



Introduction
Early optimized antidepressant therapy in patients with major depressive disorder (MDD) is important for achieving full symptomatic and functional recovery.Reference Okuda, Suzuki and Kishi 1 -Reference Oluboka, Katzman and Habert 4 Clinical guidelines recommend that patients should be assessed for improvement within 2 weeks of treatment initiation to permit early dose adjustment or change of antidepressant medication in those with inadequate response.Reference Cleare, Pariante and Young 5 -Reference Bauer, Severus, Möller and Young 7 Early response to antidepressant therapy has been shown to be predictive of remission and recovery in patients with MDD.Reference Henkel, Seemüllerm and Obermeier 8 -Reference Inoue, Fujimoto and Marumoto 13 Patients experiencing early improvement on antidepressant therapy (defined as ≥20% reduction in Hamilton Rating Scale for Depression [HAM-D] score from baseline after 2 weeks of treatment) were found to be at least three times more likely to achieve sustained response (i.e., ≥50% reduction in HAM-D score from baseline at 6 weeks) than patients showing later improvement.Reference Stassen, Angst, Hell, Scharfetter and Szegedi 14
Conversely, lingering residual symptoms appear to contribute toward a more severe, relapsing, and chronic future course of depression.Reference Judd, Akiskal and Maser 15 , Reference Judd, Schettler, Rush, Coryell, Fiedorowicz and Solomon 16 In the National Institute of Mental Health Collaborative Depression Study, patients with residual subthreshold depressive symptoms experienced shorter time to first relapse, longer and more severe episodes of depression, and greater disease burden over 10–20 years of follow-up than those who achieved asymptomatic recovery.Reference Judd, Schettler, Rush, Coryell, Fiedorowicz and Solomon 16 Similarly, patients who continue to experience functional impairment after remission of core depressive symptoms have been shown to be almost four times more likely to relapse over 12 months of follow-up than those without residual functional impairment.Reference Ishak, Greenberg and Cohen 17
Vortioxetine is a multimodal antidepressant shown to be efficacious and well tolerated for the treatment of MDD across the approved dosage range of 5–20 mg/day.Reference Gonda, Sharma and Tarazi 18 , Reference de Diego-Adeliño, Crespo and Mora 19 Vortioxetine is one of the few antidepressants shown to have a clear dose–response relationship across the spectrum of symptoms experienced by patients with MDD, including depressive, cognitive, physical, and anxiety symptoms, and functional impairment, both in randomized, controlled clinical trialsReference Baldwin, Florea, Jacobsen, Zhong and Nomikos 20 -Reference Adair, Christensen, Florea, Loft and Fagiolini 28 and in observational studies in routine clinical practice settings.Reference De Carlo, Vismara and Grancini 29 , Reference Papalexi, Galanopoulos and Kontis 30
A recent analysis of data from six pivotal clinical trials in patients with MDD showed vortioxetine 20 mg/day to be significantly more effective than vortioxetine 10 mg/day in terms of improvement in depressive symptoms assessed by the Montgomery–Åsberg Depression Rating Scale (MADRS).Reference Christensen, McIntyre, Florea, Loft and Fagiolini 26 Statistically significant and clinically meaningful improvement in depressive symptoms vs placebo was seen after 2 weeks of treatment in patients who received vortioxetine 20 mg/day; however, this was not achieved until week 4 in those who received vortioxetine 10 mg/day. This is particularly noteworthy given that patients randomized to vortioxetine 20 mg/day received the recommended starting dosage of 10 mg/day for the first week of treatment. 31 , 32 After 8 weeks of treatment, the mean change in MADRS total score from baseline was significantly greater in patients treated with vortioxetine 20 mg/day compared with vortioxetine 10 mg/day (difference between groups, −1.03 points; P < .05).Reference Christensen, McIntyre, Florea, Loft and Fagiolini 26 A difference between active treatments of at least 1 point on the MADRS is considered to be clinically significant.Reference Montgomery and Möller 33 Importantly, vortioxetine was found to be well tolerated across the approved dosage range, with no clinically relevant differences in tolerability observed between vortioxetine 20 and 10 mg/day over the 7 weeks of follow-up after vortioxetine dose up-titration from 10 to 20 mg/day.Reference Christensen, McIntyre, Florea, Loft and Fagiolini 26
This analysis was undertaken to further explore the clinical relevance of the greater and more rapid improvement in depressive symptoms previously seen with vortioxetine 20 vs 10 mg/day,Reference Christensen, McIntyre, Florea, Loft and Fagiolini 26 with particular focus on differences in the proportions of patients who achieved symptomatic response and remission and the time-points at which these outcomes were observed. Differences in the proportions of patients achieving sustained symptomatic response (i.e., response that was sustained from the specified time-point until the end of study follow-up) were also assessed. The analysis additionally considered potential vortioxetine dose effects with regard to improvement in symptoms of anxiety and overall patient functioning. The tolerability of vortioxetine 20 mg/day vs vortioxetine 10 mg/day was further assessed; specifically, the incidence of adverse events occurring during the first week of treatment, when patients randomized to receive vortioxetine 20 mg/day received the starting dose of vortioxetine 10 mg/day, compared with that during the second week of treatment immediately following vortioxetine dose up-titration in the 20 mg/day group. The incidence of adverse events related to sexual dysfunction was also analyzed.
Methods
Studies
This was an analysis of data pooled from the six short-term (8-week), randomized, double-blind, placebo-controlled, fixed-dose studies of vortioxetine in adult patients with MDD conducted by H. Lundbeck A/S and Takeda Pharmaceuticals America, Inc., that included vortioxetine 20 mg/day and used the MADRS for assessment of depressive symptoms: NCT01140906,Reference Boulenger, Loft and Olsen 34 NCT01153009,Reference Mahableshwarkar, Jacobsen, Chen, Serenko and Trivedi 35 NCT01163266,Reference Jacobsen, Mahableshwarkar, Serenko, Chan and Trivedi 36 NCT01422213,Reference McIntyre, Lophaven and Olsen 37 NCT01255787,Reference Nishimura, Aritomi, Sasai, Kitagawa and Mahableshwarkar 38 and NCT02389816.Reference Inoue, Sasai, Kitagawa, Nishimura and Inada 24 In all studies, patients randomized to vortioxetine 20 mg/day received 10 mg/day for the first week of treatment, before up-titration to their randomized dosage for the remaining 7 weeks.
Key study inclusion criteria were as follows: diagnosis of MDD according to the Diagnostic and Statistical Manual of Mental Disorders (DSM) criteria at the time that the study was undertaken (DSM 4th Edition, Text Revision, or DSM 5th Edition); current major depressive episode of at least 3 months’ duration (confirmed using the Mini International Neuropsychiatric Interview); MADRS total score of at least 26 points (i.e., at least moderately severe depression); and Clinical Global Impression Scale-Severity of Illness score of at least 4 points (i.e., at least moderately ill) at the screening and baseline visits. Patients who exhibited anxiety symptoms were allowed to participate in these studies; however, those who fulfilled DSM diagnostic criteria for a concomitant anxiety disorder were excluded.
Patients were assessed at baseline and at regular study visits (weeks 1, 2, 4, 6, and 8). In all studies, severity of depression was assessed using the MADRS. Anxiety symptoms were assessed using the Hamilton Rating Scale for Anxiety (HAM-A) in four of the studies,Reference Boulenger, Loft and Olsen 34 -Reference Jacobsen, Mahableshwarkar, Serenko, Chan and Trivedi 36 , Reference Nishimura, Aritomi, Sasai, Kitagawa and Mahableshwarkar 38 and patient functioning was assessed using the Sheehan Disability Scale (SDS) in four studies.Reference Inoue, Sasai, Kitagawa, Nishimura and Inada 24 , Reference Boulenger, Loft and Olsen 34 , Reference Jacobsen, Mahableshwarkar, Serenko, Chan and Trivedi 36 , Reference Nishimura, Aritomi, Sasai, Kitagawa and Mahableshwarkar 38
All studies were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, and were approved by the relevant research ethics committees. Patients provided written informed consent for participation.
Statistical analysis
For each endpoint, efficacy analyses were performed using data from all randomized patients who received at least one dose of study medication and had at least one valid post-baseline efficacy assessment (full analysis set). Given the well-established dose–response relationship for vortioxetine across the approved dose range of 5–20 mg/day, this analysis specifically investigated differences between vortioxetine 20 and 10 mg/day; the lower number of patients who received vortioxetine 5 and 15 mg/day precluded assessment of the outcomes of interest in those dose groups.
The proportions of patients achieving response, sustained response, and remission, as assessed by MADRS total score at weeks 2, 4, 6, and 8, were compared between treatment groups using a logistic regression model with the relevant baseline score as a covariate. Response was defined as ≥50% decrease in MADRS total score from baseline and remission as MADRS total score ≤10 points, and a last observation carried forward approach was used for patients who did not complete the study.
A patient was considered to be a “sustained responder” from a specific visit if they had achieved a ≥50% decrease in MADRS total score from baseline for the first time at the specified visit and at the last observed time-point (with only one intermediate visit allowed to show non-response) (Supplementary Table S1). To be eligible for assessment of sustained response from a specific time-point (e.g., week 2 or 4) onward, patients had to have MADRS data for both the specific time-point and at least one later visit. Patients found to be sustained responders from a specific time-point did not contribute to later time-point analyses (e.g., a patient who met the criteria for sustained response from week 2 could not also be considered a sustained responder from week 4 or 6). Patients who met the criteria for response from any time-point in addition to their last observed time-point were classed as “general sustained responders.” Kaplan–Meier curves of time to MADRS response were generated, with between-group comparisons based on a Cox regression model log-rank test.
Changes in HAM-A and SDS total scores from baseline over time were analyzed using an individual patient data meta-analytic statistical approach. This approach was chosen in order to provide granular results per week based on a unified statistical model. A mixed model for repeated measures was used, with terms for baseline values, study, visit, and treatment for the mean structure, and was applied using a completely unstructured covariance matrix.
Safety was analyzed for all eligible patients who received at least one dose of study medication (all patients treated set). Treatment-emergent adverse events (TEAEs) occurring in at least 2% of patients in any study group were summarized using Medical Dictionary for Regulatory Affairs (MedDRA Version 14.1) preferred terms. TEAEs are reported by randomized treatment group and time of onset: (i) onset before day 8 (i.e., during the first week of treatment when patients randomized to vortioxetine 20 mg/day were receiving the starting dose of 10 mg/day); and (ii) onset between day 8 and day 15 (i.e., during the second week of treatment, immediately following dose up-titration in the vortioxetine 20 mg/day group). The incidence of TEAEs specifically related to sexual dysfunction was assessed over the 8-week, double-blind treatment period. TEAEs related to sexual dysfunction were identified using a list of predefined MedDRA preferred terms (Supplementary Material S2).
Analyses were performed using SAS statistical software (version 9.4; SAS Institute Inc., Cary, NC, USA), with significance set at P < .05.
Results
Patients
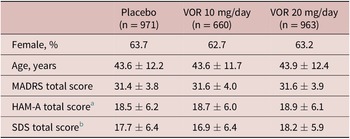
A total of 2620 patients were included in the all patients treated set (975 in the vortioxetine 20 mg/day group, 663 in the vortioxetine 10 mg/day group, and 982 in the placebo group). Of these, 2594 patients had data for MADRS total score at baseline and at least one later assessment time-point and were included in the full analysis set. Treatment groups were well matched in terms of demographic and clinical characteristics at baseline (Table 1).
Table 1. Summary of Demographic and Clinical Characteristics at Baseline in Short-Term, Randomized, Placebo-Controlled, Fixed-Dose Studies of Vortioxetine in Patients with Major Depressive Disorder (Full Analysis Set)
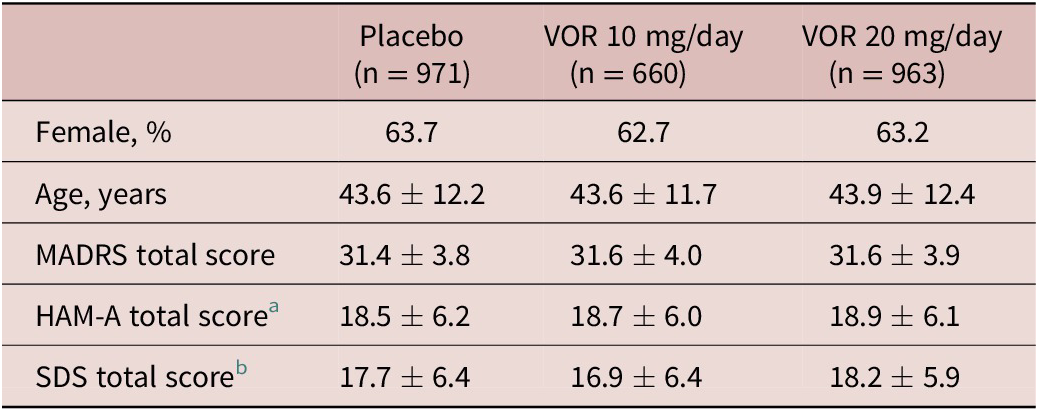
Note. All values are mean ± standard deviation unless otherwise indicated.
Abbreviations: HAM-A, Hamilton Rating Scale for Anxiety; MADRS, Montgomery–Åsberg Depression Rating Scale; SDS, Sheehan Disability Scale; VOR, vortioxetine.
a Number of randomized patients who received at least one dose of study medication and who had at least one post-baseline efficacy assessment for HAM-A total score was 533, 256, and 490 in the placebo, vortioxetine 10 mg/day, and vortioxetine 20 mg/day groups, respectively.
b Number of randomized patients who received at least one dose of study medication and who had at least one post-baseline efficacy assessment for SDS total score was 507, 344, and 491 in the placebo, vortioxetine 10 mg/day, and vortioxetine 20 mg/day groups, respectively.
Efficacy
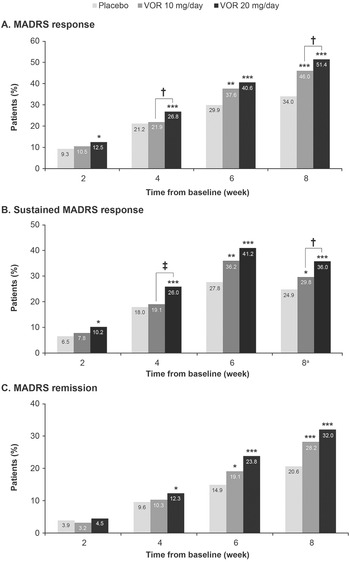
A clear dose–response relationship was observed for vortioxetine 20 mg/day vs vortioxetine 10 mg/day in terms of the proportion of patients achieving symptomatic response, sustained response, and remission over the 8 weeks of treatment (Figure 1). As shown, statistically significant differences were observed for vortioxetine 20 mg/day vs placebo from week 2 onward for the proportion of patients achieving MADRS response and sustained response and from week 4 onward for remission. However, significant differences in the proportion of patients achieving MADRS response, sustained response and remission were only observed from week 6 for vortioxetine 10 mg/day vs placebo.
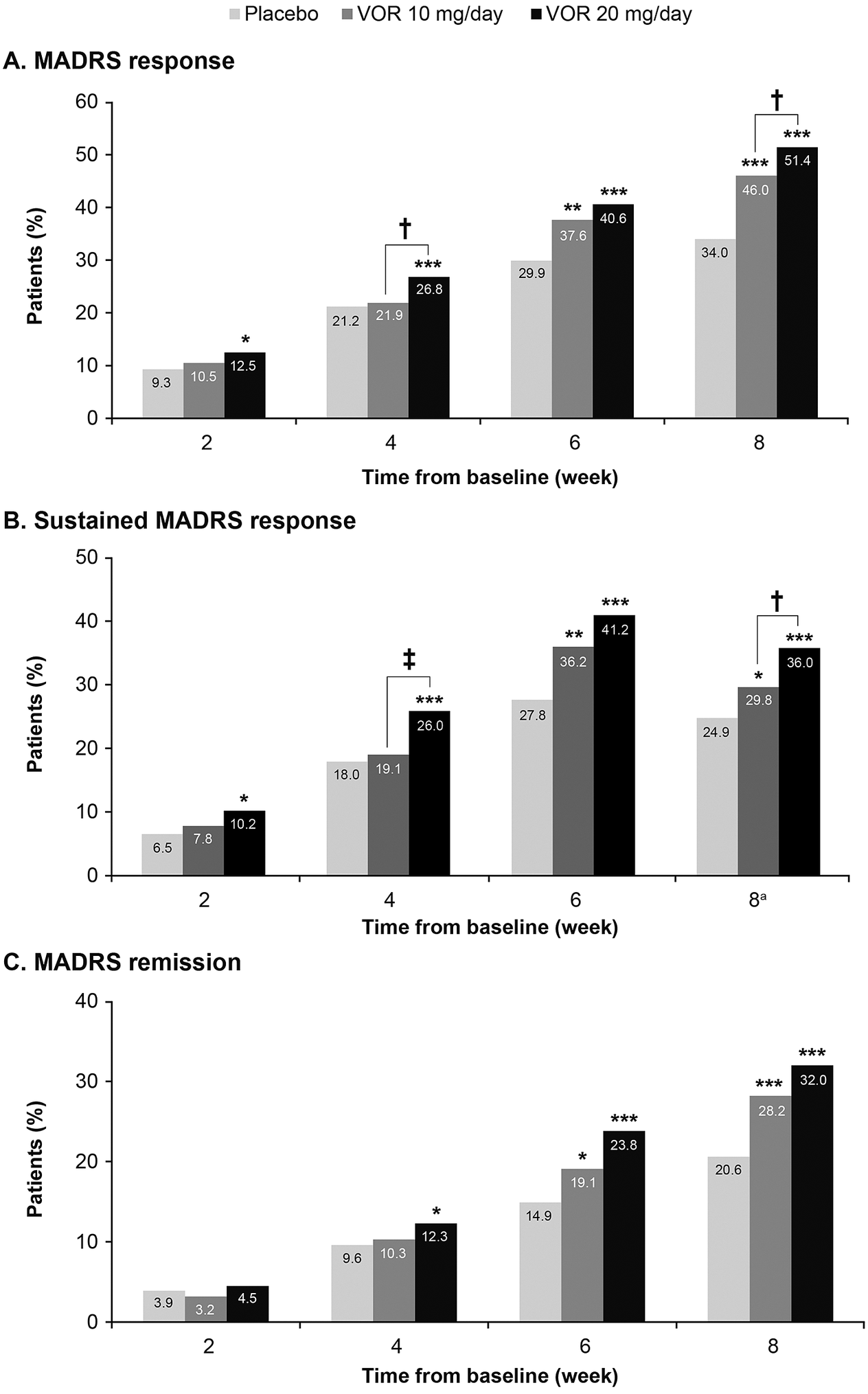
Figure 1. Proportion of patients with major depressive disorder achieving (A) response (≥50% decrease in Montgomery–Åsberg Depression Rating Scale [MADRS] total score from baseline); (B) sustained response (i.e., MADRS response at the given time-point and at the last observed time-point); and (C) remission (MADRS total score ≤10). Analysis of pooled data from short-term, randomized, placebo-controlled, fixed-dose studies of vortioxetine (VOR) in patients with major depressive disorder. Logistic regression with relevant baseline score as a covariate; P-values calculated by Wald’s test (full analysis set, last observation carried forward). *P < .05, **P < .01, and ***P < .001 for vortioxetine 20 or 10 mg/day vs placebo. †P < .05 and ‡P < .01 for vortioxetine 20 mg/day vs vortioxetine 10 mg/day. aProportion of patients achieving general sustained response, that is, MADRS response from any time-point in addition to their last observed time-point.
The proportion of patients who achieved MADRS response was 12.5% in the vortioxetine 20 mg/day group vs 10.5% in the vortioxetine 10 mg/day group at week 2 and was significantly greater in the vortioxetine 20 mg/day group than in the vortioxetine 10 mg/day group at week 4 (26.8% vs 21.9%, respectively; odds ratio [OR], 1.32 [95% confidence interval [CI]: 1.03, 1,52]; P = .0241). After 8 weeks of treatment, the proportion of patients who had achieved MADRS response was 51.4% in the vortioxetine 20 mg/day group, 46.0% in the vortioxetine 10 mg/day group, and 34.0% in the placebo group (P < .0001 for both vortioxetine groups vs placebo). The proportion of patients with MADRS response at week 8 was significantly higher for vortioxetine 20 mg/day vs vortioxetine 10 mg/day (OR, 1.25 [95% CI: 1.02, 1.52]; P = .0313).
Sustained symptomatic response from week 2 was achieved by 10.2% of patients in the vortioxetine 20 mg/day group compared with 7.8% in the vortioxetine 10 mg/day group (OR, 1.34 [95% CI: 0.88, 2.06]; P = .1760). The proportion of patients who achieved sustained symptomatic response from week 4 was significantly greater in the vortioxetine 20 mg/day group than in the vortioxetine 10 mg/day group (26.0% vs 19.1%, respectively; OR, 1.49 [95% CI: 1.15, 1.92]; P = .0025). A general sustained response (i.e., MADRS response from any time-point in addition to the patient’s last observed time-point) was also achieved by a significantly greater proportion of patients in the vortioxetine 20 mg/day group vs the vortioxetine 10 mg/day group (36.0% vs 29.8%, respectively; OR, 1.33 [95% CI: 1.07, 1.65]; P = .0103).
The proportion of patients meeting the criteria for MADRS remission at week 8 was 32.0% in the vortioxetine 20 mg/day group, 28.2% in the vortioxetine 10 mg/day group, and 20.6% in the placebo group (P < .0001 and P = .0003 vs placebo, respectively) (OR for vortioxetine 20 mg/day vs vortioxetine 10 mg/day, 1.20 [95% CI: 0.97, 1.49]; P = .0927).
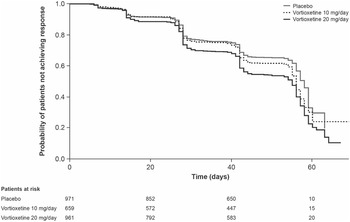
Results of the Kaplan–Meier analysis of time to MADRS response are shown in Figure 2. The median time to symptomatic response was significantly shorter in the vortioxetine 20 mg/day group than in the vortioxetine 10 mg/day group (hazard ratio [HR], 1.18 [95% CI: 1.03, 1.35]; P = .0161) and placebo group (HR, 1.49 [95% CI: 1.31, 1.70]; P < .0001).
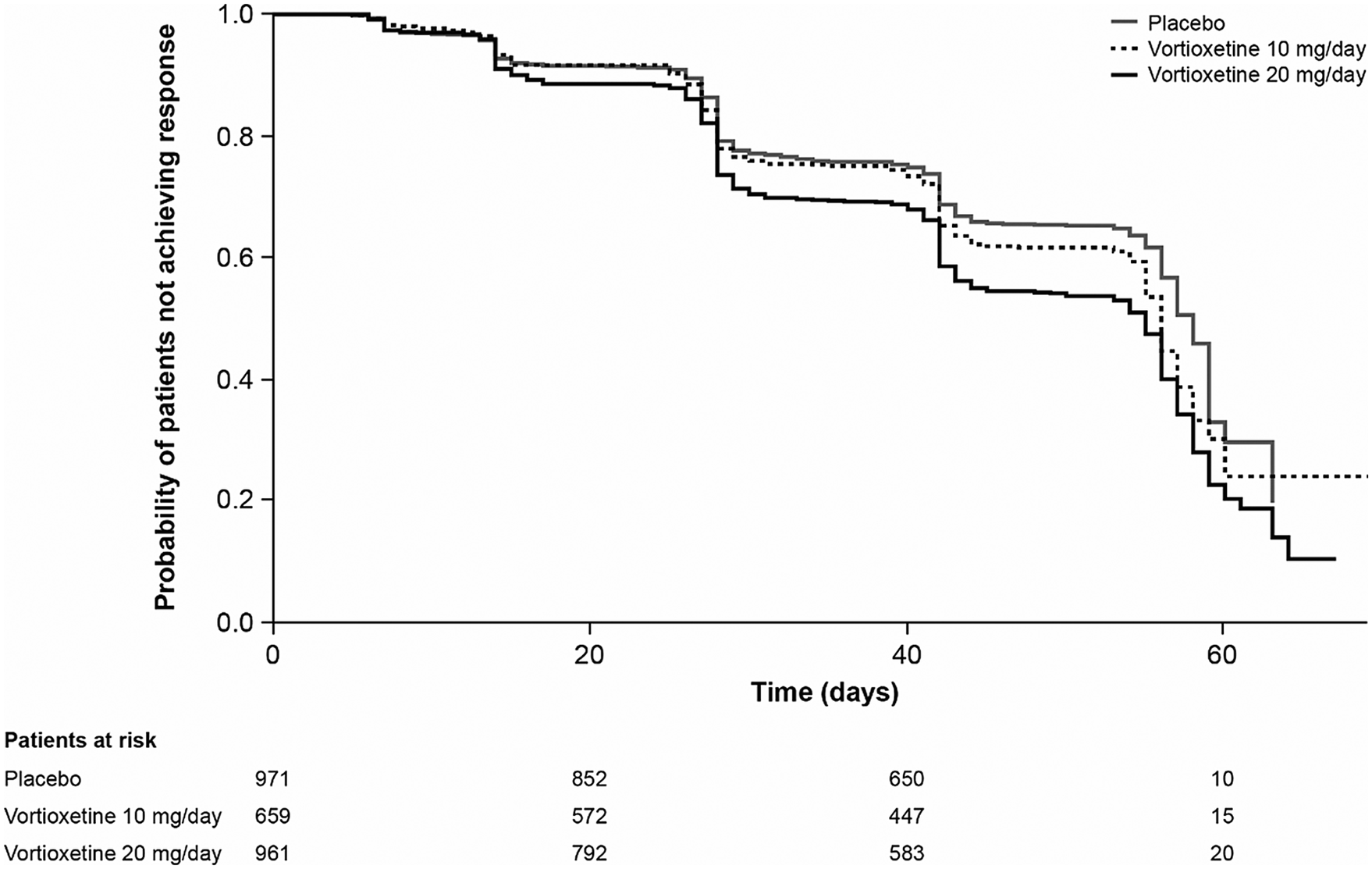
Figure 2. Kaplan–Meier estimate of time to symptomatic response (i.e., ≥50% decrease in Montgomery–Åsberg Depression Rating Scale total score from baseline) in patients with major depressive disorder treated with vortioxetine 20 mg/day, vortioxetine 10 mg/day, or placebo. Analysis of pooled data from short-term, randomized, placebo-controlled, fixed-dose studies of vortioxetine in patients with major depressive disorder (full analysis set).
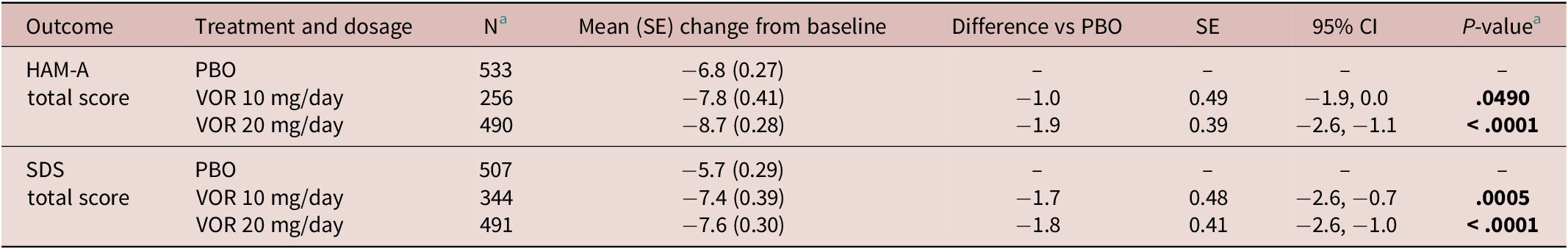
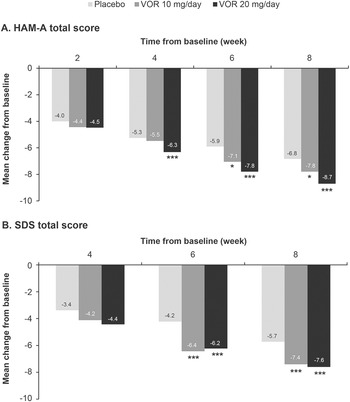
Regarding symptoms of anxiety, statistically significant differences in mean change in HAM-A total score from baseline were seen with vortioxetine 20 mg/day vs placebo from week 4 onward (P = .001 at week 4 and P < .0001 at weeks 6 and 8) (Figure 3A). For vortioxetine 10 mg/day, statistically significant differences in mean change in HAM-A total score from baseline vs placebo were only seen at weeks 6 and 8 (both P < .05). Mean (95% CI) difference in change from baseline in HAM-A total score at week 8 vs placebo was −1.9 (−2.6, −1.1) points for vortioxetine 20 mg/day (P < .0001) and −1.0 (−1.9, 0.0) points for vortioxetine 10 mg/day (P = .0490) (Table 2).
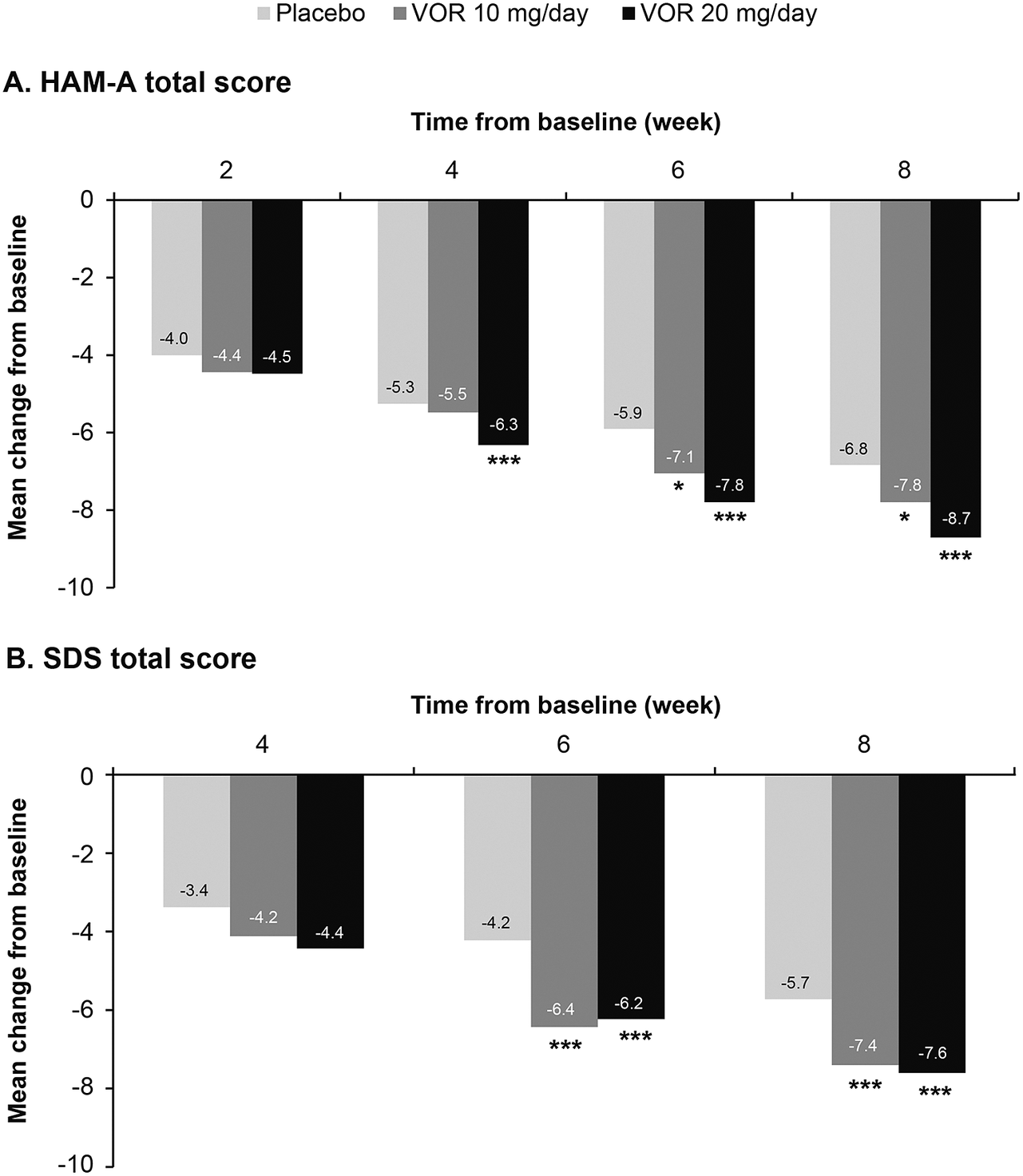
Figure 3. Mean change from baseline in (A) Hamilton Rating Scale for Anxiety (HAM-A) total score and (B) Sheehan Disability Scale (SDS) total score. Analysis of pooled data from short-term, randomized, placebo-controlled, fixed-dose studies of vortioxetine (VOR) in patients with major depressive disorder (full analysis set, mixed model for repeated measures). *P < .05 vs placebo; **P < .01 vs placebo; ***P ≤ .001 vs placebo.
Table 2. Meta-Analysis of Difference in Change from Baseline to Week 8 in HAM-A Total Score and SDS Total Score vs Placebo in Short-Term, Randomized, Placebo-Controlled, Fixed-Dose Studies of Vortioxetine in Patients with Major Depressive Disorder (Full Analysis Set, Mixed Model for Repeated Measures)
Abbreviations: CI, confidence interval; HAM-A, Hamilton Rating Scale for Anxiety; PBO, placebo; SDS, Sheehan Disability Scale; SE, standard error; VOR, vortioxetine.
a Number of randomized patients who received at least one dose of study medication and who had at least one post-baseline efficacy assessment (full analysis set).
b Bold indicates statistically significant P-values.
Statistically significant differences in mean change in SDS total score from baseline vs placebo were seen at weeks 6 and 8 for both vortioxetine 20 mg/day and vortioxetine 10 mg/day (P < .0001 and P ≤ .0009, respectively) (Figure 3B). The mean (95% CI) difference in change from baseline in SDS total score at week 8 vs placebo was −1.8 (−2.6, −1.0) points for vortioxetine 20 mg/day (P < .0001) and −1.7 (−2.6, −0.7) points for vortioxetine 10 mg/day (P = .0005) (Table 2).
Safety and tolerability
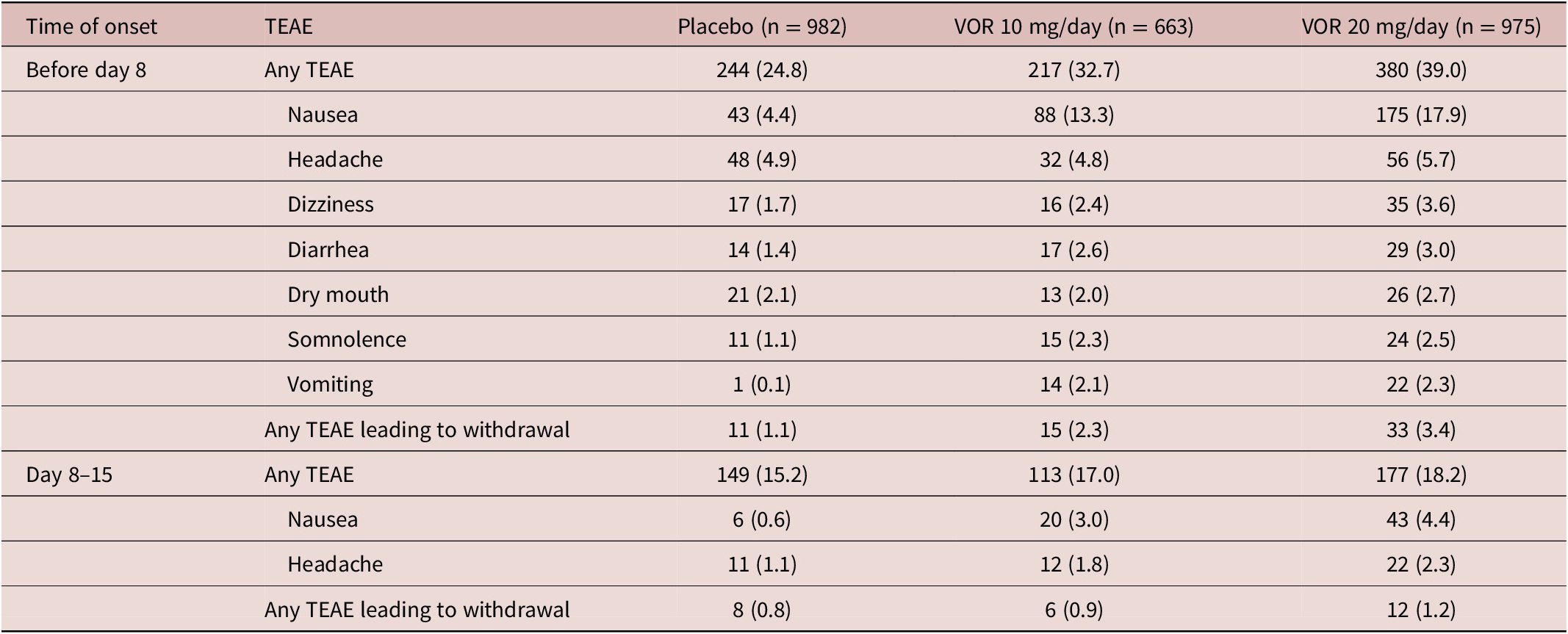
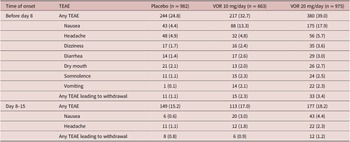
Irrespective of vortioxetine dosage and the time of TEAE onset, the most commonly reported TEAEs in vortioxetine-treated patients were nausea and headache (Table 3). During the first week of treatment (i.e., before dose up-titration in the vortioxetine 20 mg/day group), the incidence of nausea was 17.9%, 13.3%, and 4.4% in patients randomized to vortioxetine 20 mg/day, vortioxetine 10 mg/day, and placebo, respectively. During the second week of treatment, nausea was reported by 4.4% and 3.0% of patients in the vortioxetine 20 mg/day and 10 mg/day groups, respectively, and by 0.6% of patients in the placebo group. The incidence of headache during the first week of treatment was 5.7% for vortioxetine 20 mg/day, 4.8% for vortioxetine 10 mg/day, and 4.9% for placebo. Respective incidences of headache during the second week of treatment were 2.3%, 1.8%, and 1.1%.
Table 3. TEAEs by Medical Dictionary for Regulatory Affairs Preferred Terms with Incidence ≥2% in at Least One Group in Short-Term, Randomized, Placebo-Controlled, Fixed-Dose Studies of Vortioxetine in Patients with Major Depressive Disorder According to Time of Onset: (i) Before Day 8 (i.e., When Patients in the Vortioxetine 20 mg/day Group Were Receiving the Starting Dose of 10 mg/day) and (ii) Between Days 8 and 15 (i.e., During the Week Following Dose Up-Titration in the Vortioxetine 20 mg/day Group) (All Patients Treated Set)
Note. All values are n (%).
Abbreviations: TEAE, treatment-emergent adverse event; VOR, vortioxetine.
The incidence of TEAEs leading to withdrawal with onset during the first week of treatment was 3.4% in the vortioxetine 20 mg/day group, 2.3% in the vortioxetine 10 mg/day group, and 1.1% in the placebo group. The most common TEAEs with onset during the first week of treatment that led to withdrawal from treatment in more than one patient in any group were nausea (18 [1.8%], three [0.5%], and two patients [0.2%] in the vortioxetine 20 mg/day, vortioxetine 10 mg/day, and placebo groups, respectively), headache (five [0.5%], three [0.5%], and three [0.3%] patients, respectively), and vomiting (five [0.5%], three [0.5%], and one patient [0.1%], respectively).
The incidence of TEAEs leading to withdrawal with onset during the second week of treatment was 1.2% in the vortioxetine 20 mg/day group, 0.9% in the vortioxetine 10 mg/day group, and 0.8% in the placebo group. The only TEAEs with onset during the second week of treatment that led to withdrawal from treatment in more than one patient in any group were nausea (six patients [0.6%] in the vortioxetine 20 mg/day group and two patients [0.3%] in the vortioxetine 10 mg/day group) and generalized pruritus (two patients [0.3%] in the vortioxetine 10 mg/day group).
The incidence of TEAEs related to sexual dysfunction (i.e., TEAEs by MedDRA preferred terms related to sexual dysfunction) over the 8-week, double-blind treatment period was low in all treatment groups (1.8% in the vortioxetine 20 mg/day group, 0.8% in the vortioxetine 10 mg/day group, and 1.0% in the placebo group). The only TEAEs related to sexual dysfunction reported by more than one patient in any group were: decreased libido (reported by 10 patients in the vortioxetine 20 mg/day group and seven patients in the placebo group), abnormal orgasm (reported by three patients in the vortioxetine 20 mg/day group and two patients in both the vortioxetine 10 mg/day and placebo groups), delayed ejaculation (reported by two patients in the vortioxetine 20 mg/day group and one patient in the vortioxetine 10 mg/day group), and anorgasmia (reported by two patients in the vortioxetine 20 mg/day group).
Discussion
To our knowledge, this is the first study to assess the comparative efficacy of vortioxetine 20 and 10 mg/day for the achievement of sustained response in terms of reduction in depressive symptom severity in patients with MDD using data from pivotal randomized, placebo-controlled clinical trials. The results demonstrate that the significantly greater improvement in depressive symptoms vs placebo previously observed in patients with MDD after 2 weeks of treatment with vortioxetine 20 mg/day, but not vortioxetine 10 mg/day,Reference Christensen, McIntyre, Florea, Loft and Fagiolini 26 beneficially impacts on several clinically relevant endpoints and confirm that dose up-titration to 20 mg/day after 1 week of treatment is well tolerated.
Compared with vortioxetine 10 mg/day, vortioxetine 20 mg/day (increased from 10 mg/day after 1 week of treatment) was associated with more rapid and greater achievement of symptomatic response, sustained response, and remission as assessed by MADRS total score. For vortioxetine 20 mg/day, significant differences vs placebo were observed from week 2 onward (i.e., from 1 week after up-titration from the starting dose of 10 mg/day) for achievement of symptomatic response and sustained symptomatic response, and from week 4 onward for rates of remission. In contrast, for vortioxetine 10 mg/day, significant differences in rates of response, sustained response, and remission vs placebo were only observed from week 6.
After 8 weeks of treatment, significantly more patients treated with vortioxetine 20 mg/day had achieved symptomatic response (i.e., ≥50% decrease in MADRS total score from baseline) than those who had received vortioxetine 10 mg/day. Patients treated with vortioxetine 20 mg/day were also found to be significantly more likely than those who received vortioxetine 10 mg/day to achieve sustained symptomatic response (i.e., response that was sustained at the final study visit) from week 4. Patients treated with vortioxetine 20 mg/day were also significantly more likely to achieve symptomatic response that was sustained at at least one later study visit (i.e., a general sustained response). At the end of the 8-week treatment period, a numerically greater proportion of patients in the vortioxetine 20 mg/day group were in remission (i.e., MADRS total score ≤10 points) than in the vortioxetine 10 mg/day group. Our findings add to the evidence that early and sustained response to antidepressant treatment is associated with improved outcomes in patients with MDD.Reference Habert, Katzman and Oluboka 3 , Reference Oluboka, Katzman and Habert 4 , Reference Henkel, Seemüllerm and Obermeier 8 -Reference Inoue, Fujimoto and Marumoto 13
In keeping with the results of previous analyses,Reference Baldwin, Florea, Jacobsen, Zhong and Nomikos 20 , Reference Adair, Christensen, Florea, Loft and Fagiolini 28 earlier improvements in anxiety symptoms vs placebo were also achieved in patients treated with vortioxetine 20 mg/day than in those who received vortioxetine 10 mg/day. Of note, the observed dose–response relationship for vortioxetine in terms of improvement in anxiety appears more pronounced than that seen for improvement in depressive symptoms. For overall patient functioning, clinically significant improvement (i.e., increase in SDS total score ≥4 pointsReference Sheehan and Sheehan 39) vs placebo was seen in both vortioxetine dose groups from week 4 onward.
Importantly, vortioxetine dose up-titration to 20 mg/day after the first week of treatment was not associated with a higher incidence of TEAEs or higher rates of withdrawal from therapy. As previously reported,Reference Baldwin, Chrones and Florea 40 nausea was the most frequently reported adverse event reported in vortioxetine-treated patients; however, this appeared to be transient, occurring more frequently during the first week of treatment and at a lower and similar incidence during the second week of treatment in both vortioxetine dose groups. The incidence of sexual adverse events was also low and similar between the vortioxetine and placebo groups.
The broad and dose-dependent therapeutic effects of vortioxetine in patients with MDD are likely due to its multimodal mechanism of action. Vortioxetine acts as an inhibitor of the serotonin (5-HT) transporter as well as modulating the activity of several 5-HT receptor subtypes.Reference Sanchez, Asin and Artigas 41 Preclinical data show vortioxetine to be a 5-HT3, 5-HT7, and 5-HT1D receptor antagonist, a 5-HT1B receptor partial agonist, and a 5-HT1A receptor agonist.Reference Sanchez, Asin and Artigas 41 -Reference Mørk, Pehrson and Brennum 43 As such, vortioxetine directly and indirectly modulates neurotransmission across multiple systems relevant to the pathophysiology of depression.Reference Sanchez, Asin and Artigas 41 Notably, the affinity of vortioxetine has been shown to vary between 5-HT receptor types (5-HT3 > 5-HT1B > 5-HT1A ≈ 5-HT7), resulting in their dose-dependent recruitment.Reference Bang-Andersen, Ruhland and Jorgensen 42 Occupancy of the 5-HT transporter has also been shown to be related to vortioxetine dosage, ranging from ~50% for vortioxetine 5 mg to ~65% for vortioxetine 10 mg, and >80% for vortioxetine 20 mg in healthy individuals.Reference Areberg, Luntang-Jensen, Søgaard and Nilausen 44 For the treatment of MDD, 5-HT transporter occupancy of ≥80% is considered optimal.Reference Meyer 45 , Reference Sørensen, Ruhé and Munkholm 46
The transient nausea that may occur in some patients when initiating treatment with vortioxetine is likely due to its effects on 5-HT3 receptors. Vortioxetine binds at the orthosteric binding site of 5-HT3A receptors, but its mechanism of action differs from that of other 5-HT3 antagonists, inducing an initial partial agonistic response before persistent and insurmountable inhibition of receptor function.Reference Bang-Andersen, Ruhland and Jorgensen 42 , Reference Dale, Grunnet and Pehrson 47 , Reference Ladefoged, Munro and Pedersen 48 We hypothesize that the transient initial partial agonistic activity of vortioxetine at 5-HT3 receptors may contribute to nausea and vomiting in patients who experience these adverse effects and that the timing of the shift from partial agonistic to 5-HT3 functional antagonist activity―likely due to receptor down-regulation or desensitization―differs between patients.
In the experience of the authors who are in clinical practice, patients who do not experience nausea or vomiting at the starting dose of vortioxetine 10 mg/day are highly unlikely to develop these adverse events following dose up-titration. In patients who do experience nausea and vomiting following treatment initiation, vortioxetine dose up-titration is better tolerated if delayed until these initial adverse events have subsided. However, in the majority of patients (i.e., those that do not experience nausea or vomiting following treatment initiation or in whom these adverse events resolve quickly), vortioxetine dosage can be increased early in the course of treatment (i.e., from week 1 onward) in order to achieve more rapid response, without an increased risk of adverse events.
The unique mechanism of action of vortioxetine may also account for the low incidence of adverse events related to sexual dysfunction. It is likely that sexual dysfunction during treatment with selective serotonin reuptake inhibitors is mediated by increased levels of 5-HT following transporter inhibition.Reference Olivier and Olivier 49 Preclinical data suggest that the agonistic effects of vortioxetine on 5-HT1A receptors counteract any adverse effect on sexual function that may arise from 5-HT transporter inhibition, thus reducing the potential for associated adverse events.Reference Li, Pehrson, Oosting, Gulinello, Olivier and Sanchez 50
A potential study limitation is that patients treated with vortioxetine 20 mg/day did not initiate treatment at this dosage, but received the recommended starting dose of 10 mg/day for the first week of treatment. 31 , 32 It is possible that differences between the two groups may have been more marked if assessed 2 weeks after vortioxetine dose up-titration.
Conclusion
In summary, vortioxetine 20 mg/day is associated with more rapid symptomatic improvement in patients with MDD compared with vortioxetine 10 mg/day, resulting in earlier and higher rates of response, sustained response, and remission over 8 weeks of treatment. Of note, the proportion of patients who achieved symptomatic response was significantly higher in the vortioxetine 20 mg/day group than in the vortioxetine 10 mg/day group at weeks 4 and 8. Significantly more patients treated with vortioxetine 20 mg/day also achieved sustained symptomatic response from week 4 compared with patients treated with vortioxetine 10 mg/day. These findings and the favorable tolerability profile demonstrated in the present analysis suggest that, according to individual patient response and tolerability, 20 mg/day should be considered the optimal vortioxetine dosage in patients with MDD.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S1092852923002249.
Acknowledgments
Under the direction of the authors, medical writing assistance was provided by Jennifer Coward for Piper Medical Communications, funded by H. Lundbeck A/S.
Author contributions
All authors contributed to the conceptualization and interpretation of the analyses, contributed to the scientific content of the manuscript, and approved the final version of the manuscript for submission. Henrik Loft performed the statistical analyses. Employees from Lundbeck were involved in the concept of the manuscript, as well as the data analysis and interpretation of the results.
Financial support
The data reported in this paper were derived from clinical studies sponsored by H. Lundbeck A/S, Valby, Denmark, and Takeda Pharmaceuticals Inc., Deerfield, Illinois, USA. The analyses reported in this paper were funded by H. Lundbeck A/S, Valby, Denmark.
Competing interests
Drs. Christensen, Adair, Florea, and Loft are employees of H. Lundbeck A/S. Dr. McIntyre has received: research grant support from CIHR/GACD/National Natural Science Foundation of China (NSFC) and the Milken Institute; speaker/consultation fees from Lundbeck, Janssen, Alkermes, Neumora Therapeutics, Boehringer Ingelheim, Sage, Biogen, Mitsubishi Tanabe, Purdue, Pfizer, Otsuka, Takeda, Neurocrine, Sunovion, Bausch Health, Axsome, Novo Nordisk, Kris, Sanofi, Eisai, Intra-Cellular, NewBridge Pharmaceuticals, AbbVie, Atai Life Sciences; and is a CEO of Braxia Scientific Corp. Dr. Fagiolini has been a consultant and/or speaker and/or has received research grants from Allergan, Angelini, Apsen, Boehringer Ingelheim, Daiichi Sankyo Brasil Farmacêutica, DOC Generici, FB-Health, Italfarmaco, Janssen, Lundbeck, Mylan, Otsuka, Pfizer, Recordati, Sanofi Aventis, Sunovion, and Vifor Pharma.




 Open access
Open access