



Introduction
Actin is a dynamic polymer and is one of the most abundant proteins in eukaryotic cells. The monomers are globular (G-actin) and form various shaped filaments (F-actin) when polymerised. The F-actin filaments are polar double-stranded helices with two differently named ends, the barbed end and the pointed end. Actin is a major component of the cytoskeleton, which is involved in cell shape and motility as well as in exo- and endocytosis (Ref. Reference Blanchoin1).
Gelsolin (GSN) is one of the most important actin-binding proteins (ABPs) (Ref. Reference Li2) known to date. It belongs to the GSN superfamily consisting of eight members (Table 1): adseverin, adseverin D5, CapG, flightless, villin, advillin, supervillin and eponymic GSN (Ref. Reference Kwiatkowski3). GSN is involved in motility and cell shape as well as apoptosis and phagocytosis processes (for review see (Ref. Reference Silacci4)). The term ‘GSN’ is based on its capacity to transition cytosolic macrophage extracts from a gel phase to a sol phase by severing actin filaments (Ref. Reference Yin5). High levels of GSN have been found in lung and heart. Lower levels of GSN occur in skeletal muscle, testis and kidney (Ref. Reference Arai and Kwiatkowski6) (see also Fig. 1).
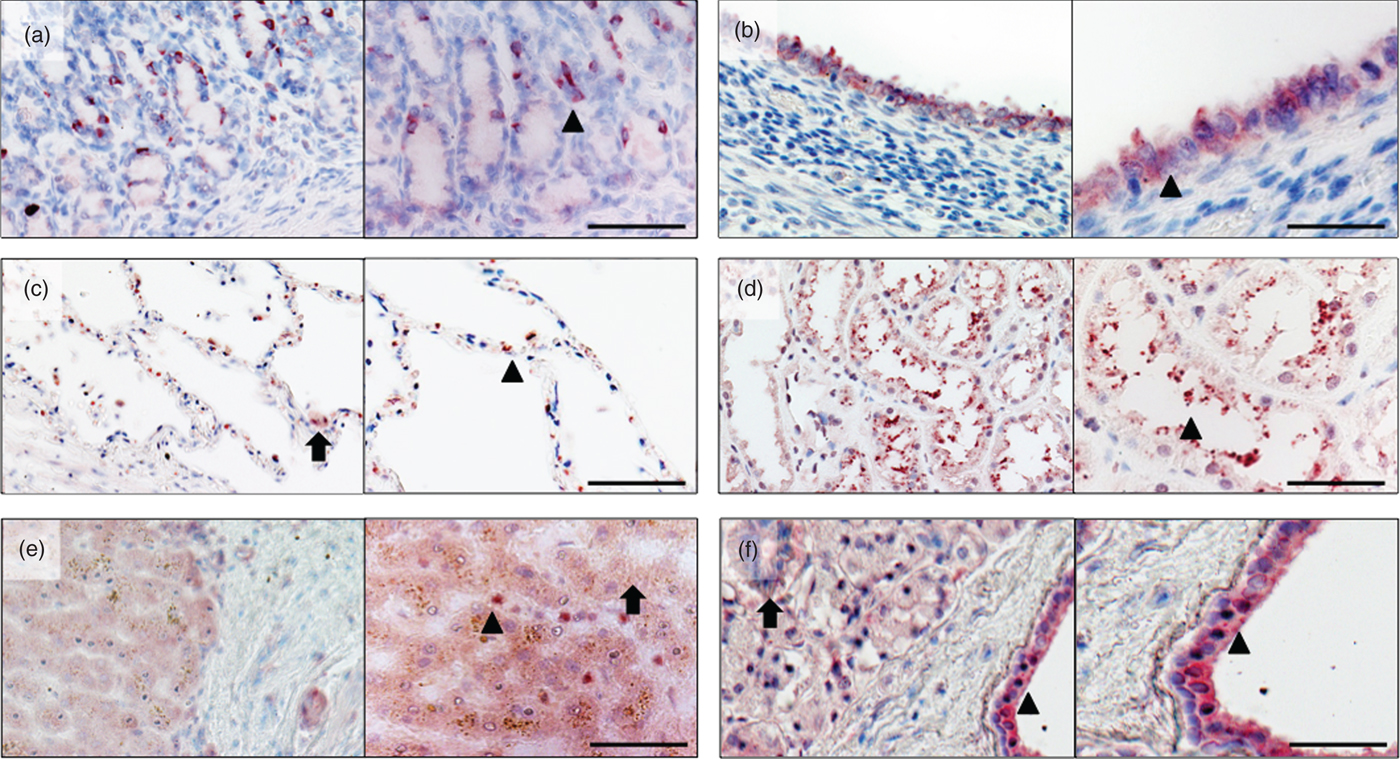
Fig. 1. Overview and detail screen of gelsolin (GSN) detection in different tissues. Immunohistochemical localisation of human GSN in stomach, uterus, lung, kidney, liver and parotid gland. (a) Stomach: GSN is detected in the tubular glands cytoplasm (arrowhead) of the foveolae gastricae (cardia). (b) Uterus: localisation of GSN in the cytoplasm of epithelial cells (arrowhead) of the uterus glands. (c) Lung: detection of GSN in pneumocytes type 2 (arrow) and pneumocytes type 1 cells (arrowhead). (d) Kidney: GSN detected within in proximal tubulus (arrowhead). (e) Liver: localisation of GSN in hepatocytes (arrow) and in the sinusoid (arrowhead). (f) Parotid gland: GSN detected in the glands, excretory duct (arrow) and cytoplasmatic in the epithelia of the interlobular excretory duct (arrowhead). Red staining indicates positive reactivity of the antibody. Right part of each picture shows magnification, scale bar 100 µm.
Table 1. Gelsolin superfamily (Ref. Reference Nag7)

GSN is one of the main regulators of the actin skeleton: It acts by severing, capping and nucleating actin filaments as well as sequestering monomers (Ref. Reference Nag7). Its activity depends on the intracellular Ca2+ concentration, intracellular pH (Ref. Reference Silacci4) and regulators like phosphatidylinositol-4,5-bisphosphate (PIP2) (Ref. Reference Janmey and Stossel8). GSN has a major impact on several diseases, such as different cancer types, Alzheimer's disease (AD) and rheumatoid arthritis, and therefore has broad implications as a target for developing novel therapeutic strategies.
Structure of GSN
GSN has a molecular weight of 82–84 kDa depending on its isoform (Refs Reference Sun9, Reference Lee and Galbraith10). Today three isoforms are known, cytoplasmic GSN (cGSN), plasma GSN (pGSN) and gelsolin-3 (Ref. Reference Nag7) (Table 2). pGSN is the extracellular isoform (Ref. Reference Silacci4) and is an abundant protein in plasma (Ref. Reference Badmalia11). It differs from cGSN with its length of 25 amino acids (Ref. Reference Kwiatkowski12). The genes of the isoforms are localised on chromosome 9 in humans and on chromosome 2 in mice (Ref. Reference Li2).
Table 2. Isoforms of gelsolin

Gelsolin-3 is the ‘youngest’ member of the GSN isoforms. It is produced by oligodendrocytes and results from an alternative splicing of the GSN gene. Because of its capacity for severing, capping and nucleating as well as initiating lamelipodia (Ref. Reference Tanaka and Sobue13), gelsolin-3 may play a role in myelogenesis (Ref. Reference Vouyiouklis and Brophy14) and myelin remodelling (Ref. Reference Tanaka, Kira and Sobue15). The GSN isoforms pGSN and cGSN have five cysteine residues, gelsolin-3, by contrast, has two more, offering further possibilities to form intracellular disulphide bonds in the protein, thus achieving a higher level of complexity (Ref. Reference Vouyiouklis and Brophy14).
GSN has two homologous halves, each consisting of three domains termed G1–G3 and G4–G6. Domains G1–G3 belong to the N-terminal half of GSN, which is Ca-free (Ref. Reference Khaitlina and Hinssen16) and its activation is independent from Ca2+ (Ref. Reference Chumnarnsilpa17), whereas domains G4–G6 are part of the C-terminal half, activation of which depends on Ca2+ (Ref. Reference Nag7, Reference McLaughlin18). The two halves are linked to each other and can be cleaved by caspase-3, an effector of apoptosis (Ref. Reference Kothakota19).
Two different calcium-binding sites have been demonstrated, the type 1 and type 2 sites. Type 1 is important for interactions with actin and can be found in domains G1 and G4 (Ref. Reference Choe20). In all six domains (G1–G6), the type 2 calcium-binding site is present and is mandatory for structural rearrangements (see Fig. 2). The G2 and G6 domains must be emphasised in the context of GSN stability (Refs Reference Choe20, Reference Nag21). These binding sites are important for the function of GSN because the concentration of free Ca2+ ions determines affinity to actin and its filaments.
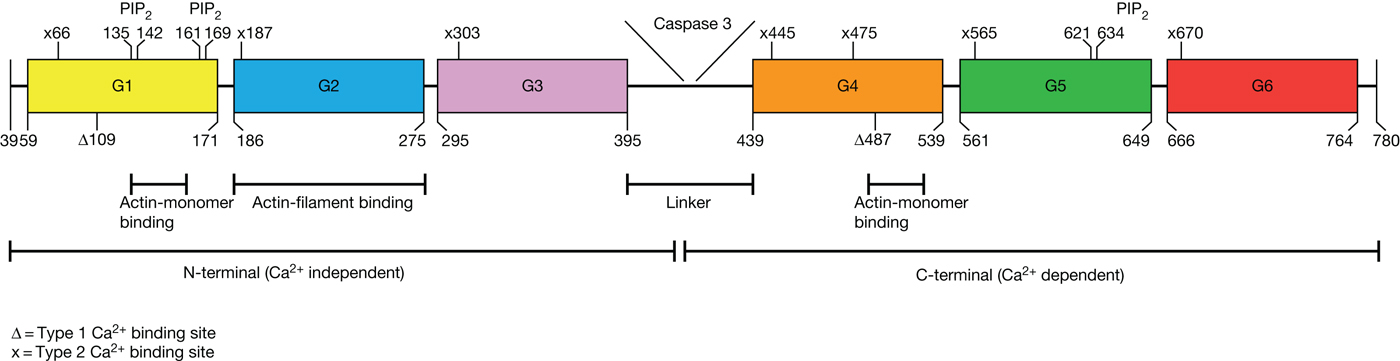
Fig. 2. Structure of gelsolin (GSN). GSN consists out of two homologous halves (N-terminal and C-terminal) each with three domains (G1–G3 and G4-G6). These two halves are bound by a linker, which can be cleaved by caspase 3. In the N-terminal halve, domains one and two can bind actin. G1 can bind actin monomers and G2 can bind actin-filaments. A second actin-monomer binding site is in domain four in the C-terminal halve. PIP2, a regulator of GSN, has three bindings sites in GSN. Each domain of GSN can bind calcium by a type 2 calcium binding site (x), a glutamic acid at the start of the helix(Ref. Reference Nag7) and an aspartic acid with a main chain carbonyl at the β-sheet (not shown in the figure, for more information see reference 7). In domains one and four, calcium can be bound by type 1 calcium binding sites (Δ). Modified out of references 7 and 9.
Without Ca2+, GSN exists in a compact, inactive conformation (Refs Reference Burtnick22, Reference Wang23) where its two halves are bound through a helical C-tail latch from G2 to G6 (Refs Reference Sun9, Reference Lueck24). In this inactive form GSN is unable to bind actin due to serial inhibition of its binding sites. A Ca2+-induced conformational change transforms GSN into its active form and enables GSN actin-binding (Ref. Reference Sun9) (see Fig. 3).

Fig. 3. Calcium-dependent opening of gelsolin (GSN). The domains are coloured as in Figure 1. (a) The inactive form of GSN is a globular form, which is bound by an α-helix (1, C-tail latch) between the second and the sixth domain. The domains one and three (G1/G3 latch) are bound by a β-sheet blend (2) as well as the domains four and six (G4/G6 latch). Because of Ca2+ the C-tail latch is released and GSN undergoes a conformational change. (b) After the C-tail latch releases the G4/G6 β-sheet blend breaks and GSN is straightened. (c) The last latch between G1 and G3 releases and GSN changed to its active form.
GSN has three actin-binding domains (G1, G2 and G4) (Ref. Reference Nag7). Of these, G1 and G4 are able to bind actin monomers (Ref. Reference Kwiatkowski12), whereas G2 in the N-terminal half can bind F-actin filaments (Ref. Reference Yin and Stossel25). GSN breaks non-covalent bonds between two actin-filaments and binds to the ‘barbed’ (+) end of the monomer, deactivating exchange and actin depolarisation (Ref. Reference Yin5). The separation is induced by conformational changes of actin and kinking by GSN (Ref. Reference Witke26), followed by interfering with the non-covalent bonds between actin filaments (Ref. Reference Liepina27). After binding on the barbed end, GSN functions as a cap and inhibits F-actin filament growth (Ref. Reference Badmalia11). GSN needs to be uncapped by PIP2 to renew actin assembly (Ref. Reference Janmey and Stossel28) (see below – PIP 2 as a regulator). PIP2, which plays a role in the recruitment, regulation and organisation of the cytoskeleton, is important in signal transduction and part of the G-protein induced cascade that releases calcium from the endoplasmic reticulum (ER).
Clearance of extracellular actin filaments released into circulation after tissue damage is important in protecting the organism from detriments due to toxic F-actin (Ref. Reference Haddad29). Therefore, the extracellular actin scavenging system, consisting of GSN and Gc-protein, is accountable (Ref. Reference Lee and Galbraith10). Actin polymerisation can also be accomplished by GSN through binding of two actin monomers at the same time. These actin monomers serve as a nucleus for filament assembly so that other monomers can bind (Refs Reference Janmey and Stossel8, Reference Janmey30–Reference Kurth and Bryan33).
Regulation of GSN
The regulation of GSN is important for its role in cell motility and cell shape and can be accomplished in different ways. The most common regulations are listed below.
Calcium as regulator
One of the main regulating factors for GSN is binding of ionised free calcium. Ca2+ binding results in an activation of GSN leading to release of three latches which are important for its active structure (Ref. Reference Nag7). The latch hypothesis states that, based on calcium, the helical tail latch, formed by an interaction of the helices of G2 and G6, (also C-tail latch, C-terminal latch, G2–G6 latch) is released and G2 then binds to actin. This binding is followed by activation of the G1 actin-binding site. After the conformational change from G2 to G6, G6 binds to the G5 domain and enables the G4 domain. Now G4 can bind calcium and actin (Ref. Reference Silacci4). The calcium-binding sites of G2 and G6 are also important in stabilisation of the calcium-free as well as the calcium-bound state of GSN (Ref. Reference Nag21).
So the activation of GSN by free Ca2+ is a gradual process: (i) the inactive globular conformation of GSN must be changed for actin binding by calcium (Ref. Reference Burtnick22). A break in the interactions between the core β-sheets of G4 and G6 enables the movement (Ref. Reference Nag7). (ii) The domains G1 and G4 must be released for actin binding and (iii) an actin-GSN-calcium complex is formed (Ref. Reference Robinson34). Calcium participates in the binding of actin to GSN and strengthens their bond (Ref. Reference Choe20) (see Fig. 3).
Calcium concentrations of 0.5 µm are needed to detect F-actin depolymerisation at physiological temperatures (37°C) (Ref. Reference Lin, Mejillano and Yin35), but binding of F-actin can be detected at calcium concentrations between 0.1 µm and 1 µm (Ref. Reference Kinosian36). The ability to sever actin depends on the calcium concentration and is not possible at submicromolar levels (Ref. Reference Yin5). At a concentration of 0.1 µm of Ca2+, cross-linking between two actin monomers and GSN takes place (Ref. Reference Harris37). Chelating calcium with ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) can stop the severing activity of GSN (Ref. Reference Kinosian36).
Regulation by temperature
A recent study states that GSN is also regulated by temperature in the absence of Ca2+. Small-angle X-ray scattering analysis as well as circular dichroism showed that temperatures between 30° and 40°C alone could open the G1 domain (without Ca2+) (Ref. Reference Badmalia11). A previous study shows that at lower temperatures higher calcium concentrations are needed to activate actin depolymerisation by GSN (Ref. Reference Lin, Mejillano and Yin35). The C-tail latch between G2 and G6 is Ca2+-sensitive and is not affected by low pH or temperature, so that free calcium ions alone can lead to a completely open and active structure (Ref. Reference Garg38). Thus the calcium concentration needed for activation of GSN depends on the temperature chosen for the experiments, differing within a 10-fold range from 24°C to physiological temperatures (37°C) (Ref. Reference Badmalia11).
Regulation by intracellular pH
A change in intracellular pH may occur in different scenarios. A decrease of pH is a sign for apoptosis and associated processes in the cell (Ref. Reference Garg38). Recent studies demonstrated that low pH values can activate GSN even in the absence of calcium. A pH under 6 lowers the diffusion coefficient of GSN in comparison with a physiological pH of 7.5–8 (Ref. Reference Garg38). Furthermore, a pH of 5 leads to an opening of the inactive conformation of GSN and an interaction of the N-terminal half with actin. This conformation differs from a calcium-activated shape and is comparable with changes that take place at pH 8 and a calcium concentration of 40 nm (for more information see (Ref. Reference Garg38)). It was demonstrated that the formation rate of the GSN-actin complex is controlled by intracellular pH and that a reduction from pH 8 to pH 6 increases the complex forming rate 5-fold (Ref. Reference van Loon and Schatz39). Moreover, it was shown that in EGTA solutions (pH 7) no severing activity of GSN is detectable but at a lower pH (e.g. 6) an increase in severing occurs (Ref. Reference Lamb40). As mentioned above, there are differences between the calcium and pH-activated conformations of GSN. One of the major differences is the affinity of the G4 domain to actin, which is in a high state when calcium is activated but significantly lower in pH-activated GSN. A conformational change in the G2 domain is also initiated at pH 6, but in the opposite direction, as though calcium-activated, and could be a reason for the binding of G2 with G1 or G3 under low pH instead of binding with G6 (Ref. Reference Lagarrigue41).
PIP2 as a regulator
When GSN has formed a complex with actin it must be uncapped for further polymerisation (Ref. Reference Janmey and Stossel8). This uncapping is accomplished by PIP2 through inhibition of actin binding to GSN. Furthermore, existing bonds between actin and GSN can be dissipated (Ref. Reference Janmey and Stossel8). GSN residues from 135 to 142 and 150–169 are important for PIP2 binding, which is localised in the cell membrane. These residues build a β-strand with G1 and G2 (Ref. Reference Liepina27). Between the N-terminal half of GSN and the phosphate groups of PIP2 salt bridges occur and hydrophobic bonds are formed between the peptide and the lipid (Ref. Reference Liepina27). A high affinity of GSN to PIP2 can be seen and half-maximal inhibition is reached at a concentration of 1.7 µm PIP2 relative to 86 nM GSN (Ref. Reference Janmey and Stossel8). Overexpression of PIP2 leads to actin assembly and inhibition causes depolymeriszation (Ref. Reference Yin and Janmey42).
GSN as a component of cellular interactions
Besides its functions in actin regulation, GSN also acts as a regulator of cell metabolism (Ref. Reference Li2). The role of GSN in the modulation of ion channels was shown in vivo. GSN−/− neurons reveal a higher concentration of filamentous actin and the N-methyl-D-aspartate (NMDA) receptor-mediated influx of CA2+ is reduced by actin depolarisation-activated GSN (Ref. Reference Furukawa43). NMDA receptors and their hyper- and hypofunctions are associated with pathological neuronal processes (Ref. Reference Newcomer, Farber and Olney44). Apart from this, GSN is involved in the regulation of neuronal growth and the formation of cones. GSN participates in the retraction process of filopodia. Gsn− mice show a higher number of filopodia and it is suggested that in these mice an increase of filopodia extrusion occurs (Ref. Reference Lu45). However, GSN is not needed in the process of myelin sheet development, but is involved in the remyelination process of sciatic nerves after injuries. An increase of macrophages expressing GSN was found at the injury site. After 6 months, no significant difference in myelination was observed between mice with a sciatic nerve injury and control mice. This suggests that GSN is needed for recruitment of macrophages to injury sites (Ref. Reference Goncalves46).
Small GTPases like Ras and Rac are known to be central downstream components in membrane ruffling of fibroblasts (Ref. Reference Malliri47). It was demonstrated that GSN is a downstream regulator of Rac and that in absence of GSN actin dynamics are modulated. GSN− cells therefore show a 50% reduced motility as well as an overexpression of Rac. Additionally, cultured fibroblasts revealed large bundles of actin stress fibres (Ref. Reference Azuma48).
GSN in the immune response
There are two different ways the isoforms of GSN act in the immune system. Extracellular GSN takes part in the recognition of bacterial wall molecules and assaults on immune components. Besides this, intracellular GSN is important for macrophage recruitment and motility (Ref. Reference Goncalves46).
One of the different immune processes GSN is involved in is phagocytosis. Phagocytosis is an important instrument of our immune system to depredate microorganisms and damaged cells. This process is also involved in remodelling mechanisms of connective tissue matrices (Ref. Reference Arora49). Because remodelling processes in cells are based on rearrangements of actin and its filaments, one is inclined to accept that GSN, as an ABP, will be involved in this process (Ref. Reference Arora49).
Collagen-induced phagocytosis, which is related to α2β1-intergins, can be modulated by cGSN. In GSN− mice cells, collagen binding was 60% lower than in wild-type (WT) mice cells but could be restored after transfection with GSN. In the study of Arora et al. it is demonstrated that actin assembly by cGSN is an essential step in α2β1-intergin mediated phagocytosis (Ref. Reference Arora49).
The role of GSN in neutrophils was also studied in GSN-null mice, which were infected with complement opsonised or IgG-opsonised yeast. The phagocytosis of the complement-opsonised yeast was nearly the same as in WT mice but IgG-opsonised phagocytosis was decreased. This suggests that cGSN is essential for IgG-opsonised phagocytosis and additionally that the process of attachment and ingestion is actin-dependent. Other processes such as granule exocytosis, phagosome processing and activation of Nicotinamide Adenine Dinucleotide Phosphate Hydrogen (NADPH)-oxidases are not modulated in GSN-null mice. Therefore GSN draws a distinction between complement and IgG-mediated phagocytosis (Ref. Reference Serrander50). Besides this, the inhibition of GSN is also important for phagocytosis. The internalisation of collagen fibrils depends on actin-assembly steps in collagen phagocytosis. To achieve this assembly, GSN requires regulation by PIP2. This was demonstrated in GSN− fibroblasts, which were transfected with GSN-severing mutants (Ref. Reference Arora51).
During phagocytosis, GSN interacts with various immune system cell types. Macrophages that are released after development as monocytes from the bone marrow into the blood circulation leave the blood after a couple of hours (1–2 days), migrating into different tissues and organs. It was demonstrated that GSN is needed for this migration, at least in vitro. For recruitment to injury sites, GSN also plays a role in macrophages (Ref. Reference Goncalves46). When macrophages respond to pathogens, like Streptococcus species, it was observed that this response leads to an increase of GSN in the organism (Ref. Reference Fettucciari52).
Beside macrophages, other defense cells are also influenced by GSN, such as neutrophils. Neutrophils are part of the innate immune system and are a major component of the leucocyte pool. Activation of Rac by G-protein-coupled receptors in neutrophils promotes uncapping of actin filaments (Ref. Reference Arcaro53). GSN also takes part in the regulation and adhesion of neutrophils through β1 integrins (Ref. Reference Langereis54).
Another important cell type of the immune system is T-lymphocytes (T-cells). T-lymphocytes recognise antigens presented by antigen-presenting cells in the organism. Burn-induced suppression of T-cells is facilitated by treatment with recombinant human GSN. In addition, pGSN is also exogenously able to enhance the function of peripheral T-cells. In this context it was shown that treatment with GSN leads to a decrease of inflammatory cytokines like IL-1β and IL-6 in the brain (Ref. Reference Zhang55).
In mice, a subtype of adseverin (adseverin D5, a member of the GSN superfamily) was found that was induced by IL-9 producing T-cells. IL-9 therefore could upregulate the expression of GSN in T-cells (Ref. Reference Robbens56).
Apoptosis
GSN is involved in apoptosis in different ways. It can be inhibitory or supportive depending on the surrounding conditions and cell types (Refs Reference Geng57, Reference Koya58). Caspase 3, as mentioned above, can cleave GSN at its linker into two fragments: an N-terminal half with 39 kDa and a C-terminal half with 41 kDa (Ref. Reference Kothakota19). N-terminal fragments thus generated are able to sever actin but are also involved in the structural cellular changes that occur during apoptosis (Ref. Reference Kothakota19).
An important enzyme for apoptosis is DNase 1, which is mainly involved in DNA degradation (Ref. Reference Mannherz59). It was shown that GSN interacts with DNase 1 and forms a complex together with actin. In contrast, the N-terminal fragment can promote apoptotic activity by interference with the DNase-1-actin interaction (Ref. Reference Chhabra, Nosworthy and dos Remedios60).
GSN can also enhance apoptotic activity via the GSN-hypoxia-inducible factor 1 (HIF-1)-α-DNase 1 pathway. Through this pathway, GSN and the HIF-1 can interact, resulting in an increase of DNase 1 (Ref. Reference Li61).
In contrast to this, the C-terminal fragment of GSN in combination with PIP2 shows anti-apoptotic behaviour (Ref. Reference Li2). GSN was also found in processes of mitochondrial-dependent cell death with its ability to bind voltage-dependent anion channels resulting in the closure of these channels. The closure prevents cytochrome C release and inhibits caspase-3, resulting in an anti-apoptotic effect (Ref. Reference Li2). In this context it was shown that smooth muscle cells from GSN-null mice show resistance to apoptosis induced by inflammatory cytokines (Ref. Reference Geng57).
Knockout mice Gsn−/−
GSN knockout mice do not express any amount of pGSN or cGSN (Ref. Reference Witke26). One study shows, that it is not lethal for mice having a lacking GSN expression (Ref. Reference Li2), in another study the pups of BALB/c and C57/B6 die within a few days when a GSN gene defect was bred into their background (Ref. Reference Kwiatkowski3). In addition a study shows that in comparison with control mice, Gsn−/− mice have a decreased mortality rate, which contrasts with findings described in (Ref. Reference Kwiatkowski3).
Gsn−/− mice show a delay in the development of postnatal mammary glands (Ref. Reference Crowley62). Other impairments have also been found in mice lacking GSN, such as impaired platelet function and decreased migration of fibroblasts. The fibroblasts show more actin stress fibres and contractility. In addition, Gsn−/− mice do not show the formation of podosomes, which are dynamic and actin-rich structures formed by the plasma membrane of animal cells, and exhibit an inhibited motility of osteoclasts (Ref. Reference Chellaiah63). In GSN− mice also show a decreased risk of coronary artery hypertrophies. Cardiac function is increased in these mice and they show a reduced infarction rate as well as interstitial fibrosis (Ref. Reference Li61).
Because of these findings, GSN has been reported to be essential for processes such as wound healing, homeostasis and inflammation (Ref. Reference Witke26). Suggestions have also been made that other members of the GSN superfamily compensate GSN knockout in mice (Ref. Reference Silacci4).
Role of GSN with regard to major diseases
GSN is an interesting protein in current research. Because of its various roles in many cellular pathways, cell shape and viability in general, GSN is involved in several diseases and is targeted to understand disease-related mechanisms. In healthy mammals, pGSN has a level of 200 ± 50 mg/l in blood plasma (Ref. Reference Osborn64), but is reduced in the course of several diseases (Refs Reference Lee65, Reference Suhler66) as described below.
GSN and cancer
Reflecting the regulatory effects of GSN in cell motility, growth and apoptosis, it is a much-discussed topic in current cancer studies. Nevertheless, many results generated to date are controversial regarding the role of GSN in different carcinomas.
In breast cancer, cGSN seems to promote the growth of metastases when overexpressed, whereas a combined overexpression of cGSN and Nm23-H1 (a metastasis suppressor gene) significantly reduces the motility of tumour cells and growth of metastases in murine breast cancer cells. Interactions between Nm23-H1 and cGSN plus an additional overexpression of Nm23-H1 have a significant impact on the severing and modifying of actin fibres through GSN (Ref. Reference Marino67). However, the activity of cGSN promoters is lower in breast cancer cells than in healthy controls (Ref. Reference Tanaka68). Overexpression of cGSN further results in a variation of cell proliferation and cell cycle (Ref. Reference Chen69). In contrast to previously mentioned publications, it was shown in a clinical trial that rising tumour grades of breast cancer are detectable with decreasing concentrations of cGSN (Ref. Reference Baig70).
Expression of GSN is nearly undetectable in human bladder cancer. Overexpression of cGSN in such cells leads to a loss of tumourigenicity. As a result of this, it is suggested that cGSN functions as a tumour suppressor in urinary bladder carcinomas (Ref. Reference Tanaka71). On the other hand, overexpression of cGSN in hepatocellular carcinomas (HCC) promotes cell proliferation in tumour cells. Moreover, knockdown by siRNA results in decreased cell invasion and viability in these cells (Ref. Reference Deng72). mRNA levels of GSN are significantly higher in HCC metastasis tissue than in controls. GSN is increased in HCC tumours and is therefore suggested as a potential oncogene for HCC (Ref. Reference Deng72).
Consistent with this, increased cGSN levels are found in patients with chemoresistance to gynaecological cancers and a higher expression of cGSN decreases the survival rate (Ref. Reference Abedini73). Furthermore, studies of renal cell carcinomas reveal for cGSN, in the context of p53 and NF-KBcyt, a correlation with aggressive tumour behaviour (Ref. Reference Kankaya74).
These different studies show that the role of GSN in cancer depends on the carcinoma type under investigation. It has therefore been suggested that GSN functions as both tumour suppressor gene and oncogene. In any case, both roles mark GSN as a promising target for therapeutic interventions.
GSN and the eye
Vision is one of the most important senses for modern humans. The cornea, the ‘window’ of the eye, is transparent. The cornea consists of epithelium, stroma and endothelium (Wittmann 2018, in revision). Several diseases or injuries can impair the cornea, such as Steven-Johnson syndrome, a systemic inflammatory disorder that can impair the re-epithelialisation of the cornea (Ref. 75). Because of the special role of GSN in cell motility and wound healing, it is of interest how GSN acts at the ocular surface.
GSN was found concentrated in the corneal epithelium and presents as a prevalent water-soluble protein in the transparent cornea of zebra fish. It has been suggested that cGSN might have a ‘crystalline-like role’ in the cornea that is important for vision and transparency (Ref. Reference Xu76). Moreover, it was found that GSN is needed for optimal dorsalisation in zebra fish during embryogenesis (Ref. Reference Kanungo77).
Studies in rabbits revealed immuneoreactivity of GSN in retinal photoreceptors. It was also suggested that GSN plays a role in neuronal morphogenesis (Ref. Reference Fried and Risling78). Previous studies also showed that GSN is a component of oligodendrocytes of rabbits and is built into the medullary ray region of the retina (Ref. Reference Fried and Risling78).
GSN and amyloidosis
Interestingly, GSN can also be associated with distinct ocular diseases in humans. Type 2 lattice corneal dystrophy is caused by a mutation in the GSN gene that is relevant for the G2 domain. This mutation leads to a release of amyloid fragments inside the eye (Ref. Reference Carrwik and Stenevi79). Another form of an amyloidosis is the familial Finnish type (Meretoja's syndrome, FAF). This syndrome is characterised by mask-like expressions of the patients and corneal lattice amyloid deposits (also known as familial amyloid polyneuropathy type IV). GSN is associated with the disease based on a mutation at position 187 in the GSN gene. This form of amyloidosis and others were tested for their GSN immunoreactivity (Ref. Reference Loeffler, Edward and Tso80). Controls revealed moderate or higher immunoreactivity of anti-GSN in the epithelium and endothelium of the cornea. Different types of amyloidosis showed of strong anti-GSN labelling in the corneal epi- and endothelium. It was therefore concluded that GSN must be present in the amyloid deposits on different types of corneal amyloidosis (Ref. Reference Loeffler, Edward and Tso80). Patients diagnosed with FAF tested positive for GSN-c68, a 68 kDa fragment of GSN, in their cerebrospinal fluid (CSF) (Refs Reference Maury81, Reference Paunio82). The autosomal, dominantly, inherited form of amyloidosis, hereditary GSN amyloidosis, is also caused by a mutation of GSN (Ref. Reference Kiuru-Enari and Haltia83).
Our own investigations demonstrated GSN production in human and murine cell lines of the ocular surface (cornea, conjunctiva, meibomian gland) as well as in different tissues of the ocular surface (i.e. lacrimal gland, cornea, conjunctiva, efferent tear ducts, meibomian gland). The GSN concentration was significantly higher than in samples from lung, liver or stomach. Moreover, it was found that the pGSN level in tears from patients suffering from aqueous-deficient dry eye or evaporative dry eye was significantly higher than in tears from healthy volunteers. Focusing on the cellular proliferative functions of pGSN it was demonstrated that pGSN promotes cell proliferation and supports wound healing in a murine cornea defect model (alkaline burn). These studies showed the supportive effect of recombinant (r) human (hu)-pGSN during corneal wound healing, suggesting a great potential of rhu-pGSN in the treatment of ocular surface diseases such as dry eye disease (Wittmann 2018, in revision). An important regulatory factor of wound healing is transforming growth factor β (TGF-β), which also significantly impacts the genetic expression of GSN (Ref. Reference Chen69). Thus these two aspects must be considered together in the context of wound healing.
GSN in AD
AD is a degenerative disease of the brain affecting mainly elderly persons. Amyloid-β-protein is a soluble protein under normal conditions; in pathological conditions the protein becomes fibrillated and forms plaques in the brain (Ref. Reference Yang84). These plaques are formed by the amyloid-β-fibres Aβ 1–40 and Aβ 1–42 (Ref. Reference Yang84). Formation of amyloid-β-plaques (Aβ) in the brain is a characteristic sign of AD that represents serious progress of the disease (Ref. Reference Antequera85). Studies of AD in combination with GSN revealed that GSN acts as an anti-amyloidogenic protein (Ref. Reference Yang86). This is interesting as Aβ has toxic effects on mitochondria and can induce apoptosis. GSN binds to this Aβ and inhibits induction of apoptosis. Moreover, it was shown that cGSN, as well as pGSN, clearly influences the distribution of Aβ in the brain (Ref. Reference Antequera85).
GSN can bind to the amyloid-beta protein, inhibiting its fibrillation and also dissolving fibrils that have already formed (Ref. Reference Yang86) as well as contributing to the removal of Aβ from the brain (Ref. Reference Yang84). In combination with trichostain A, a histone deacetylase inhibitor, the expression of GSN increases significantly (Ref. Reference Yang86). In female AD transgenic mice, pGSN levels increase by 82% and by 76% in male mice (Ref. Reference Yang84). The plasma levels of Aβ 1–40 and Aβ1–42 correlate with plasma levels of GSN, leading to the suggestion that pGSN can be a helpful target to clear Aβ out of the brain and other related tissues (Ref. Reference Yang84). It was also shown that pGSN forms complexes with Aβ in the plasma (Ref. Reference Chauhan, Ji and Chauhan87). Oxidative stress due to excessive amounts of free radicals in the metabolic process, also results in an increase of GS, suggesting that GSN functions as an antioxidant.
Patients with AD have a 30% lower concentration of GSN in the choroid plexus than controls (Ref. Reference Antequera85) and both pGSN and metalloproteinase 3 (MMP3) are found in blood plasma of AD patients. It is thought that because of reduced GSN levels and increased MMP3 levels in AD patients, these proteins could function as a biomarker in AD (Ref. Reference Peng, Jia and Qin88). For specific binding of Aβ, GSN has two binding sites (Ref. Reference Chauhan, Ji and Chauhan87) and is therefore suggested as a means of evaluating Aβ levels in AD (Ref. Reference Yu89). Additionally, MMP3 can cleave GSN into fragments of 43–48 kDa (Ref. Reference Li61). A fragment at 47 kDa was also just recently detected in a study of GSN functions at the ocular surface (Wittmann 2018, in revision).
Further studies suggest that GSN can be a therapeutic target with regard to its role in fibrillation of Aβ. It was shown that the import of plasmid DNA, which codes for pGSN, has a positive effect on Aβ pathology. However, it must be taken into consideration that an increase of pGSN could be associated with neurotoxicity based on microglia activation and the oligomeric forms of amyloid (Ref. Reference Hirko90).
GSN in arthritis
Chronic autoimmune diseases like rheumatoid arthritis (RA) are a common problem in industrialised nations. RA affects the synovial lining of diarthrodial joints followed by chronic and persistent joint inflammation. There are two other forms of the disease besides RA: osteoarthritis and the spondylarthritis (Ref. Reference Casey91). Arthritis can be a chronic disease that can also be induced by a bacterial or viral infection (Ref. Reference Casey91).
Reduced levels of pGSN have been detected in patients suffering from RA in synovial fluid and in their blood plasma. It has been suggested that pGSN is depleted in the inflamed joint and that a high actin concentration is the reason for this decrease of pGSN levels (Ref. Reference Osborn64). This high actin concentration is caused by an injury, which releases actin into the extracellular matrix (where it forms filaments). These filaments induce downstream inflammatory processes. Therefore, the clearance of the filaments is important (Ref. Reference Osborn64). Furthermore, it is suggested that pGSN may be a ‘broad-spectrum anti-inflammatory buffer’ in this context (Ref. Reference Osborn64).
pGSN levels show a negative correlation with analysed levels of C-reactive protein (CRP) (Ref. Reference Osborn64). CRP is an important component of the immune system based on its role in cellular and humoral defense mechanisms. In this context, levels of pGSN were tested in correlation with IL-6, but no significant correlation was determined. Furthermore, it was shown that pGSN activity was reduced in synovial fluid of RA patients (Ref. Reference Osborn64). In comparison with healthy controls, RA patients revealed higher concentrations of actin and GSN-actin complexes in synovial fluid (Ref. Reference Osborn64).
In contrast to these results it was demonstrated that GSN expression is increased 5-fold in the synovial fluid of RA patients compared with patients suffering from osteoarthritis (OA) (Ref. Reference Biswas92). Also, CRP concentrations were 2–12 times higher in non-erosive RA patients and 42–147 times higher in erosive RA patients than in OA patients (Ref. Reference Liao93). Furthermore, it was shown that serum amyloid A is even more sensitive in inflammatory lesions found in RA (Ref. Reference Chambers94). However, present experts believe that the pGSN level decreases in RA patients (Ref. Reference Wood95).
Gsn−/− mice that suffer from experimentally induced RA show synovial membrane hyperplasia and overall exacerbation of RA. It is suggested that cytoskeletal rearrangements by GSN may act as a determinant for RA (Ref. Reference Aidinis96).
GSN in other diseases
Involvement of GSN has been described in many different diseases. Here, we review some distinct implications of GSN in chronic kidney disease (CKD), diabetes type 2, critically ill patients and multiple sclerosis.
In CKD, low pGSN levels are detectable in hemodialysed blood. This raised the question as to whether circulating pGSN levels can serve as a biomarker since pGSN levels correlate negatively with the degree of systemic inflammation and muscle wasting (Ref. Reference Lee, Bhan and Thadhani97). Further studies revealed that the survival rate of hemodialysis patients decreases when low pGSN levels are detected (Ref. Reference Lee98). Another disease where decreasing levels of pGSN play a role is type 2 diabetes. Such results were obtained in a mouse model of type 2 diabetes as well as in human patients (Ref. Reference Khatri99).
Interestingly, it has been shown that surgically treated patients who are critically ill are at higher risk of dying if their pGSN levels are low. A concentration below 61 mg/l pGSN is thought to be critical and is considered a landmark for higher mortality (Ref. Reference Lee100).
Depletion of pGSN can also be found in patients suffering from multiple sclerosis (MS) (Ref. Reference Li-ChunHsieh101). CSF of MS patients tested positive for pGSN, even if only low levels were detected (2.1 ± 0.7 µg/ml) compared with other diseases that do not affect CSF (7.2 ± 4.3 µg/ml) (Ref. Reference Kulakowska102). This leads to the suggestion of a depletion of GSN, albeit not accompanied by an increase of circulating actin in the CSF (Ref. Reference Kulakowska102). Because of this it is suggested that GSN in CSF could function as a marker for neuropathologies (Ref. Reference Kulakowska102).
Other studies show that GSN is also important for therapeutic applications in MS. Vitamin D, a therapeutic tool for MS, causes down-regulation of GSN in rats suffering from experimental autoimmune encephalomyelitis (EAE), the animal model of MS. Therefore, overexpressing GSN-lentiviruses are used to increase GSN levels in rats. Treatments with these viruses show a positive effect on the EAE course. Furthermore, the combination of vitamin D and overexpressing GSN-lentiviruses causes a reduction of inflammation and even a delay in the onset of EAE in tested rats (Ref. Reference Gao103).
GSN as a therapeutic target
As mentioned above, GSN can have various functions in different diseases and can therefore act as a therapeutic target. However, as described for GSN involvement in cancer, the therapeutic applicability of GSN depends on the cancer subtype. Overexpression or depletion of GSN might reveal future therapeutic potentials. In Ras-induced tumours, it was shown that mutated cGSN can suppress tumour growth (Ref. Reference Mullauer104). Nevertheless, much more research with regard to cancer is needed to clarify the possible roles of GSN as a therapeutic agent.
GSN shows considerable potential for treatments of other diseases. In studies related to type 2 diabetes it was observed that in mice a daily dose of pGSN could keep sugar levels within a normal range. Furthermore, improvements of pGSN levels and reduction of blood glucose levels were observed with sitaglipitin, a hyperglycaemic managing drug, (Ref. Reference Khatri99). GSN treatment also reveals significant improvement in mice suffering from hyperoxia in comparison with control groups (Ref. Reference Christofidou-Solomidou105). Another positive effect of rhuGSN was found in the treatment of inflammation-induced lung injuries (Ref. Reference Rothenbach106).
Initial clinical results in patients treated with GSN were published in 2017 for therapeutic applications in RA and Achilles tendinopathy. The method is based on an increase of GSN and other proteins, cytokines and growth factors. First clinical results revealed effects in patients suffering from Achilles tendinopathy. Following treatment the pain scores of patients were significantly lowered in the longer term. Full regeneration of the tendon was seen after 1 year in all patients treated (Ref. Reference Schneider, Wallich, Felmet and Murrel107).
Conspectus
GSN is a multifunctional protein that is involved in many cellular mechanisms and processes in organisms. It has important functions in phagocytosis and apoptosis as well as in signalling and cell regulation. Interaction of GSN with specific immune system cells reveals the complexity of this system and shows how one protein can change various cell processes. This is also apparent in certain diseases in which GSN is involved. In cancer, overexpression or depletion of GSN can worsen the course of the disease. Low pGSN expression levels are found in AD and other diseases, suggesting a function of GSN as a biomarker or potential therapeutic target. First approaches are underway to using GSN as a therapeutic target and developing new therapies for diseases that have no cures to date.
All in all, GSN has the potential to become a useful therapeutic tool for many diseases (see Fig. 4). However, further research is required and compensatory functions of GSN superfamily members must be considered in the development of new medical products.

Fig. 4. Summary of gelsolin functions, related diseases and molecular structure.
Author ORCIDs
Jessica Feldt, 0000-0002-4205-263X.
Acknowledgements
The authors would like to thank Hong Nguyen and Jörg Perkarsky for their help with the pictures and figures. This research received no specific grant from any funding agency, commercial or not-for-profit sectors. The authors declare no conflict of interest.
Useful resources
For more information about the 3D structure and the genetic neighbourhood of gelsolin, please visit: www.string-db.org
If you wish to inform yourself about cellular processes gelsolin is involved in, please visit: www.uniprot.org.






