



Introduction
Adipose tissue is an endocrine and metabolically active organ, which regulates feeding and metabolism in response to energy variations(Reference Lee, Mottillo and Granneman1,Reference Sanchez-Gurmaches and Guertin2) . Adipose tissue dysfunction causes obesity, which is associated with metabolic disorders such as the metabolic syndrome, representing one of the most important public health problems worldwide(Reference Pellegrinelli, Carobbio and Vidal-Puig3).
The prevalence of obesity around the world has increased at an alarming rate, particularly in children and adolescents. The WHO estimates that if current trends continue, the number of overweight infants and young children globally will increase to 70 million by 2025(Reference Blüher4).
During childhood, the most rapid weight gain occurs between the ages of 2 and 6 years, and 90 % of children with obesity at the age of 3 years present overweight or obesity during adolescence(Reference Whitlock and Lewington5). Thus, an increasing prevalence of childhood obesity is associated with the emergence of co-morbidities previously considered to be ‘adult’ diseases, including type 2 diabetes mellitus(Reference Taveras, Rifas-Shiman and Sherry6).
Currently, a large number of pregnant women are affected by overweight or obesity(Reference Chen, Xu and Yan7). Pregnancy in an obesogenic environment and fetal exposure to malnutrition leads to an increased risk of developing metabolic complications in the future, both for the pregnant women and their offspring(Reference Young, Johnson and Krebs8–Reference Usta, Usta and Yildiz10). Consequently, genetic predisposition, nutrient-regulated gene expression and epigenetic modifications together with environmental factors during intra-uterine and postnatal development play an important role in the development of adult diseases(Reference McMillen, Muhlhausler and Duffield11–Reference Barker13). This concept, now termed as the ‘developmental origins of health and disease’ (DOHaD) proposes that the homeostatic system affected during gestational and postnatal development impedes the ability to regulate body weight after birth, particularly in the face of high energy intake, resulting in adult obesity and metabolic diseases(Reference Barker12,Reference Kelishadi, Poursafa and Jamshidi14) .
In this line, we have conducted an extensive review based on adipose tissue as an endocrine organ and its physiology and development, with the current knowledge of maternal influences during the perinatal period that may lead to obesity in early postnatal life and adulthood.
The biology of adipose tissue
All animal species, from Caenorhabditis elegans to Homo sapiens, store energy in the form of fat(Reference Gesta, Tseng and Kahn15). Although adipose tissue was classically considered an energy-storing site, it is now widely recognised as an endocrine organ capable of regulating feeding and metabolism through hormone and cytokine secretion(Reference Sethi and Vidal-Puig16). Adipose tissue is composed of adipocytes as well as stromal cells, immune cells, endothelial cells, blood vessels and adipocyte precursors(Reference Sethi and Vidal-Puig16,Reference Stephens17) .
Two well-characterised types of adipose tissue coexist in mammals: white and brown. White adipose tissue is composed mostly of adipocytes containing a large unilocular lipid droplet. Brown adipose tissue specialises in energy expenditure; its adipocytes are rich in mitochondria and contain multiple smaller (multilocular) lipid droplets(Reference Sanchez-Gurmaches and Guertin2). Recently, a third type of adipose tissue was described: the inducible ‘brown-like’ adipocyte, also named ‘brite’/‘beige’, ‘browning’ or induced brown adipose tissue. Such adipocytes are inserted into the white adipose tissue of human and mice(Reference Di Franco, Guasti and Mazzanti18,Reference Nedergaard, Bengtsson and Cannon19) . ‘Beige’ adipocytes have an overlapping, but also a distinct gene expression pattern compared with classic brown adipocytes. Both brown and beige adipocytes express a core programme of thermogenic and mitochondrial genes, including Ucp1 (uncoupling protein 1), but murine beige cells also express the surface markers CD137 (TNF receptor superfamily member 9) and Tmem26 (transmembrane protein 26). Other marker genes, such as Zic1 (Zn finger of the cerebellum family member 1), are expressed in classic brown adipocytes but not beige cells(Reference Waldén, Hansen and Timmons20,Reference Beranger, Karbiener and Barquissau21) . To date, the origin or precursor cells of beige adipocytes is still unclear(Reference Di Franco, Guasti and Mazzanti18).
Adipose tissue distribution is variable between individuals as a function of genetics, sensitivity to hormones, sex and race(Reference Sanchez-Gurmaches and Guertin2,Reference Rosen and Spiegelman22) . Additionally, the cellular composition of adipose tissue, adipocyte morphology and the adipose tissue fat depot location define its metabolic and endocrine functions(Reference Pellegrinelli, Carobbio and Vidal-Puig3,Reference Macotela, Emanuelli and Mori23) . At the molecular and biochemical levels, adipocytes are well equipped with the machinery to respond to both hormonal (for example, insulin) and sympathetic (for example, adrenergic) stimulation(Reference Bartness, Kay Song and Shi24,Reference Bartness, Liu and Shrestha25) . Moreover, adipose tissue has high plasticity, being the only tissue in the body that can markedly change its mass after adult size is reached(Reference Kozak, Koza and Anunciado-Koza26).
White adipose tissue is organised in discrete anatomical depots identified as subcutaneous and visceral adipose tissue; the first is distributed below the skin, while visceral adipose tissue is located in the trunk cavity in humans and mice(Reference Sanchez-Gurmaches and Guertin2). During times of increased food intake and/or decreased energy expenditure, surplus energy is deposited efficiently in white adipose tissue in the form of neutral TAG. When food is scarce and/or energy expenditure requirements increase, lipid reserves are released to provide fuel for energy generation. TAG from adipose tissue break down into glycerol and fatty acids, which are transported in the blood to the liver, muscle and brown adipose tissue, where they are used in fatty acid oxidation. As such, white adipose tissue plays a key role not only by ensuring efficient energy storage but also by rapidly mobilising lipids to ensure peripheral demands through a coordinated endocrine regulation between the nervous system(Reference Sethi and Vidal-Puig16).
On the other hand, brown adipose tissue is distributed in axillary, cervical, perirenal, periadrenal and interscapular regions, actively participating in basal and inducible thermogenesis(Reference Cypess, White and Vernochet27). For a long time, it was thought to be present only in rodents and newborn humans. However, recent investigations using 18F-fluorodeoxyglucose positron emission tomography after cold exposure, lactation and exercise re-discovered the presence of brown adipose tissues (brown adipose tissue and induced brown adipose tissue) in adult humans(Reference Cypess, White and Vernochet27–Reference Cypess, Lehman and Williams29).
Several studies demonstrated that increasing brown/beige adipose mass could be a potential therapeutic approach to treat obesity(Reference Cypess, Lehman and Williams29). The confirmation that adult humans display highly metabolically active brown adipose tissue raises the possibility of controlling its growth and/or energy expenditure in obesity(Reference Daquinag, Tseng and Salameh30,Reference Schulz, Huang and Tran31) . Nevertheless, this conception may be limited by the uncertainty regarding the identity and origins of adipocytes from different depots and poor information available about how obesity-associated changes in cellularity influence white adipose tissue plasticity(Reference Pellegrinelli, Carobbio and Vidal-Puig3).
Development of adipose tissue
Cellular expansion of adipose tissue is associated with hypertrophy (increase in size) and hyperplasia (increase in number, ‘adipogenesis’). Both phenomena occur in normal growth throughout life and during obesity development(Reference Gesta, Tseng and Kahn15,Reference Hausman, DiGirolamo and Bartness32) . Adipogenesis is the result of the differentiation of new adipocytes from precursor cells in the adipose tissue. Adipogenesis comprises several steps orchestrated by a transcriptional cascade involving the nuclear PPARγ: the ‘master regulator’ of adipocyte differentiation along with the members of the enhancer binding proteins family (C/EBP), bone morphogenetic proteins (BMP) and Zn finger proteins (ZFP). Two interesting reviews on this field are by Ma et al. (Reference Ma, Lee and Chisholm33) and Ghaben & Scherer(Reference Ghaben and Scherer34).
Both brown and white adipocytes start their development in utero. However, the timing and rate of adipose tissue formation varies somewhat between species(Reference Nakagami35). In humans, Poissonnet et al. (Reference Poissonnet, Burdi and Bookstein36,Reference Poissonnet, Burdi and Garn37) found that adipose tissue (in the form of fat lobules with no lipid storage) first appears during early pregnancy in both sexes, between 14 and 24 weeks of gestation (start of the second trimester). The precise timing may depend to some degree on fetal size or weight, with larger fetuses developing identifiable adipocytes earlier than smaller ones. By the third trimester (28 weeks), adipose tissue depots are already established(Reference Poissonnet, Burdi and Garn37). In particular, the first evidence of brown adipose tissue depots is from 20 weeks of gestation, with a maximal time of 26 weeks, being stabilised at 35 weeks(Reference Moragas and Torán38).
In rodents, adipose tissue mainly develops between late gestation and 4 weeks of postnatal age(Reference Greenwood and Hirsch39). A study in the mouse model called ‘AdipoChaser’ – a system that can detect the already present adipocytes from those that are newly formed – revealed that subcutaneous adipose tissue development occurs early during embryogenesis, in embryonic days 14–18. In contrast, epididymal adipocytes preferentially differentiate postnatally(Reference Wang, Tao and Gupta40). Another study using sensitive reporters revealed the expression of adipose-specific markers in the subcutaneous region as early as embryonic days 16·5–17·5, after lipid-filled subdermal adipocytes. This research also shows that the epididymal adipose tissue was formed postnatally(Reference Birsoy, Berry and Wang41). Both lines of evidence coincide with Han et al. (Reference Han, Lee and Jin42), who demonstrate that precursor cells are not found in the nascent epididymal pad until postnatal day 4.
Adipose tissue in both humans and rodents remains to some extent expandable later in life(Reference Spalding, Arner and Westermark43). Generally, adipocyte expansion ceases at adolescence. In humans, roughly 8 % of adipocytes turn over approximately every year, while in mice, 0·6 % of adipocytes are renewed each day(Reference Spalding, Arner and Westermark43,Reference Rigamonti, Brennand and Lau44) . Interestingly, the absolute number of turn-over adipocytes in individuals with obesity is approximately twice compared with lean individuals(Reference Spalding, Arner and Westermark43). However, the major development of adipose tissue coincides with plastic periods of pregnancy and lactation, when hormonal, nutritional and epigenetic signals influenced by the mother probably programme permanent changes in the offspring’s adipose tissue(Reference Liang, Yang and Fu45).
Adipose tissue as an endocrine organ
Adipose tissue secretes numerous peptides, hormones and molecules (called adipokines), which act in auto-, para- and endocrine manners. Adipokines participate in signalling the functional status of the adipose tissue to target cells in the brain, liver, pancreas, vasculature, muscle and other tissues(Reference Fasshauer and Blüher46,Reference Lehr, Hartwig and Sell47) (Fig. 1).

Fig. 1. Interaction between white adipose tissue with other organs contributes to maintaining energy balance. The development of either normal or altered adipokine secretion may contribute to whole-body homeostasis during health and disease.
Adipokines participate in the regulation of several physiological processes in the adipose tissue and at a systemic level. Adipokine secretion, mainly leptin and adiponectin, modulate appetite, fuel metabolism, innate immune function and reproduction (Table 1). Moreover, abnormal adipokine secretion contributes to a spectrum of obesity-associated diseases(Reference Ghaben and Scherer34). The evidence of hormones and growth factors that modulate the adipose function is summarised in Table 2.
Table 1. Adipokines and their potential role in adipose tissue programming

LGA, large for gestational age; HOMA-IR, homeostatic model assessment of insulin resistance; HFD, high-fat diet; MCP-1, monocyte chemoattractant protein 1; RBP4, retinol binding protein 4; GDM, gestational diabetes mellitus.
Table 2. Endocrine regulators of adipose tissue physiology and their potential role in the developmental origins of health and disease (DOHaD)

IGF, insulin-like growth factor; IUGR, intra-uterine growth restriction; FGF, fibroblast growth factor; GDM, gestational diabetes mellitus; T3, triiodothyronine; THRα-1, thyroid hormone receptor α-1; fT3, free triiodothyronine; Dio2, deiodinase iodothyronine type II; WAT, white adipose tissue; HFD, high-fat diet; fT4, free thyroxine; THRβ-1, thyroid hormone receptor β-1; GH, growth hormone; SGA, small for gestational age; 11β-HSD2, 11-β hydroxysteroid dehydrogenase type 2; AGA, appropriate for gestational age; ERα, oestrogen receptor α; ERβ, oestrogen receptor β; hCG, human chorionic gonadotropin.
Hormonal signals during pregnancy modulating fetal growth and adiposity
Due to the obesity epidemic, attention has turned to potential influences of maternal obesity (BMI ≥30 kg/m2) and the risk of disease in the offspring(Reference Patel, Pasupathy and Poston48). Maternal obesity and diabetes are associated with increased birth weight, excessive nutrition in neonates, and rapid ‘catch-up growth’ that predispose the offspring to fat accumulation(Reference Calkins and Devaskar49,Reference Shankar, Harrell and Liu50) . The mechanisms linking pregnancy obesity to altered fetal growth and programming of adult disease are not well established. However, during the gestational period and birth, studies have shown a correlation between maternal hormones and birth weight(Reference Jansson, Nilsfelt and Gellerstedt51).
An important element associated with birth weight in the function of gestational age is insulin-like growth factor (IGF)-1(Reference Leger, Noel and Limal52). Maternal IGF-1 is stimulated by hormonal signals, promoting placental growth and function(Reference Murphy, Smith and Giles53,Reference Forbes and Westwood54) . In both animals and humans, circulating IGF-1 is reduced in intra-uterine growth restriction (IUGR) neonates(Reference Leger, Noel and Limal52,Reference Luo, Nuyt and Delvin55) . However, when IGF-1 is administered to mothers of IUGR fetuses, it promotes placental nutrient transfer to enhance fetal growth. IGF-1 levels are correlated with weight gain and increased nutrient intake after intra-uterine nutrient deprivation(Reference Leger, Noel and Limal52). Notably, maternal IGF-1 treatment in the late pregnant ewe is associated with enhanced glucose delivery to the fetus(Reference Wali, de Boo and Derraik56). This was also observed in a mouse model of IUGR, where placental GLUT expression was increased after an intraplacental injection of adenovirus-mediated IGF-1, thus restoring fetal weight(Reference Dimasuay, Boeuf and Powell57). In addition, reduced maternal circulating IGF-1 is associated with small-for-gestational-age and growth-restricted babies(Reference Hernandez-Valencia, Zarate and Ochoa58). Therefore, tissues chronically depleted of insulin and IGF-1 throughout fetal life and suddenly exposed to increased concentrations of those hormones shortly after birth may counteract the actions of insulin by developing insulin resistance as a defence mechanism against hypoglycaemia(Reference Dimasuay, Boeuf and Powell57,Reference Brenseke, Prater and Bahamonde59) .
Obesity in pregnancy is characterised by elevated maternal serum insulin and leptin hormones that also stimulate placental amino acid transporters in vitro (Reference Dimasuay, Boeuf and Powell57). High levels of these hormones activate placental insulin/IGF-1/mTOR (mammalian target of rapamycin) and leptin signalling pathways, thus increasing placental amino acid transport capacity and determining fetal overgrowth in a maternal obesity mouse model(Reference Rosario, Powell and Jansson60).
Additionally, leptin has a broader range of actions, particularly during growth and development(Reference Vickers, Gluckman and Coveny61). Besides adult(Reference Blüher and Mantzoros62) and fetal adipose tissue(Reference Gómez, Carrascosa and Yeste63), the placenta is an important site of leptin production(Reference Henson and Castracane64). The human leptin gene has a placental-specific upstream enhancer, which mediates its placental expression(Reference Vickers, Gluckman and Coveny61,Reference Bi, Gavrilova and Gong65) . Furthermore, leptin may play a role in implantation, placental endocrine function and fetal development(Reference Ashworth, Hoggard and Thomas66). Circulating maternal leptin increases during pregnancy and evidence indicates that leptin may participate in placental angiogenesis, immunomodulation, nutrient transport and growth(Reference Henson and Castracane64,Reference Ashworth, Hoggard and Thomas66) . Co-localisation of leptin and its receptor in the syncytiotrophoblasts/cytotrophoblasts (one of the three layers of placenta villi) at the maternal interface is evidence that placental leptin is an important fetal growth factor(Reference Ashworth, Hoggard and Thomas66). Therefore, the temporal co-expression of the long isoforms of the leptin receptor and its ligands in mesenchymal tissues during fetal development leads to leptin acting as a paracrine or autocrine factor during fetal life(Reference Vickers, Gluckman and Coveny61). Supporting that evidence, placental leptin concentration is increased in small-for-gestational-age newborns and correlates negatively with placental weight. Moreover, the leptin receptor isoforms LEPRα (short form) and LEPRβ (long form) are differentially expressed in the placenta according to birth weight(Reference Lazo-de-la-Vega-Monroy, González-Domínguez and Zaina67). The umbilical cord leptin correlates positively with placental weight and the newborn’s weight(Reference Lazo-de-la-Vega-Monroy, González-Domínguez and Zaina67), explaining up to 21 % of birth weight variation(Reference Karakosta, Chatzi and Plana68). In addition, umbilical cord blood leptin has a significant correlation with fetal insulin resistance(Reference Catalano, Presley and Minium69). Thus, both IGF-1 and leptin have crucial roles in activating pathways that lead to the increase of placental amino acid transport capacity and may importantly determine fetal overgrowth in maternal obesity.
Furthermore, both insulin and leptin may also activate adipogenesis. Hence, maternal obesity and elevated circulation levels of insulin and leptin may enhance adipogenesis and lipogenesis, resulting in higher white adipose tissue mass and adipocyte hypertrophy in offspring(Reference Murabayashi, Sugiyama and Zhang70,Reference Lecoutre, Deracinois and Laborie71) . In agreement with this concept, increased circulating leptin levels in macrosomic fetuses and decreased leptin levels in growth-restricted fetuses have been reported(Reference Vela-Huerta, San Vicente-Santoscoy and Guizar-Mendoza72). The significance of altered fetal leptin levels in circulation is relevant for fetal physiology, since those levels are proportionate to insulin levels and adiposity(Reference Tessier, Ferraro and Gruslin73). These findings align with the fact that fetuses from mothers with obesity develop insulin resistance in utero and are born with increased body fat compared with newborns from lean, healthy mothers(Reference Catalano, Presley and Minium69). Importantly, leptin treatment from postnatal days 3–13 in a rodent model of maternal undernutrition resulted in a transient slowing of neonatal weight gain in the offspring and normalised energy intake, locomotion activity, body weight, fat mass, and fasting plasma glucose, insulin and leptin concentrations(Reference Vickers, Gluckman and Coveny61). Consequently, it is possible that increased circulating concentrations of maternal hormones, such as leptin and insulin, constitute a mechanistic link between maternal overweight or obesity and fetal overgrowth, mediated by modulation of placental nutrient transport and placental growth(Reference Lecoutre and Breton74).
Adiponectin may also play a role in modulating growth and adiposity during pregnancy(Reference Aye, Powell and Jansson75,Reference Aye, Rosario and Powell76) . There are controversial reports on which tissue produces adiponectin during pregnancy. In physiological conditions (non-pregnant), adiponectin is produced exclusively in the adipose tissue. Reports indicate that during pregnancy, the placenta secretes adiponectin(Reference Caminos, Nogueiras and Gallego77). In contrast, other studies have not confirmed these findings(Reference Ichida, Moriyama and Morita78) and suggest that it is likely that the adiponectin influencing placental function predominantly originates from the maternal adipose tissue(Reference Aye, Powell and Jansson75).
The role of adiponectin in pregnancy is related to placental transporters(Reference Aye, Powell and Jansson75,Reference Aye, Rosario and Powell76) . In maternal obesity, adiponectin levels are reduced and placental amino acid nutrient transporters are up-regulated(Reference Jones, Jansson and Powell79). Interestingly, adiponectin has insulin-sensitising actions in liver and muscle(Reference Aye, Powell and Jansson75). However, in primary human trophoblast cells, adiponectin attenuates insulin signalling. As a result, adiponectin inhibits insulin-stimulated amino acid transport, which has an important role in placental nutrient transport and fetal growth during pregnancy(Reference Jones, Jansson and Powell79). In addition, adiponectin supplementation in dams with obesity from embryonic day 14·5 to 18·5 reverse the adverse effects of maternal obesity on placental function and fetal growth(Reference Aye, Rosario and Powell76).
Another important hormone produced during pregnancy is human chorionic gonadotropin. Its concentrations increase exponentially during the first 2 months of pregnancy and decrease abruptly to reach basal values at the end of the third month, a period in which a significant change in the depot-specific fat mass is evident, and, particularly, an accumulation of fat mass to cover the energy demands of both the fetus and the mother occurs(Reference Sidebottom, Brown and Jacobs80). However, little is known concerning the role of human chorionic gonadotropin in adipose tissue metabolism, especially when this hormone is actively secreted.
During pregnancy, other hormones such as the sex hormones progesterone, oestrogens and oestradiol play a key role in adipose tissue metabolism(Reference Jansson, Nilsfelt and Gellerstedt51,Reference Lacasa, Le Liepvre and Ferre81) . However, the role of hormonal status during pregnancy is still a broad field to investigate. Further elucidation about the physiology of actively secreted hormones between the mother and progeny during pregnancy may offer novel strategies for therapy and prevention of birth weight alterations in the offspring.
Nutritional signals modulating fetal growth and adiposity during pregnancy
It is well know that both over- and undernutrition result in endocrine changes associated with adult adiposity(Reference McMillen, Muhlhausler and Duffield11). These opposite paradigms have been used to study the long-term effects of nutritional manipulations during prenatal and early postnatal life. Several studies in both human and mouse models have demonstrated that perturbation of circulating factors, such as nutrients and/or hormones induced by diet during fetal development, increase the risk of obesity during childhood and adulthood(Reference Lecoutre and Breton82,Reference Ross and Desai83) . Furthermore, other investigations have shown that the programming of appetite dysregulation contributes to the obesity phenotype in IUGR offspring(Reference Lecoutre and Breton74). IUGR and small-for-gestational-age newborns typically present accelerated postnatal growth, defined as ‘catch-up’ growth, within the first years of life(Reference Singhal84). This suggests that accelerated infant and childhood weight gain is associated with increased energy intake and diminished satiety response(Reference van Deutekom, Chinapaw and Vrijkotte85). In addition, Desai et al. (Reference Desai and Guang Han86) found that IUGR rats present hypertrophic adipocytes and increased de novo lipogenesis, factors that predispose increased fat storage. Moreover, compelling evidence indicates that faster infant growth in humans is associated with obesity later in life, independently of country income, gestational age, birth weight or breast-feeding(Reference Singhal84).
Maternal obesity, excessive nutrition and accelerated neonatal growth have been shown to sensitise offspring to obesity(Reference Desai, Jellyman and Han87). In a rodent model, maternal high-fat diet (HFD) during pregnancy increased maternal adiposity and circulating leptin and decreased adiponectin, causing a 43 % increase in fetal growth compared with controls(Reference Tessier, Ferraro and Gruslin73). Male neonates from mothers fed a HFD before and during gestation and lactation exhibit a rapid weight gain during lactation, concordant with a key period of adipose tissue development in rodents(Reference Greenwood and Hirsch39). In addition, male offspring from HFD mothers have higher expression of Zn finger protein 423 (zfp423) and lower DNA methylation of its promoter in epididymal fat progenitors compared with control lean offspring, coincidentally with enhanced adipose tissue differentiation in this period(Reference Liang, Yang and Fu45). Moreover, adult male offspring from HFD mothers are predisposed to fat accumulation, showing increased visceral, gonadal and perirenal fat depots, together with hyperleptinaemia. The perirenal adipose tissue depot exhibits elevated sterol regulatory element-binding protein 1 (SREBP1), fatty acid synthase (FAS), leptin, and diminished PPARγ mRNA levels(Reference Lecoutre, Deracinois and Laborie71). Further, maternal HFD during pregnancy alone programmes increased offspring adiposity with normal body weight, whereas maternal HFD during lactation increases both body weight and adiposity(Reference Desai, Jellyman and Han87). The reasons behind this discrepancy are not understood but may depend on differences in the nutritional intervention such as the duration of maternal HFD feeding, lipid content in diet, and/or in the different genetic background of rodents.
Adult rat offspring from mothers obese by cafeteria-diet feeding during gestation and lactation exhibit an increase in adipose tissue TAG content with elevated lipogenic enzyme activities, along with abnormalities in fatty acid composition(Reference Bayol, Simbi and Stickland88). These findings indicate that perinatal exposure to a diabetic milieu characterised by increased glucose and/or insulin levels can programme developmental processes such as adipogenesis(Reference Lecoutre and Breton74).
The consumption of fructose-rich diets is on the rise and is thought to be associated with obesity and cardiometabolic diseases. Recently, fructose has been used as a sweetener in many food items(Reference Stephan, Wells and Brayne89). Feeding pregnant C57BL/6J dams with fructose at 10 % (w/v) (similar to most fructose-sweetened soft drinks) makes the offspring present different risks for disease profiles in a sex-dependent manner. Males become hypertensive and develop insulin resistance, while females develop additional abnormalities including obesity, increased adiposity, fatty liver, high serum leptin and low serum adiponectin(Reference Saad, Dickerson and Kechichian90). It is also known that male and female placentas react differently to the perinatal environment, and differential effects of fructose on the placental transport system have been reported(Reference Vickers, Clayton and Yap91). For example, male placentas are glucocorticoid resistant, whereas female placentas respond to different glucocorticoid levels by altering cortisol metabolism, placental protein/gene expression, placental growth factor pathways and placental immunity(Reference Clifton, Bisits and Zarzycki92,Reference Geary, Pringle and Rodeck93) .
The influence of overnutrition is not the only nutritional factor determining the programming of adipose tissue dysregulation. For example, the offspring of low-protein diet (LPD)-fed dams during gestation and lactation have persistent smaller adipocytes due to a decrease in adipose cell size(Reference Lecoutre and Breton74,Reference Ferland-McCollough, Fernandez-Twinn and Cannell94) . However, in adulthood, offspring from LPD-fed mothers exhibited increased mRNA expression levels of C/EBPα and PPARγ, suggesting enhanced adipogenesis(Reference Guan, Arany and van Beek95). Furthermore, this model also exhibited increased adipose tissue expression of miRNA-483-3p, known to regulate in vitro differentiation and lipid storage of adipocytes. In contrast, the offspring of LPD-fed dams, which had 50 % food restriction during gestation, exhibited low birth weight and enhanced adipogenic factors, such as PPARγ and C/EBPα, and presented small adipocytes(Reference Ferland-McCollough, Fernandez-Twinn and Cannell94). Nevertheless, if maternal protein was 70 % food restricted during gestation, the persistent feature was the appearance of hypertrophic adipocytes(Reference Lukaszewski, Mayeur and Fajardy96). The reasons of these findings are completely uncertain. However, according to the DOHaD concept, a single genotype could produce many phenotypes, depending on intervention characteristics and timing, environmental influences and/or epigenetic changes, leading to alterations in the fetus or neonate in long-term programming and responses particularly against a metabolic challenge in adulthood(Reference Levian, Ruiz and Yang97,Reference Dolinoy, Weidman and Jirtle98) .
Epigenetic changes during pregnancy modulating fetal growth and adiposity
Studies in both human and animal models provide evidence of programmed adiposity and metabolic diseases resulting from early nutritional exposures, suggesting that epigenetic modification may be a major contributing factor(Reference Desai, Jellyman and Ross99).
Indeed, maternal (and/or paternal) perinatal nutritional interventions can cause epigenetic modifications such as differential gene promoter methylation (i.e. in CpG sites), chromatin remodelling, histone acetylation/methylation, and post-transcriptional changes such as differential mRNA expression in offspring(Reference Lecoutre and Breton74,Reference Desai, Jellyman and Han100) . Notably, exposure to either maternal famine or obesity reduces DNA methylation of the imprinted IGF-2 gene in the offspring. Studies in old adults who were prenatally exposed to famine showed hypomethylation on the IGF-2 gene, and hypermethylation in two major obesity-related non-imprinted genes: TNFα and leptin(Reference Morales, Groom and Lawlor101). In addition, placental leptin gene methylation is increased in gestational diabetes(Reference Soubry, Murphy and Wang102). Moreover, alterations in methylation at specific sites in weight loss have been observed(Reference Aronica103), and several associations between methylation marks at birth and later-life obesity were found(Reference Godfrey, Sheppard and Gluckman104,Reference Clarke-Harris, Wilkin and Hosking105) . Other reports have evidenced DNA methylation of genes such as IGF-2/H19 in the offspring of mothers with an unbalanced diet during pregnancy(Reference Drake, McPherson and Godfrey106,Reference Huang, Galati and Burrows107) . These modifications in methylation are associated with increased weight, waist circumference, BMI, blood pressure, fat accumulation, and obesity in the offspring(Reference Rohde, Keller and la Cour Poulsen108).
On the other hand, HFD feeding during pregnancy in mice increases the expression of zfp423, a key transcription factor responsible for adipogenic lineage commitment during fetal development in the offspring. Accordingly, repressive histone methylation (H3K27me3) was lower in the zfp423 promoter in fetal tissues from dams with obesity(Reference Yang, Liang and Rogers109). Offspring from dams with obesity, which were fed a HFD from birth until 3 months of age, showed an increased proportion of adipocyte progenitor density compared with mice fed a HFD postnatally but unexposed to maternal obesity(Reference Liang, Yang and Fu45). This evidence suggests that maternal obesity may epigenetically limit the expansion capacity of offspring adipose tissue. In addition, alterations in DNA methylation of CpG sites and CGI shores of pro-adipogenic factors, such as Zfp423 or C/EBP-β, have also been demonstrated in young rats from mothers with obesity during pregnancy(Reference Borengasser, Zhong and Kang110). Obesity-prone weanling rat from dams with induced obesity by intragastric feeding of a HFD present greater ex vivo adipocyte differentiation, associated with increased mRNA expression levels of PPARγ and C/EBPβ, together with alterations in DNA methylation of CpG sites(Reference Borengasser, Zhong and Kang110).
Other perinatal food interventions can cause epigenetic modifications in offspring. For example, adult mice from LPD-fed dams presented removal of CpG methylation in the leptin promoter in white adipose tissue, affecting leptin expression dynamics in response to a meal(Reference Jousse, Parry and Lambert-Langlais111). A LPD during pregnancy also produces an increase in miRNA-483-3p expression levels with a decrease in GDF3 (growth differentiation factor 3; a member of the bone morphogenetic protein/transforming growth factor β (BMP/TGF-β) family) protein content, a factor that impairs late stages of adipocyte differentiation in rat offspring(Reference Ferland-McCollough, Fernandez-Twinn and Cannell94).
Maternal microbiota modulates fetal growth and adiposity
It has been largely demonstrated that the gut microbiota plays a key role in health and disease, including obesity and its metabolic complications(Reference Wang, Xu and Xia112,Reference Kvit and Kharchenko113) . However, the presence of an intra-uterine microbiome is still under debate. While certain studies have shown the presence of bacteria in placenta(Reference Aagaard, Ma and Antony114–Reference Collado, Rautava and Aakko116) and amniotic fluid(Reference Collado, Rautava and Aakko116), others attribute these findings to sample contaminations(Reference Lauder, Roche and Sherrill-Mix117,Reference Lim, Rodriguez and Holtz118) . To date, a newborn’s colonisation at birth is the only mechanism of neonatal microbiome origin(Reference Perez-Muñoz, Arrieta and Ramer-Tait119).
Several maternal nutritional and pathological conditions, including maternal obesity, have been associated with changes in maternal and neonatal gut-microbiota dysbiosis, which could affect microbial composition and promote metabolic disturbances in the offspring (reviewed in Calatayud et al. (Reference Calatayud, Koren and Collado120)). A study by Koren et al. (Reference Koren, Goodrich and Cullender121) that analysed the faecal microbiota in ninety-one healthy pregnant women from the first to the third trimester found that significant changes in microbial diversity occurred during the third trimester, whereas in early pregnancy, the microbiota was similar compared with the normal controls of the Human Microbiome Project. Interestingly, these changes are associated with the increase in proteobacteria population, which correlates with levels of proinflammatory cytokines interferon-γ, IL-2, IL-6 and TNF-α(Reference Zhou and Xiao122,Reference Mukhopadhya, Hansen and El-Omar123) . In addition, the transference of the microbiota from healthy pregnancies in the third trimester to germ-free mice increased adiposity, insulin resistance and inflammation compared with mice receiving first-trimester microbiota(Reference Koren, Goodrich and Cullender121). Therefore, the gut microbial community may have a critical role in maternal metabolic adaptations, which may influence fetal growth and development during normal pregnancy. Conversely, another study demonstrated that gut microbial diversity was stable throughout pregnancy(Reference DiGiulio, Callahan and McMurdie124). Although the altered composition of gut microbiota is still controversial among different studies, discrepancies may be partly attributed to genetics, BMI, maternal nutrition and/or gestational age of the woman studied(Reference Zhou and Xiao122).
In an animal model study, about twenty-six genera of gut-microbiota were significantly different in HFD-fed pregnant mice compared with normal diet-fed mice(Reference Cani, Possemiers and Van de Wiele125,Reference Paul, Bomhof and Vogel126) . Other studies also analysed the association between gut-microbiota composition and maternal metabolic parameters, and its changes with maternal preconceptional diet(Reference Santacruz, Collado and García-Valdés127,Reference Gomez-Arango, Barrett and Wilkinson128) . Furthermore, the increased Staphylococcus population positively correlated with increased plasma cholesterol levels, whereas a decrease in Bacteroides was associated with lower HDL-cholesterol and higher serum TAG levels(Reference Gohir, Whelan and Surette129). Thus, these alterations in the gut microbiota could predict the changes in metabolic pathways(Reference Zhou and Xiao122). Interestingly, a significant reduction in Bacteroides was also observed in the neonatal gut microbiota from HFD-fed mothers during gestation, which persisted until 6 weeks of age(Reference Chu, Antony and Ma130). Hence, the increased risk of obesity for children from mothers with obesity could be partially explained by the transmission of maternal obesogenic intestinal microbes(Reference Kozyrskyj, Kalu and Koleva131).
Maternal weight has been found to be a major influencing factor determining milk bacterial composition(Reference Kozyrskyj, Kalu and Koleva131). Higher abundance of Staphylococcus aureus was reported in breast milk over the first 6 months of lactation, as well as that of Lactobacillus in the first month of women with obesity compared with mothers with normal weight(Reference Cabrera-Rubio, Collado and Laitinen132,Reference Nuriel-Ohayon, Neuman and Koren133) . Interestingly, high levels of S. aureus are also found in the gut microbiota of overweight children(Reference Kalliomäki, Collado and Salminen134). However, the concrete effects of these findings are still unclear. Therefore, future research is needed to determine whether milk microbial population is associated with maternal and offspring adiposity.
Major changes in maternal microbiota during pregnancy have also been shown to correlate with adipose tissue development in humans(Reference Poissonnet, Burdi and Garn37), in which, as described above, alterations could be determining adipose tissue dysfunction in both the mother and the offspring(Reference Shankar, Harrell and Liu50). Additionally, modification in specific metabolic hormones including insulin, gastric inhibitory polypeptide, and adipokines were also found to be correlated with alterations in bacterial population, supporting a connection between microbial diversity and metabolic hormones during pregnancy(Reference Gomez-Arango, Barrett and McIntyre135).
Overweight and obesity during pregnancy result in obvious changes in microbiota composition and concomitant metabolic disorders in the mother. Maternal microbial transfer to the neonate could represent the first colonisation of gut microbiota in early life. Therefore, gut microbiota might be a critical factor supporting the DOHaD hypothesis and play a crucial role in programming. More studies oriented to characterise human microbiota during pregnancy and its complications – and associations with its effects on fetal development – will surely shed light on the potential role of the microbiome in programming obesity in childhood and adulthood.
Adiposity regulation in the early postnatal period
In rodents, adipose tissue development is particularly active during the perinatal period, especially the last week of gestation and early postnatal life(Reference Poissonnet, Burdi and Bookstein36,Reference Poissonnet, Burdi and Garn37,Reference Greenwood and Hirsch39) . Offspring from undernourished or overnourished mothers present alterations in the adipose tissue, such as changes in fat distribution/composition, enhanced adipogenesis, altered hormone levels and sensitivity to them (especially the glucocorticoid system), environmental influences, and epigenetic mechanisms, all of which modify adipose tissue programming during the perinatal period(Reference Desai, Jellyman and Ross99,Reference Lukaszewski, Eberlé and Vieau136) .
Critical periods to body composition establishment during the neonatal period and early childhood may be predicted by maternal pre-pregnancy BMI, adiposity, gestational weight gain, maternal nutrition, maternal TAG concentrations, maternal smoking, and inflammation during pregnancy(Reference Jansson, Nilsfelt and Gellerstedt51,Reference Weden, Brownell and Rendall137) . About 15–45 % of babies born to diabetic mothers are macrosomic. Weight gain rate during the first year, particularly peripheral fat, is an indicator of trunk fat mass and affects the rate of weight gain during the first 6 months of life(Reference McMillen, Muhlhausler and Duffield11). The molecular mechanisms driving the correlation between excess weight gain in the first 6 months and later obesity remain unknown.
Several studies have associated early infant growth with later risk for overweight, suggesting that early infancy is a crucial window of metabolic programming(Reference Young, Johnson and Krebs8). It is even suggested that the first weeks and months of life are particularly associated with later weight status(Reference Gillman138), which are a critical time when metabolic programming can occur, similar to the in utero period, because the infants’ organs still maintain considerable plasticity for adaptation to nutritional and environmental exposures(Reference Weden, Brownell and Rendall137,Reference Chandler-Laney, Gower and Fields139) . Thus, this early age provides a critical opportunity for interventions directed to have an impact on future metabolic health.
Breast milk represents the main source of nutrients in the first months of life for breastfed infants, providing different nutrients and bioactive factors, especially hormones and growth factors such as leptin, ghrelin, insulin and IGF-1. It also plays a role in food intake regulation, metabolism and body composition(Reference Fang, Shi and Yu140–Reference Savino and Lupica142).
Breast milk composition is dynamic and variable among women. Fat is a major digestible component, comprising over 50 % of the energy of breast milk(Reference Hassiotou, Hepworth and Williams143). Macronutrient intake, hormonal and behavioural mechanisms related to breast milk composition (i.e. protein, PUFA, oligosaccharides, cytokines and hormones, in particular leptin, adiponectin and resistin), together with the breast-feeding practice itself, can influence the infant’s feeding behaviour and regulation of growth and appetite control later in life. On the other hand, serum levels of leptin vary dramatically during intra-uterine and early postnatal life, with a 5- to 10-fold increase in leptin occurring between postnatal days 4 and 10 in female mice(Reference Ahima, Prabakaran and Flier144). Moreover, breast milk leptin may participate in postnatal development, possibly contributing to elevated circulating levels of this hormone in the neonate(Reference Vickers, Gluckman and Coveny61). However, research is needed to confirm the hormonal biological effects.
In addition, fatty acid composition of human milk is influenced by maternal diet. For example, trans-fatty acid content in the maternal diet increases the risk of increased adiposity in the neonate, and the associated chronic diseases later in life. Overall maternal dietary trans-fatty acid intake may influence adiposity in mothers and their infants(Reference Anderson, McDougald and Steiner-Asiedu145). However, several studies support the hypothesis that breast-feeding might confer protection against obesity later in life(Reference Arenz, Rückerl and Koletzko146,Reference Grummer-Strawn and Mei147) . A meta-analysis showed that the length of the breast-feeding period was associated with a decreased risk of childhood obesity. Children breastfed for ≥7 months were significantly less likely to have obesity compared with those breastfed <3 months(Reference Yan, Liu and Zhu148). However, other studies have reported conflicting results(Reference Victora, Bahl and Barros149,Reference Dewey150) , which could be due to forms of interpretation depending on the type of study, postpartum period, group size, or a dose-dependent effect of breast-feeding duration on the prevalence of obesity. Moreover, various studies agree on the need to expand the research about the effects of breast milk composition on childhood adiposity and overweight(Reference Anderson, McDougald and Steiner-Asiedu145,Reference Grummer-Strawn and Mei147) . Interestingly, obesity-prone rat offspring artificially raised by intragastric canula (‘pup in the cup’ model) with a high-carbohydrate milk formula exhibited hyperinsulinaemia, hypertrophic adipocytes and enhanced adipose tissue lipogenic enzyme activities compared with pups fed rat milk where the major source of energy was fat(Reference Srinivasan, Mitrani and Sadhanandan151).
A recent study evidenced that prolactin, one of the most abundant hormones secreted during lactation and present in high quantities in maternal milk(Reference Melo, Pérez-Ledezma and Clapp152), was reduced in dams fed a HFD during lactation, showing altered mammary gland structure and function compared with control-fed dams. Moreover, offspring from HFD-fed dams had increased body weight and adiposity, developing fatty liver, hyperinsulinaemia and insulin resistance at weaning. Interestingly, increasing prolactin levels in HFD-fed mothers improved mammary gland function and reduced visceral adiposity, ameliorated fatty liver and improved insulin sensitivity in offspring(Reference de Los Ríos, Ruiz-Herrera and Tinoco-Pantoja153).
Thus, modification of the maternal diet has a high potential for childhood obesity reduction, by improving the maternal and fetal metabolic environment. In addition, there is also evidence to suggest that the benefit of these interventions may extend to the adult health of the offspring(Reference Patel, Pasupathy and Poston48).
Transgenerational programming of obesity
During the past two decades, there has been a marked increase in the global prevalence of adult and childhood obesity(Reference Blüher4). Notably, the obesity phenotype has occurred in a relatively short period (one or two generations)(Reference Desai, Jellyman and Ross99). Since the 1980s decade, Beach et al. (Reference Beach, Gershwin and Hurley154) drove attention to the physiological response of the offspring across two or more generations, a term now called ‘transgenerational programming’(Reference Vickers155). More recently, offspring from rats treated with dexamethasone during pregnancy presented retarded fetal growth, hypertension and glucose intolerance which, in turn, had effects on glucose homeostasis that persisted for two more generations(Reference McMillen, Muhlhausler and Duffield11,Reference Drake, Walker and Seckl156) .
In another study, maternal high-fat feeding caused a sex-specific insulin-resistant phenotype in second- and third-generation mice offspring(Reference Zambrano, Martínez-Samayoa and Bautista157). In addition, Dunn & Bale(Reference Dunn and Bale158) previously reported that maternal HFD exposure in mice results in an increase in body size and reduced insulin sensitivity, which persisted to the F2 generation via both maternal and paternal lineages. In mice, it has been shown that both male and female offspring present increased body weight and adiposity over generations exposed to multigenerational HFD feeding(Reference Lecoutre and Breton74). Thus, a vicious cycle develops, as females born to women with obesity may have an increased risk of obesity and give birth to subsequent generations with the same risks(Reference Desai, Jellyman and Ross99). In contrast, the transmission of programming effects to subsequent generations perpetuates a cycle of obesity and metabolic disorders. However, the mechanisms responsible for these transgenerational effects remain to be elucidated.
White adipose tissue represents a prime target of metabolic programming induced by maternal obesity(Reference Liang, Yang and Fu45). Perturbations to the perinatal nutrient supply may affect adipocyte development, conducing to persistent alterations in adipocyte number and physiology(Reference Lecoutre and Breton82). Indeed, in fetuses and neonates, adipocytes are sensitive to maternal factors. In humans, it is well characterised that an increase in the number of adipocytes occurs early in life, which is a determinant for fat mass in adulthood(Reference Poissonnet, Burdi and Garn37). Conversely, in rodents, adipose tissue growth and adipogenesis mainly take place during the last week of gestation and accelerate throughout lactation(Reference Muhlhausler and Smith159). Despite the evidence that adipogenesis occurs throughout life, maternal obesity at conception may predispose to adipose tissue development, resulting in higher white adipose tissue mass and hypertrophied adipocytes in children and adults(Reference Borengasser, Zhong and Kang110).
Overnutrition (i.e. HFD feeding) during lactation and/or postweaning periods leads to accelerated growth and alterations in adipogenesis and lipogenesis programming(Reference Masuyama and Hiramatsu160). This correlates with an up-regulation of the key adipogenic factor PPARγ, characteristic of fat expansion in the offspring from mothers with obesity(Reference Guan, Arany and van Beek95,Reference Desai, Jellyman and Han100) . These observations agree with the ‘adipose tissue expandability hypothesis’, which proposes that insufficient precursor availability to enable increased adipogenesis, when challenged with a high-energy diet, limits adipocyte hyperplasia and hampers replacement of dead adipocytes. As a result, the adipose tissue of maternal obesity offspring has the impaired ability to maintain the progenitor pool. The premature differentiation of offspring progenitor cells limits the corresponding adipose tissue expandability, leading to adipocyte hypertrophy, a cause of hypoxia and inflammation, two major characteristics of type 2 diabetes mellitus(Reference Liang, Yang and Fu45).
Conclusions
Adipocytes play a critical role in whole-body energy homeostasis and endocrine regulation in all stages of life. Gestation and early postnatal life are critical plastic periods for programming adipose tissue biology, as adipocyte development and physiology may be modelled by several maternal factors such as genetics, environmental influences, epigenetic changes, nutritional status, hormonal signalling and energy balance. This programming may lead to obesity and its co-morbidities in the progeny, with further transgenerational transmission and development of chronic diseases later in life (Fig. 2).
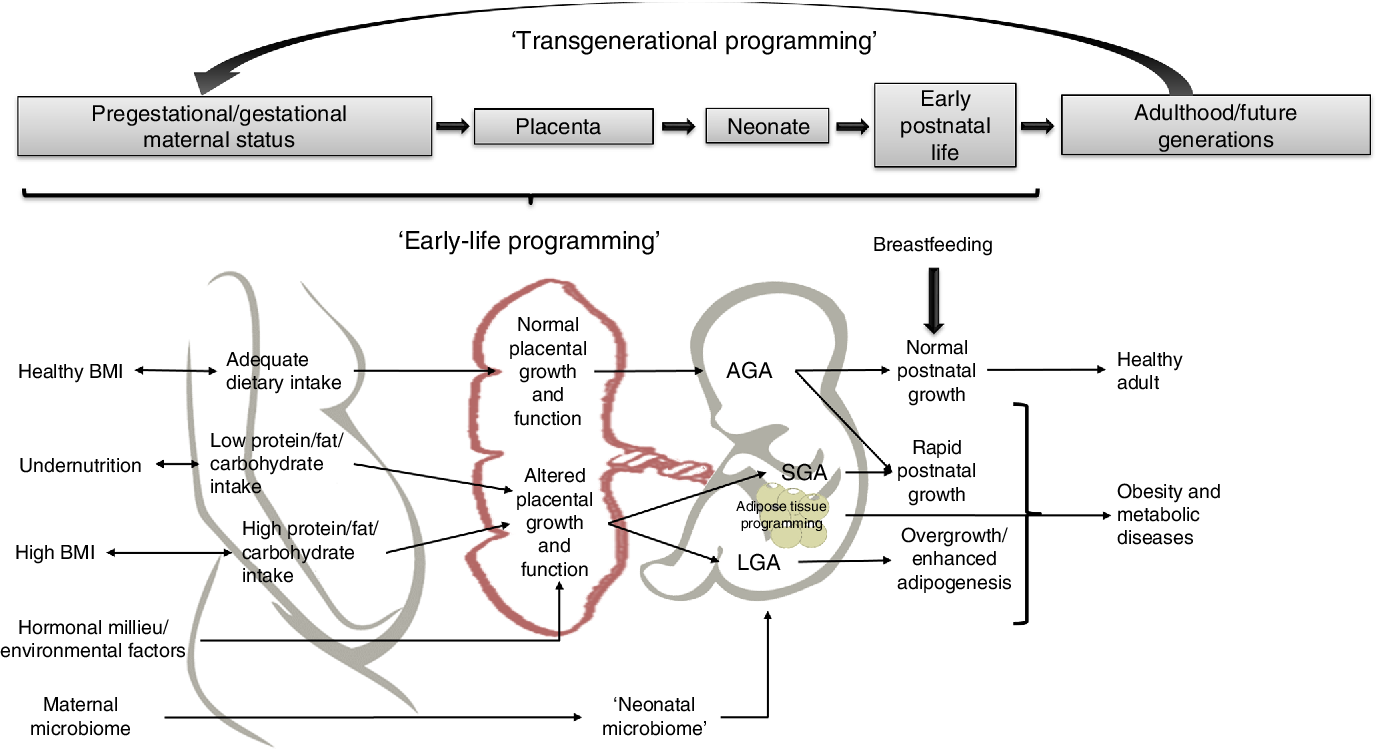
Fig. 2. Potential mechanisms linking early-life factors during pregnancy that may lead to obesity in children and adults. AGA, appropriate for gestational age; SGA, small for gestational age.
The complexity of adipose tissue results in numerous challenges for this study area. Therefore, the mechanisms by which adipose tissue development and physiology change in response to environmental demands are now being elucidated. Some of the questions in this topic that must be addressed that could greatly contribute to the DOHaD field are:
-
(a) How is placental development and physiology influencing fetal adiposity and how is the placenta programmed by maternal and/or paternal pregestational conditions, such as obesity or metabolic status?
-
(b) How is adipose tissue development and location programmed by specific macro- and micronutrient intake during gestation?
-
(c) Which molecular mechanisms and/or specific genes are involved in the epigenetic programming of fetal adiposity?
-
(d) What is the role of maternal gut and breast milk microbiota in adipose tissue programming of the offspring?
-
(e) How does breast milk composition change in response to maternal hormonal, nutritional and metabolic status, and how can these changes influence adipose tissue development in the neonate?
-
(f) Which are the precise mechanisms behind transgenerational programming of obesity in humans?
The potential impact of these findings for public health is vital, because pregnancy and early infancy represent critical windows of opportunity during which parents are most willing to adopt lifestyle changes, which, in turn, may have significant health implications across multiple generations.
Acknowledgements
The present review was supported by the Dirección de Apoyo a la Investigación y al Posgrado, Universidad de Guanajuato (M.-L. L.-V.-M., CIIC 056/2019), Consejo Nacional de Ciencia y Tecnología (CONACYT) (C. F.-M., 219787), Fondos Federales (C. F.-M., 074/2013) and Dirección General de Asuntos del Personal Académico (C. F.-M., PAPIIT: IN 206617), Universidad Nacional Autónoma de México. E. M.-M. is a PhD student from ‘Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México’, and was a recipient of two CONACYT fellowships (CVU/Becario: 383325/255765 and Becario: 27664). S. Q.-F. was a recipient of a post-doctoral CONACYT fellowship (Becario: 295457). The funding providers had no role in the design, analysis or writing of this article.
E. M.-M., S. Q.-F., C. F.-M. and M.-L. L.-V.-M conceived the idea and reviewed the literature. S. Q.-F. and C. F.-M. outlined and scripted the basis of the manuscript. E. M.-M. and S. Q.-F. had a role in the design, analysis and writing of the article. E. M.-M. and M.-L. L.-V.-M. designed the figures. M.-L. L.-V.-M. drafted the manuscript. All authors critically reviewed the manuscript and approved the final version.
The authors declare no conflicts of interest.




