


Mycobacterium avium complex (MAC) comprises eight bacterial species and a number of subspecies with a different degree of pathogenicity, host preference and environmental distribution [Reference Cayrou1]. Among them, Mycobacterium avium subsp. hominissuis (MAH) [Reference Mijs2] is the most widely distributed and it has been isolated from many host species and environmental samples [Reference Biet3]. MAH is an opportunistic pathogen that has acquired an increasing importance in public health in recent decades due to its ability to cause pulmonary disease, lymphadenitis in children and disseminated infections in immuncompromised patients [Reference Falkinham4].
Although MAH can infect a wide variety of animals, swine is its primary animal host species, causing granulomatous lesions mainly in lymph nodes of the digestive tract [Reference Thorel5] which can reduce the value of carcasses. MAH has been recovered from swine samples worldwide [Reference Matlova6–Reference Komijn8], although due to the absence of clinical disease, it is normally detected during meat inspection in abattoirs. This results in economic losses due to condemnation of meat of animals with macroscopical lesions (Regulation 2004/854/EC) and possible restrictions on the sale and movement of animals from infected farms [Reference Matlova9]. Differential diagnosis with M. tuberculosis complex infection should be performed when granulomatous lesions in lymph nodes are observed at slaughterhouses. Finally, the potential risk of infection of immunocompromised patients with this zoonotic emerging pathogen through consumption of insufficiently cooked pork meat remains to be determined [Reference Pate10]. In spite of the fact that Spain is currently one of the main pork producers in the European Union and holds 16·3% of the total European swine census [11], we are unaware of any reports regarding incidence of infection in this animal species. Official statistics only revealed MAH involvement in 16 samples with lesions out of 27 investigated samples in 2008; in 2007 no MAH isolation was achieved from 218 samples with lesions (Source: Spanish Ministry of the Environment and Rural and Marine Affairs).
Due to the pathogen's wide environmental distribution many possible sources of infection for swine can be often identified, usually making epidemiology of MAH infections complex. For this reason the application of molecular characterization techniques in order to compare clinical and environmental isolates is a powerful epidemiological tool that can sometimes clarify the origin of infection, and has also demonstrated that MAH is the most variable subspecies of MAC [Reference Mijs2, Reference van Soolingen12, Reference Turenne13]. Among typing methods, restriction fragment length polymorphism analysis (RFLP) using insertion sequence IS1245 has been one of the most widely applied tools [Reference Komijn8, Reference Matlova9]. However, the existence of MAH strains that harbour low numbers (or none) of this element [Reference Ritacco7, Reference Komijn8, Reference Alvarez14] can impair the discriminatory power of this test. An alternative characterization technique, pulsed-field gel electrophoresis (PFGE), which is independent of insertion sequences, has also been widely applied on MAH isolates [Reference Tobin-D'Angelo15]. Still, both techniques share the common disadvantage that large amounts of DNA are required for their performance. For this reason PCR-based tools have been developed for identification and typing of MAC isolates, these include hsp65 sequencing [Reference Turenne13], detection of long sequence polymorphisms [Reference Semret16] and the study of variable-number tandem repeats (VNTRs) [Reference Thibault17]. These techniques are fast and more convenient to perform but can have lower discriminatory power. Some of these techniques have reported close genetic relatedness between human and porcine MAH isolates [Reference Komijn8, Reference Pate10] suggesting either a common source of infection or a possible transmission from pigs to humans, although this possibility has never been demonstrated.
The current study describes the application of several molecular characterization techniques to describe the epidemiology of an outbreak involving 10 related pig farms in order to identify the sources of infection and to introduce corrective measures. From November 2007 to March 2008 granulomatous lesions in submandibular and mesenteric lymph nodes were detected at abattoir inspection in pigs from six fattening farms (1–6) located in central Spain. All these animals were born and weaned on four breeding farms (I–IV) sharing the same veterinary team and food suppliers. Heads and in some instances whole carcasses were condemned, causing severe economic losses to the farmers [i.e. up to 27% (60/220) of the carcasses sent to the slaughterhouse were rejected].
Fifteen animals coming from the affected fattening farms were sampled at the abattoir, and samples were submitted to our laboratory to identify the causative agent of the outbreak. Samples were collected from affected lymph nodes and were processed for culture as described previously [Reference Corner and Trajstman18] and inoculated onto blood agar, Coletsos, Löwestein–Jensen and Herrold's egg-yolk media (bioMérieux España, Spain). Isolates were identified by acid-fast staining and amplification of Mycobacterium genus and MAC-specific DNA targets [Reference Boddinghaus19] and insertion sequences IS901 [Reference Kunze20] and IS1245 [Reference Guerrero21].
After incubation for a period of up to 3 months, acid-fast rod growth was observed in 13/15 cultured clinical samples (Table 1). All isolates were identified as MAH by detection of specific DNA fragments of 16S rDNA, absence of IS901 and presence of the IS1245 element.
Table 1. Molecular characterization results from the 16 isolates cultured from clinical (n=13) and environmental (n=3) samples
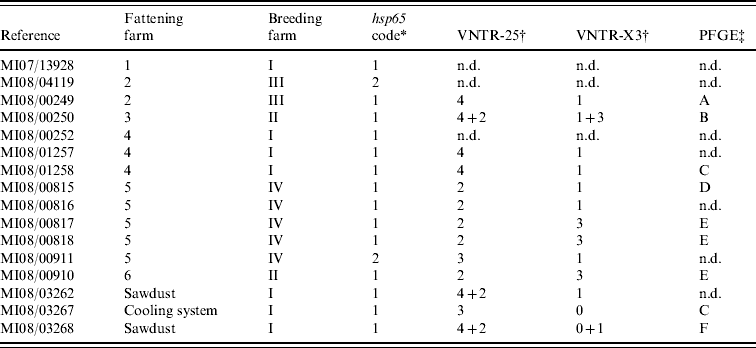
n.d., Not determined.
* According to Turenne et al. [Reference Turenne13].
† Number of repetitions found at each loci where more than one pattern was found.
‡ Unrelated profiles were defined according to Tenover et al. [Reference Tenover24].
Once all isolates from animal samples were identified, an environmental sampling was performed in one breeding farm (farm I) and one fattening farm (no. 4) that received piglets from this breeding farm in order to evaluate potential sources of infection for animals. Samples collected (n=15) included feed, sawdust and water from different locations, and from several humidified cellulose sheets acting as filters in cooling systems. Samples and isolates were analysed as described above. Positive cultures were obtained from four samples collected at breeding farm I: three were MAH isolates from sawdust (n=2) and cooling system (n=1) samples; the fourth isolate, cultured from a drinking trough sample, was identified as an M. chelonae based on sequencing of the 16S rDNA gene [Reference Boddinghaus19] and hsp65 gene [Reference Swanson22]. All samples from farm 4 were negative.
The 3' region of the hsp65 gene was amplified and sequenced on all MAH isolates as described previously [Reference Turenne13]. Tandem repeats (VNTRs) analysis was performed on a subset of clinical (n=11) and environmental (n=3) isolates (Table 1) as described by Frothingham & Meeker-O'Connell with slight modifications [Reference Frothingham and Meeker-O'Connell23] using four loci (X3, 25, 32, 292) that had been previously described as polymorphic in M. avium isolates [Reference Thibault17]. The resulting PCR amplicons were analysed by direct visualization on a 2·5% agarose gel to analyse polymorphisms in the number of tandem repeats, and four of the PCR amplicons were sequenced to confirm the number of repetitions present in the different amplicons.
In addition all clinical (n=13) and environmental (n=3) MAH isolates were subcultured on broth (Middlebrook 7H10 Agar, Becton Dickinson and Company, Spain) and subjected to PFGE analysis as described previously [Reference Alvarez14] using the restriction enzyme XbaI. The profiles obtained were visualized in gels stained with SYBR green (Invitrogen S.A., Spain) and interpreted according to the criteria proposed by Tenover et al. [Reference Tenover24]: profiles were considered closely related if differences between them involved no more than 2–3 bands.
hsp65 sequencing performed on clinical isolates revealed two sequevars (Table 1): code 1 was sequenced from isolates of all fattening farms and two isolates from different farms were code 2 sequevar. All three environmental isolates presented a code 1 sequevar. MAH strains containing hsp65 sequevars codes 1 and 2 have already been isolated from human, swine and environmental samples [Reference Turenne13, Reference Alvarez14], highlighting their wide distribution.
In VNTR analysis, all isolates showed the same number of repetitions at loci 292 (two repetitions) and 32 (eight repetitions). Therefore all polymorphisms were limited to loci 25 and X3 (three different profiles in each one, yielding eight different possible patterns in combination). In three isolates double profiles were obtained (Table 1). VNTR analysis showed good discriminatory power, as previously reported [Reference Thibault17], although no variability was observed in two loci. Although VNTR technique is fast and easy to perform, it has not been much applied on MAH strains, making it difficult to interpret the results in some cases.
From the 16 isolates analysed by PFGE, readable patterns were obtained for nine (Table 1), as due to the necessity of large amounts of high-quality bacterial DNA it was impossible to type five of the isolates. From the six different profiles identified (patterns A–F), only two were present in more than one isolate: pattern E was observed in three clinical samples from two different farms and pattern C in one clinical and one environmental strain from the cooling system. PFGE was able to discover differences in isolates belonging to the same VNTR group (MI08/00249 and MI08/01258); conversely, VNTR analysis differentiated two cultures with the same PFGE pattern (MI08/01258 and MI08/03267), showing the complementarity of these techniques. The variability found in this panel of isolates using VNTR analysis and PFGE confirms the high genetic variability described previously in this subspecies [Reference Turenne25].
The comparison of the molecular profiles obtained by the characterization techniques applied to the clinical and environmental isolates revealed certain shared features. If mixed profiles are considered, VNTR profile 4–1 could be observed in two isolates cultured from sawdust from farm I and in two pigs born on this farm, suggesting a possible epidemiological link. Whereas identical PFGE profiles were observed in a strain isolated from the cooling system and one isolate cultured from a pig born on the same farm, the VNTR profiles of these two isolates were different. However, based on the high discriminatory power of XbaI PFGE these two strains are most likely epidemiologically related, and the variation in the two loci studied by VNTR analysis may simply reflect high variability rates already described in certain targets of MAH strains [Reference Pestel-Caron and Arbeit26].
Clinical isolates from animals from fattening farms 2 and 4 showed the same VNTR profile (4–1) (Table 1), although two cultures from the two farms typed by PFGE had different patterns (A and C), suggesting that at least two different strains were involved on both farms. Three VNTRs and two PFGE profiles were observed on five isolates from farm 5 (Table 1), indicating a complex epidemiological situation. There was only one strain (VNTR ‘2-3’ and PFGE ‘E’) isolated from clinical samples from two different farms (nos. 5 and 6). The high discriminatory power obtained using PFGE with the restriction enzyme XbaI combined with VNTR analysis indicates the possibility that these infections had a common source, even though pigs came from different production and fattening farms.
The fact that MAH was isolated from almost all clinical samples (Table 1) confirms that this is the MAC member usually associated with the production of granulomatous lesions in swine. Moreover, isolation of MAH from three samples from a panel of environmental samples revealed that a number of potential sources of infection were present in breeding farm I. Certain genes that could be related with virulence in MAH strains have been recently characterized [Reference ackenzie27, Reference Jha28]; further studies involving investigation on the presence of these genes in both environmental and clinical strains would be necessary in order to determine if MAH isolates cultured from clinical samples show some potential virulence markers. The implication of sawdust in the origin of outbreaks due to MAC members has already been described [Reference Matlova6]. Our results are in agreement with that report, as in the current study MAH was isolated from two different batches of stored sawdust that was later used for the bedding of newborn piglets. Therefore piglets were exposed to environmental MAH on their first days of life, when they are more susceptible to bacterial infection. Moreover, the isolation of another strain from the cellulose used in one of the cooling systems reveals the importance of this kind of equipment in the dissemination of the disease, since the fan could spread this strain to an entire group of piglets. This represents an important risk factor, as coolers are used in many areas of Spain due to the high temperatures during summer months. Finally, the isolation of M. chelonae, a conditionally pathogenic mycobacterium, from one drinking trough revealed another possible risk for animals. M. chelonae has been reported as an occasional causative agent of granulomatous lesions in pigs [Reference Matlova6], and its presence in the drinking water reveals insufficient disinfection of the water distribution system or an environmental contamination of the drinking troughs.
The current report describes an outbreak due to MAH infection affecting ten swine farms in central Spain. Lack of data regarding involvement of this bacterial species in large outbreaks in Spain (causing severe economic losses) made this case unusual. Application of different molecular characterization techniques suggests a large number of strains circulating in these pig farms, some of which could cause macroscopical lesions, and excluded a possible role of zoonotic M. tuberculosis complex members in the causation of granulomatous lesions. Comparison with environmental isolates cultured from one of the breeding farms involved in the outbreak revealed the potential sources of mycobacterial infection for piglets, as environmental isolates shared certain genetic features with clinical strains, therefore highlighting their possible implication in the epidemiology of the outbreak. The identification of these potential sources of infection allowed their removal or disinfection, this enabled the control of the outbreak, as no more lesions were subsequently observed in pigs from these farms at abattoir inspection. Our results show the need for implementation of good hygiene measures on pig farms in order to minimize contamination of the environment due to MAH, and therefore decrease the risk of infection for animals.
ACKNOWLEDGEMENTS
This research was funded by the EU project ParaTBTools FP6-2004-FOOD-3B-023106. We thank the farmers and veterinary practitioners involved in the management of the outbreak and collection of samples for their assistance, and appreciate the technical help of F. Lozano and N. Moya. J. Álvarez, is currently affiliated with Instituto de Investigación en Recursos Cinegéticos IREC, Ciudad Real, Spain.
DECLARATION OF INTEREST
None.

