


Tobacco use is one of the leading preventable risk factors of premature death, accounting for 18% of deaths in high-income countries (World Health Organization, 2009). Tobacco use also increases morbidity of multiple somatic diseases, such as respiratory disease, heart disease, diabetes, and cancer (World Health Organization, 2009). In addition to somatic diseases, tobacco use is associated with other addictions and mental disorders (Royal College of Physicians, 2013), though the causal and mechanistic nature of the association is not well established. The heritability of susceptibility to smoking behaviors is estimated to be 40–60%, the heritability estimates generally being higher for smoking quantity and nicotine dependence than for susceptibility to smoking initiation (Rose et al., Reference Rose, Broms, Korhonen, Dick, Kaprio and Kim2009).
Depression, ranging from mild depressed mood to major depressive disorder (MDD; Nolen-Hoeksema, Reference Nolen-Hoeksema2007), is estimated to be the second leading cause of disability worldwide by 2020 (Dome et al., Reference Dome, Lazary, Kalapos and Rihmer2010). Approximately 8–13% of the general population experience clinical depression during their lifetime (World Health Organization, 2004). Interindividual genetic differences are estimated to account for about 40% of variability in the liability of developing MDD (Goldberg, Reference Goldberg2006).
Depression is a major comorbid condition associated with smoking (Jane-Llopis & Matytsina, Reference Jane-Llopis and Matytsina2006). Smokers have a greater likelihood of lifetime depression or current depressive symptoms than non-smokers, and those with depressive disorders tend more often to be smokers than are healthy controls (Dani & Harris, Reference Dani and Harris2005; Jane-Llopis & Matytsina, Reference Jane-Llopis and Matytsina2006; Morrell & Cohen, Reference Morrell and Cohen2006). The prevalence of current smoking among US adults with history of major depression was double compared with prevalence among those without history of depression (Lasser et al., Reference Lasser, Boyd, Woolhandler, Himmelstein, McCormick and Bor2000). The association is well established, but there are competing hypotheses for its explanation. First, pre-existing depression may predict onset of smoking or progression to nicotine dependence (Breslau et al., Reference Breslau, Novak and Kessler2004; Fergusson et al., Reference Fergusson, Goodwin and Horwood2003; Murphy et al., Reference Murphy, Horton, Monson, Laird, Sobol and Leighton2003). In other words, depressive symptoms may lead to self-medication and foster initiation or acceleration of smoking (Windle & Windle, Reference Windle and Windle2001). Second, long-term persistent smoking may increase the risk of depression (Morrell & Cohen, Reference Morrell and Cohen2006), as shown by longitudinal studies of adolescents (Goodman & Capitman, Reference Goodman and Capitman2000; Wu & Anthony, Reference Wu and Anthony1999) and adults (Klungsoyr et al., Reference Klungsoyr, Nygard, Sorensen and Sandanger2006; Korhonen et al., Reference Korhonen, Broms, Varjonen, Romanov, Koskenvuo, Kinnunen and Kaprio2007). Bidirectional predictive associations have been demonstrated in adolescent samples (Breslau et al., Reference Breslau, Peterson, Schultz, Chilcoat and Andreski1998; Tjora et al., Reference Tjora, Hetland, Aarø, Wold, Wiium and Øverland2014; Windle & Windle, Reference Windle and Windle2001). Successful smoking cessation results in improved mood, according to a recent meta-analysis (Taylor et al., Reference Taylor, McNeill, Girling, Farley, Lindson-Hawley and Aveyard2014).
As both depression and smoking behaviors are influenced by both genetic and environmental factors, shared factors may enhance vulnerability to smoking co-occurring with depression (Williams & Ziedonis, Reference Williams and Ziedonis2004). Correlations between genetic components of smoking and depression (r g) have been investigated with conflicting results. Kendler et al. (Reference Kendler, Neale, MacLean, Heath, Eaves and Kessler1993) reported that comorbidity of smoking and MDD largely arose from genetic factors (r g = 0.56) in adult twin sisters. However, other twin studies have reported lower and hardly significant genetic correlations. McCaffery and co-authors (Reference McCaffery, Niaura, Swan and Carmelli2003) found among male twins that unique environmental factors accounted for most of the covariation between liability to smoking and depression, with a low genetic correlation (r g = 0.17). In a longitudinal twin study, Korhonen et al. (Reference Korhonen, Broms, Varjonen, Romanov, Koskenvuo, Kinnunen and Kaprio2007) found that, after controlling for familial factors, smoking remained a gender-sensitive predictor of depressive symptoms. The stronger association in men was modestly accounted for by underlying shared genes (r g = 0.25). Finally, Fu et al. (Reference Fu, Heath, Bucholz, Lyons, Tsuang, True and Eisen2007) reported significant shared genetic vulnerability to nicotine dependence and MDD in a cross-sectional study of twin brothers. However, after controlling for genetic influences on conduct and antisocial disorders, that genetic correlation approached zero (r G = 0.06).
To summarize the earlier literature, the genetic correlations for smoking and depression have varied extensively, and in many studies those correlations have been relatively low. The nature and direction of the association between smoking and depression also remains unclear; it is very challenging to state whether smoking causes depression or depression causes smoking, or whether such association can be explained by shared genes, because the interaction between these two phenotypes may be more complex. Thus, it would be necessary to investigate GxE underlying these phenotypes.
The aim of this study was to investigate genetic and environmental interactions between depressive symptoms measured by the BDI and number of CPD using quantitative genetic modeling of twin data.
Materials and Methods
Data
The data were drawn from the Older Finnish Twin cohort's third study wave collected in 1990. The cohort was established to examine the genetic, environmental, and psychological determinants of chronic diseases and health behaviors. This population-based cohort was compiled from the Central Population Registry, consisting of all same-sex twin pairs born in Finland before 1958 with both co-twins alive in 1967 (altogether 13,888 pairs of known zygosity). The first questionnaire survey was conducted in 1975 (response rate 84%), and the second in 1981. The data collection procedures have been described in more detail elsewhere (Kaprio & Koskenvuo, Reference Kaprio and Koskenvuo2002). In the third data collection wave, the questionnaires were sent in 1990 to same-sex twin pairs born between 1930 and 1957 who participated in at least one earlier survey (n = 16,179). Only the third data collection wave was considered in the analyses as the depression measure was not included in the earlier data waves. The response rate was 77%, with 12,502 respondents. The zygosity of the twins was determined by means of a validated questionnaire (Sarna et al., Reference Sarna, Kaprio, Sistonen and Koskenvuo1978). The Finnish Twin Cohort study was approved by the Ethical Committee of the University of Helsinki.
Analyses were conducted using only individuals who provided self-reported depression measure in 1990. Data with depression measure on total of 12,063 individuals (5,512 men, 6,551 women, aged 33–60, mean age 43.8 years, SD 7.7 years) were available for analyses. This included 1,465 full monozygotic (genetically identical) and 2,779 dizygotic (non-identical) twin pairs. The data included 5,578 never-smokers, 404 occasional smokers, 2,693 former smokers, and 3,057 current daily smokers. Smoking status was either unknown or inconsistently reported by 331 individuals.
Measures
Beck depression inventory
Depressiveness was measured using the BDI (Beck & Beamesderfer, Reference Beck and Beamesderfer1974; Beck et al., Reference Beck, Ward, Mendelson, Mock and Erbaugh1961; Varjonen et al., Reference Varjonen, Romanov, Kaprio, Heikkilä and Koskenvuo1997). The BDI assesses depressive symptoms rather than making a clinical depression diagnosis. The questionnaire has 21 items describing symptoms and attitudes, which the participants rate as they perceive them ‘right now’. Each item is rated in intensity from 0 to 3 points (theoretical range as a sum score 0–63). The sum score can be categorized into three classes as follows: (1) 0–9 points corresponding to none or minimal depression, (2) 10–16 points equivalent to mild depression, and (3) more than 16 points, considered to be at least moderate depression (Korhonen et al., Reference Korhonen, Broms, Varjonen, Romanov, Koskenvuo, Kinnunen and Kaprio2007; Varjonen et al., Reference Varjonen, Romanov, Kaprio, Heikkilä and Koskenvuo1997). The frequencies of these depression categories in our sample are presented in Table 1. In the present analysis we used BDI as a continuous variable. For descriptive purposes of comparing ‘healthy’ with ‘depressed’ participants, the variable was further dichotomized to classes 0–9 points (i.e., healthy) and more than 9 points (depressed; including 567 persons with at least moderate depression).
TABLE 1 Frequencies (%) of Depression Classifications (BDI Category), Smoking Status, and Number of Cigarettes Smoked Daily (CPD) by Sex and Zygosity

Smoking quantity
A detailed smoking history was used to classify smoking status in 1990 as ‘never smokers’ (i.e., having smoked less than 100 cigarettes lifetime), ‘former smokers’ (i.e., having smoked at least 100 cigarettes lifetime but did not smoke at the time of the survey), and ‘current smokers’ (i.e., occasional or daily smokers). The amount smoked by current daily smokers and occasional smokers smoking nearly daily was assessed as CPD. The CPD was inquired as an ordinal variable with the following classes: 1 = 0 CPD, 2 = 1–4 CPD, 3 = 5–9 CPD, 4 = 10–14 CPD, 5 = 15–19 CPD, 6 = 20–24 CPD, 7 = 25–39 CPD, and 8 = >40 CPD. All non-smokers were assigned to the CPD category of 1 (i.e., smoking zero CPD). The frequencies of smoking status and CPD categories in this sample are presented in Table 1.
Data Analysis
Classical twin modeling was used to study the genetically informative data. Genetic modeling was carried out with the Mx statistical package, version 1.7 (Neale & Maes, Reference Neale and Maes2006). We treated the variables as continuous phenotypes in the analyses. Age and sex were used as covariates in all models. First, we studied the variance components (additive genetic A, common environment E, unshared environment E) influencing each trait and the genetic and environmental components shared by the traits by a bivariate Cholesky decomposition.
To study GxE, we used twin models incorporating moderation effects (Purcell, Reference Purcell2002). Non-linear moderation was allowed with the addition of a quadratic term of moderation effect (βxM2). The GxE models were built in two ways as follows: (1) using CPD as the trait and BDI as the moderator and (2) vice versa. In these models, the standard paths a, c, and e each include a ‘b’-term, indicating the significance of potential moderator variable ‘M’ on each of these paths. The moderation model is presented in Figure 1. The value M is the value of the measured moderation effect (CPD or BDI) for each individual, thus changing from individual to individual. As the β coefficient is estimated separately for each variance component, the model allows testing which of the effects are changing as a linear function of the measured M variable.

FIGURE 1 Partial path diagram for non-linear ACE moderation model, shown for one twin. Circles represent the latent unmeasured variables: A = additive genetic influences, C = common environmental influences, and E = unshared environmental influences. Triangle represents the mean for the trait (T). The standard paths a, c, and e indicate the magnitude of the effect of each latent variable on the trait. Each includes a β term, which indicates the significance of a measured moderator variable M on each of the genetic and environmental influences.
As GxE models are complex, it may be challenging to find a stable model, that is, to retrieve the same estimates when running the model several times. In order to find a more stable model, we first ran a univariate linear GxE model and tested it using a jiggle-option in Mx (jiggles start values) to ensure we get to the same, stable model (instead of a saddle point) with different start values. Then, we used the values obtained from this linear model as starting values in the model, including a quadratic term, and set the starting value to quadratic terms to zero. This way, we were able to find stable (i.e., getting to the same model solution even with different starting values using jiggle or TH options) non-linear models. In GxE models, we first tested dropping all the moderator effects. However, this worsened the model fit significantly for both sets of models implying that GxE are influencing the variation of both traits.
We conducted all the models using the raw data option in Mx (Neale et al., Reference Neale, Boker, Xie and Maes2006). The significance of each parameter in the model was tested by dropping the parameter and evaluating the change in the χ2 statistics and Akaike Information Criterion (AIC) between the initial model and the nested submodel.
Results
The BDI sum scores in our data ranged from 0 to 49 (theoretical range 0–63) with a mean value of 5.2. Thus, the majority of our participants were classified as non-depressed. The number of CPD ranged on our ordinal scale from 1 (=0 CPD) to 8 (≥40 CPD), mean being 2.0, which corresponds to 1–4 CPD. The two measures correlated only weakly (Pearson r = 0.09). When the participants were classified as healthy (0–9 points on BDI) or depressed (more than 9 points on BDI), the depressed participants smoked more (mean CPD score 2.3) than the healthy participants (mean CPD score 1.9, tdf =2594 = 7.4, p < .0001). Moreover, 32.6 % of depressed persons were current smokers compared to 24.8% of non-depressed persons. Ever-smokers had a mean BDI score of 5.3 compared to the mean score of 4.9 among never smokers.
Bivariate Cholesky decomposition revealed that the variance and covariance of traits was explained by additive genetic (A) and unshared environmental (E) components (shown in Figure 2). The heritability (proportion of variance explained by genetic effects) of CPD was 50% and that of BDI, 36%. The phenotypic correlation (r = 0.09) between BDI and CPD was explained by shared genetic (r g = 0.18, which explains 63% of the covariation) and environmental (r e = 0.08, which explains 37% of the covariation) factors.

FIGURE 2 The Cholesky decomposition showing the variances and covariances of the traits for one twin individual.
In the model where CPD was used as a trait and BDI was the moderator, the full model, with linear and quadratic moderation effects on all variance components and mean included, provided the best fit (Table 2). Especially, the influence of genetic effects on CPD was increased, with higher BDI score implying that the influence of genetic effects on smoking quantity is potentiated among individuals with an elevated depression score (Figure 3).
TABLE 2 Fit Statistics to the Full Moderation Model and Submodels Testing the Significance of Dropping One or More Components From the Model of CPD Variance Being Moderated by BDI Score
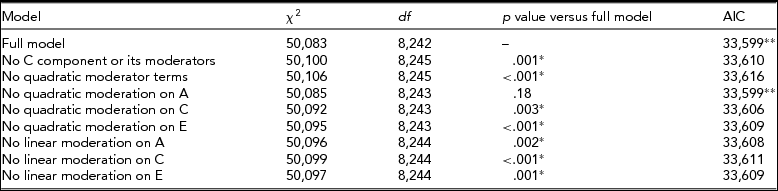
The model chosen to provide the best fit is in bold type. *Significant difference compared to the full model, that is, the term cannot be dropped from the model. **Lowest Akaike's Information Criterion (AIC).

FIGURE 3 Proportions of changing variance in additive genetic effects (A), common environmental effects (C), and unique environmental effects (E) on smoking quantity across increasing Beck Depression Inventory (BDI) scores.
When BDI was used as the trait and CPD as moderator, the best fit came from a model without C effects or its linear/quadratic moderations (Table 3). Again, the influence of genetic effects on depression increased with increasing moderator value, that his, greater amount smoked (Figure 4). Thus, the more individuals smoke, the larger is the role that the genes appear to play in depressive symptom variance.
TABLE 3 Fit Statistics to the Full Moderation Model and Submodels Testing the Significance of Dropping One or More Components From the Model of BDI Score Variance Being Moderated by CPD
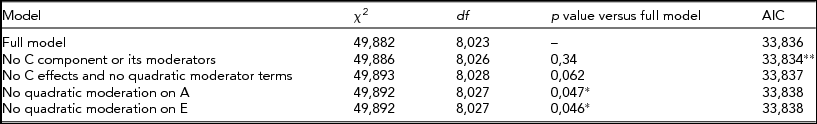
The model chosen to provide the best fit is in bold type. *Significant difference compared to the full model, that is, the term cannot be dropped from the model. **Lowest Akaike's Information Criterion (AIC).

FIGURE 4 Changing variance in additive genetic effects (A) and unique environmental effects (E) on Beck Depression Inventory (BDI) across increasing smoking quantity 1 = 0 CPD, 2 = 1–4 CPD, 3 = 5–9 CPD, 4 = 10–14 CPD, 5 = 15–19 CPD, 6 = 20–24 CPD, 7 = 25–39 CPD, and 8 = >40 CPD.
Discussion
The aim of this study was to investigate gene by environmental interactions between depressive symptoms and smoking quantity using quantitative genetic modeling of twin data. Our results showed that the more individuals smoke, the larger the role genes play in depressive symptom variance. And vice versa, the heritability of smoking is higher among the more depressed individuals. Our findings indicate that the comorbidity of smoking and depression may be even more complicated than the earlier studies have implied. Not only do partly common genetic and environmental factors influence the variation in both traits, but one trait can modify the genetic liability to another trait. It is plausible that compared to mentally healthy persons, depressed individuals may find it harder to refrain from smoking when they are genetically prone to smoking/nicotine dependence and/or surrounded by smoking peers. As both smoking and depression may vary over time, we decided to concentrate in the analyses on their state at the moment of data collection. Modeling their past relationships using GxE based on a cross-sectional data set is challenging, if not impossible to model with current techniques and datasets available. A longitudinal dataset with repeated measures of depressiveness and smoking behavior would be ideal for such analyses, as transitions between different smoking states could be accurately assessed, and the inherent variability in mood considered.
Our bivariate analysis revealed that the heritability of smoking quantity was 50% and that of depressive symptoms was 36% while the genetic correlation was 0.18. Earlier studies have shown quite varying genetic correlations between smoking and depression. The highest correlation was reported among American women (r g = 0.56; Kendler et al., Reference Kendler, Neale, MacLean, Heath, Eaves and Kessler1993), whereas a clearly lower correlation was observed among American men (r g = 0.17; McCaffery et al., Reference McCaffery, Niaura, Swan and Carmelli2003), and a somewhat higher correlation among Finnish men (r g = 0.25; Korhonen et al., Reference Korhonen, Broms, Varjonen, Romanov, Koskenvuo, Kinnunen and Kaprio2007). Further, after controlling for genetic influences on conduct and antisocial disorders, Fu et al. (Reference Fu, Heath, Bucholz, Lyons, Tsuang, True and Eisen2007) demonstrated in an American male sample that genetic correlation was close to zero (r g = 0.06). When comparing these genetic correlations, it seems that our study shows one of the lowest correlations. Because in general these genetic correlations vary greatly, the evidence on shared underlying genes in the association between depression and smoking seems to be inconsistent. There may be several reasons for a great variation in the genetic correlations across different studies. Findings may be sensitive to definitions of phenotypes, study designs, sample size, and sampling used in each analysis, as well as to characteristics of each sample (e.g., age, sex, and race distributions; Edwards & Kendler, Reference Edwards and Kendler2012; Edwards et al., Reference Edwards, Maes, Pendersen and Kendler2011) and the environments where the participants come from (e.g., presence or absence of smoking restrictions). It has also been observed that Individuals with a familial vulnerability for major depression, even without a personal history of major depression, are more likely to smoke (Lyons et al., Reference Lyons, Hitsman, Xian, Panizzon, Jerskey, Santangelo and Tsuang2008). Explorations of common genetic liability for comorbid conditions, such as smoking and depression, seem to be still at an early stage (Rose et al., Reference Rose, Broms, Korhonen, Dick, Kaprio and Kim2009).
Our analysis could not reveal what might be the mechanism underlying the GxE between the traits under investigation here. One could hypothesize that both smoking and depression could lead to changes in gene expression. Another mechanism might be that there are genes that modify susceptibility to smoking only in depressed individuals. A couple of studies have suggested that dopamine receptor genes are involved in the interaction of smoking and depression. For example, stimulation smoking and negative-affect smoking have been shown to be heightened in depressed smokers who are homozygous for the short alleles of dopamine D4 receptor gene, but not in individuals with other genotypes of the gene (Lerman et al., Reference Lerman, Caporaso, Main, Audrain, Boyd, Bowman and Shields1998). In addition, it has been shown that among adolescents, the likelihood of rapid increase in smoking is associated to the dopamine D2 receptor genes A1 allele and this effect is significantly potentiated by depression symptoms (Audrain-McGovern et al., Reference Audrain-McGovern, Lerman, Wileyto, Rodriguez and Shields2004).
Methodological Considerations
This study was based on a population-based cohort, where the twin pairs have been recruited to the study from the Central Population Register of Finland (Kaprio & Koskenvuo, Reference Kaprio and Koskenvuo2002), which can be considered as a methodological strength. It has to be noted that the data was collected in 1990. The environmental pressure to quit smoking has probably increased since then, and the proportion of smokers in the population has decreased, especially among men. The way in which depression was ascertained by means of a questionnaire may be a limitation. Optimally, we would have conducted a psychiatric interview to have diagnostic phenotype for depression, but this was not logistically feasible for over 12,000 participants. On the other hand, the BDI is a well-recognized measure of depression with good properties for screening cases in the population (Lasa et al., Reference Lasa, Ayuso-Mateos, Vazquez-Barquero, Diez-Manrique and Dowrick2000). Here, we used the continuous BDI score for genetic modeling. For descriptive purposes, we compared the depressed versus healthy individuals using a cut-off value of BDI >9 for the cases, including participants with varying degrees of severity. We have successfully applied this cut-off in our earlier study investigating persistent smoking as a predictor of depressive symptoms (Korhonen et al., Reference Korhonen, Broms, Varjonen, Romanov, Koskenvuo, Kinnunen and Kaprio2007). As the phenotype of smoking heaviness, we applied smoking quantity at the time of the study. This was self-reported CPD on an ordinal scale but used as a continuous measure, given that category sizes were fairly constant. A more sophisticated measure would have been blood or urine cotinine level, which reflects nicotine intake more accurately than self-reported CPD. Unfortunately; in this data set we did not have access to any biomarkers of smoking. Although the data were collected 25 years ago, we consider that the underlying biological relationships between depression and tobacco use are likely to hold true nowadays and the results are informative.
The classical twin design relies on the assumption of random mating in terms of the trait(s) studied. However, in case of smoking, at least, assortative mating has been shown to occur; in a study of female twins and their male spouses, women who regularly smoked were more likely to marry men who also smoked (Agrawal et al., Reference Agrawal, Heath, Grant, Pergadia, Statham, Bucholz and Madden2006). The non-random mating may result in dizygotic twins sharing a greater percentage of the genes influencing the trait than expected. Non-normal distribution of data is another limitation of our study. As the data were derived from a population-based cohort, both CPD and BDI scores encounter a floor effect, that is, most individuals are not depressed and do not smoke.
Recently, the GxE model used in this study has been criticized for providing false positive moderation effects (van der Sluis et al., Reference van der Sluis, Posthuma and Dolan2012). However, the elevation of false positive rate has only been shown in cases where moderator (M) and trait (T) are strongly correlated. In our data, BDI and CPD were only very modestly correlated (Pearson r = 0.09). In addition, the criticism highlights that the increase in false positive moderation effects is most prominent in cases where the correlation between the trait and the moderator runs fully or predominantly via unshared environmental effects (E). In our data, the correlation between smoking and depression was explained mostly via genetic correlation. Thus, we feel that using the simpler univariate moderation model is justified in our dataset. We also did not consider alternate models, including those with non-linear main effects such as Zheng et al. (Reference Zheng, Van Hulle and Rathouz2015) and van Hulle and Rathouz (Reference Van Hulle and Rathouz2015), and therefore our findings need to be interpreted with caution.
In conclusion, these results suggest that both shared genetic and environmental factors, as well as GxE, underlie the comorbidity of smoking and depression.
Acknowledgments
Supported by the Academy of Finland (141054, 265240, 263278, 264146) and by grants from the Department of Public Health, University of Helsinki (TK & KV).
Conflicts of Interest
Tellervo Korhonen and Jaakko Kaprio have consulted for Pfizer on nicotine dependence from 2012 to 2015.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Ethical Committee of the University of Helsinki and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. For this type of study written consent is not required and return of the questionnaire is considered to indicate consent given that information about the study is given in the cover letter, and participants are also informed that they may withdraw from the study at any point.







