α-synuclein neurotoxicity in flies (Ref. Reference Auluck, Meulener and Bonini105). Geldanamycin derivatives are now being considered for the development of a potential drug treatment for neurodegenerative diseases that involve protein aggregation.
Recently, Whitworth and colleagues showed that the drug Rapamycin, which is used in some transplant patients to prevent immune rejection, protects cells against the damaging effects of two of the mutant genes that cause inherited forms of PD (Ref. Reference Tain53). This is an interesting observation if one considers that the current pharmacological interventions for PD are designed to replace or mimic the effects of dopamine, rather than actually change the course of the condition.
Research in progress and outstanding research questions
Recent work has highlighted the importance of protein translational switches during times of cellular and environmental stress, and the role of 4E-BP in this process. Upon dietary restriction, a translational switch to nuclear-encoded mitochondrial genes occurs in a 4E-BP-dependent manner (Ref. Reference Zid106). This suggests an important role for enhanced mitochondrial function in conditions of cellular stress. It remains to be determined whether a 4E-BP-dependent rescue of mitochondrial dysfunction in models of PD is mechanistically linked to the translational activation of nuclear encoded mitochondrial genes. It has recently been shown that inhibition of cap-dependent translation via 4E-BP rescues phenotypes related to parkinsonism (Ref. Reference Tain53). In addition, the oxidation-sensitive PD protein DJ-1 binds to mRNAs of several mitochondrial genes, including 4E-BP, which are released under conditions of oxidative stress (Ref. Reference van der Brug107). These observations suggest that cellular responses to oxidative stress might involve a general switch in protein translation that leads to the selective upregulation of mitochondrial proteins. The molecular dissection of pathways controlling expression of mitochondrial genes is proceeding in earnest and is likely to lead to a better understanding of novel mitochondria-protection mechanisms. Such understanding will hopefully lead to a better knowledge of the disease processes involving mitochondrial dysfunction, as well as more efficient treatment avenues.
Finally, one other potential route for treatment of neurodegenerative disease is the drug Metformin, which is already in common use for the treatment of type 2 diabetes, particularly in overweight and obese people. Metformin is also frequently used for research with the AMP analogue AICAR as an AMPK agonist. AMPK activation is one of the mechanisms by which mTOR can be suppressed, and studies that implicate AMPK in the regulation of mTOR showed that pharmacological activation of AMPK by AICAR causes a dramatic reduction of 4E-BP phosphorylation (Ref. Reference Corradetti and Guan108). Given that both diabetes and PD are reported to have a deregulation of the mTOR pathway, and that Metformin is one of the most prescribed drugs in the world, it would be interesting to see whether patients receiving the drug have a reduced risk of developing PD.
Acknowledgements
Inês Pimenta de Castro is recipient of a PhD grant (SFRH/BD/41682/2007) from FCT Portugal. We thank our anonymous peer reviewers for their constructive comments on this article.







 Open access
Open access