


INTRODUCTION
The word metagenome was first used in its modern sense in 1998 to refer to the collection of all microbial genomes found in a sample of soil, including sequences from organisms that could not cultured (Handelsman et al. Reference Handelsman, Rondon, Brady, Clardy and Goodman1998). The term has since been generalized to cover any set of multiple genomes found in an environmental or clinical sample.
In its strictest sense, the term metagenomics is used to describe the recovery of information from metagenomes via the creation of shotgun sequence libraries. Such libraries can then be sequenced or, if cloned in an expression system, screened for functional activities of interest. In 2004, Venter and his colleagues described a landmark study on the metagenomics of the Sargasso Sea that established the utility of this approach in exploring not just microbial communities but also taxonomic and sequence space (Venter et al. Reference Venter, Remington, Heidelberg, Halpern, Rusch, Eisen, Wu, Paulsen, Nelson, Nelson, Fouts, Levy, Knap, Lomas, Nealson, White, Peterson, Hoffman, Parsons, Baden-Tillson, Pfannkoch, Rogers and Smith2004).
Unfortunately, the term metagenomics is often misleadingly used loosely to cover any culture-independent sequence-based profiling of microbial communities, particularly amplification and sequencing of molecular barcodes, such as sequences from rRNA genes. To make it clear that one is using the term in its stricter sense, the phrase shotgun metagenomics is often used.
Here, I review the progress of and prospects for the use of shotgun metagenomics in the discovery and detection of microbial pathogens in clinical samples: an approach I call diagnostic metagenomics. Due to constraints of space, I cannot provide a detailed description of all the relevant sequencing and bioinformatics protocols. These are summarized in Fig. 1 and are described in more depth in other recent reviews (Wooley et al. Reference Wooley, Godzik and Friedberg2010; Loman et al. Reference Loman, Constantinidou, Chan, Halachev, Sergeant, Penn, Robinson and Pallen2012; Morgan and Huttenhower, Reference Morgan and Huttenhower2012; Weinstock, Reference Weinstock2012).

Fig. 1. Comparisons between culture, targeted molecular assays and diagnostic metagenomics.
PROBLEMS WITH CURRENT APPROACHES
Before discussing diagnostic metagenomics, it is worth briefly reviewing the problems with existing approaches. Even though we are now well into the 21st century, diagnostic bacteriology is still largely reliant on techniques developed over a century ago in the 1880s: in particular, the detection and characterization of bacteria under the microscope using the staining technique developed by Hans Christian Gram and their propagation as colonies on solid growth media, as pioneered by Robert Koch (Koch, Reference Koch1881; Gram, Reference Gram1884). As different bacteria have different growth requirements, this leads to a complex set of workflows for handling samples in the clinical microbiology laboratory, which in turn requires input from a skilled workforce. However, one advantage of the Gram–Koch paradigm in diagnosis is that it is to a large degree open-ended, in that it will detect a wide range of pathogens, including those that are unsuspected. For this reason, microscopy and/or culture have also been used widely in other branches of diagnostic microbiology, including virology and parasitology. For example, microscopy of blood films is still used to diagnose malaria.
In situations where microscopy is cumbersome or unrewarding and culture proves difficult or even impossible, culture-independent approaches to pathogen detection have been developed, including immunoassays and detection of nucleic acid sequences. However, these approaches are generally target-specific and thus lack the ability to detect unsuspected pathogens. This means a battery of tests may have to be applied to each sample, each of which requires onerous optimization and standardization.
One attempt to combine the open-endedness of culture with the ease of culture-independent molecular methods is the use of a molecular bar code, such as rRNA gene sequences, that can be amplified en masse from a sample using primers targeting conserved sequences, with subsequent analyses focused on taxon-specific sequences flanked by the conserved regions. However, there are several problems with this approach, which have led to it being dubbed ‘the one-eyed king’ or ‘scratched lens’ (Forney et al. Reference Forney, Zhou and Brown2004; Temperton and Giovannoni, Reference Temperton and Giovannoni2012). For example, so-called ‘universal primers’ may not in fact detect all organisms, taxon counts may be inflated with some sequencing technologies and taxonomic resolution is generally poor. Thus, a 16S rRNA gene sequence may tell you that Escherichia coli is present in a sample, but will give no clue as to a strain's potential for virulence or antibiotic resistance. In addition, an entirely different set of primers will be needed to amplify homologous 18S rRNA barcodes from eukaryotes and there are no broad-range barcodes for viruses.
MICROBIOMES: A DISTRACTION FROM DIAGNOSTIC METAGENOMICS
In recent years, concepts and techniques from environmental microbial ecology have entered clinical microbiology, largely due to recognition of the importance of the human microbiome, the complex community of microbes and their genomes associated with the human body. Culture-independent sequence-based approaches to the detection and enumeration of the components of the human microbiome have been widely adopted because they provide greater ease of use and higher throughput than culture-based approaches. These efforts have been energized by steady improvements in high-throughput sequencing, with the result that large sums of money have been spent on high-profile projects to characterize the human microbiome through whole-genome sequencing of cultured isolates and through culture-independent sequence-based profiling of host-associated microbial communities (Turnbaugh et al. Reference Turnbaugh, Ley, Hamady, Fraser-Liggett, Knight and Gordon2007; Peterson et al. Reference Peterson, Garges, Giovanni, McInnes, Wang, Schloss, Bonazzi, McEwen, Wetterstrand, Deal, Baker, Di Francesco, Howcroft, Karp, Lunsford, Wellington, Belachew, Wright, Giblin, David, Mills, Salomon, Mullins, Akolkar, Begg, Davis, Grandison, Humble, Khalsa, Little, Peavy, Pontzer, Portnoy, Sayre, Starke-Reed, Zakhari, Read, Watson and Guyer2009; Arumugam et al. Reference Arumugam, Raes, Pelletier, Le Paslier, Yamada, Mende, Fernandes, Tap, Bruls, Batto, Bertalan, Borruel, Casellas, Fernandez, Gautier, Hansen, Hattori, Hayashi, Kleerebezem, Kurokawa, Leclerc, Levenez, Manichanh, Nielsen, Nielsen, Pons, Poulain, Qin, Sicheritz-Ponten, Tims, Torrents, Ugarte, Zoetendal, Wang, Guarner, Pedersen, de Vos, Brunak, Dore, Antolin, Artiguenave, Blottiere, Almeida, Brechot, Cara, Chervaux, Cultrone, Delorme, Denariaz, Dervyn, Foerstner, Friss, van de Guchte, Guedon, Haimet, Huber, van Hylckama-Vlieg, Jamet, Juste, Kaci, Knol, Lakhdari, Layec, Le Roux, Maguin, Merieux, Melo Minardi, M'rini, Muller, Oozeer, Parkhill, Renault, Rescigno, Sanchez, Sunagawa, Torrejon, Turner, Vandemeulebrouck, Varela, Winogradsky, Zeller, Weissenbach, Ehrlich and Bork2011; Le Chatelier et al. Reference Le Chatelier, Nielsen, Qin, Prifti, Hildebrand, Falony, Almeida, Arumugam, Batto, Kennedy, Leonard, Li, Burgdorf, Grarup, Jorgensen, Brandslund, Nielsen, Juncker, Bertalan, Levenez, Pons, Rasmussen, Sunagawa, Tap, Tims, Zoetendal, Brunak, Clement, Dore, Kleerebezem, Kristiansen, Renault, Sicheritz-Ponten, de Vos, Zucker, Raes, Hansen, Bork, Wang, Ehrlich, Pedersen, Guedon, Delorme, Layec, Khaci, van de Guchte, Vandemeulebrouck, Jamet, Dervyn, Sanchez, Maguin, Haimet, Winogradski, Cultrone, Leclerc, Juste, Blottiere, Pelletier, LePaslier, Artiguenave, Bruls, Weissenbach, Turner, Parkhill, Antolin, Manichanh, Casellas, Boruel, Varela, Torrejon, Guarner, Denariaz, Derrien, van Hylckama Vlieg, Veiga, Oozeer, Knol, Rescigno, Brechot, M'Rini, Merieux and Yamada2013).
It is worth highlighting three themes that have emerged from the application of microbial ecology to clinical microbiology. First is the implicit assumption of the ‘uncultured microbial majority’ (Rappe and Giovannoni, Reference Rappe and Giovannoni2003) – i.e. that most bacteria cannot be isolated in the laboratory in pure culture and so molecular methods will report a wider range of organisms than culture and are generally more sensitive. While most researchers accept these assumptions, in a provocative counter-blast, Raoult and his colleagues have claimed that adoption of a wide range of cultural approaches (which they term ‘culturomics’) is actually more sensitive than sequence-based approaches (Lagier et al. Reference Lagier, Armougom, Million, Hugon, Pagnier, Robert, Bittar, Fournous, Gimenez, Maraninchi, Trape, Koonin, La Scola and Raoult2012). By contrast, Dowd and his colleagues claim that management of infection is improved when culture-independent approaches are adopted (Dowd et al. Reference Dowd, Wolcott, Kennedy, Jones and Cox2011).
A second theme is the recognition that differences in host-associated microbial communities can influence the balance between health and disease in conditions not normally thought of as microbial or infectious in origin: for example, inflammatory bowel disease, cancer or obesity (Kinross et al. Reference Kinross, Darzi and Nicholson2011; Lozupone et al. Reference Lozupone, Stombaugh, Gordon, Jansson and Knight2012).
The third emerging theme is that it may not be sufficient to focus diagnostic efforts on single ‘headline pathogens’ in clinical samples that are thought single-handedly to cause disease. Instead, it is now recognized that interactions between organisms in a community can influence disease outcome and in some cases it might even be appropriate to treat a whole microbial community as a pathogenic entity (Rogers et al. Reference Rogers, Stressmann, Walker, Carroll and Bruce2010).
Although community profiling using molecular barcodes still predominates in the field of microbiome studies, there have now been several shotgun metagenomic studies of human-derived samples, chiefly focused on the characterization of such communities and their genes in healthy volunteers and non-infectious diseases (Qin et al. Reference Qin, Li, Raes, Arumugam, Burgdorf, Manichanh, Nielsen, Pons, Levenez, Yamada, Mende, Li, Xu, Li, Li, Cao, Wang, Liang, Zheng, Xie, Tap, Lepage, Bertalan, Batto, Hansen, Le Paslier, Linneberg, Nielsen, Pelletier, Renault, Sicheritz-Ponten, Turner, Zhu, Yu, Li, Jian, Zhou, Li, Zhang, Li, Qin, Yang, Wang, Brunak, Dore, Guarner, Kristiansen, Pedersen, Parkhill, Weissenbach, Bork, Ehrlich and Wang2010, Reference Qin, Li, Cai, Li, Zhu, Zhang, Liang, Zhang, Guan, Shen, Peng, Zhang, Jie, Wu, Qin, Xue, Li, Han, Lu, Wu, Dai, Sun, Li, Tang, Zhong, Li, Chen, Xu, Wang, Feng, Gong, Yu, Zhang, Zhang, Hansen, Sanchez, Raes, Falony, Okuda, Almeida, LeChatelier, Renault, Pons, Batto, Zhang, Chen, Yang, Zheng, Li, Yang, Wang, Ehrlich, Nielsen, Pedersen, Kristiansen and Wang2012; Arumugam et al. Reference Arumugam, Raes, Pelletier, Le Paslier, Yamada, Mende, Fernandes, Tap, Bruls, Batto, Bertalan, Borruel, Casellas, Fernandez, Gautier, Hansen, Hattori, Hayashi, Kleerebezem, Kurokawa, Leclerc, Levenez, Manichanh, Nielsen, Nielsen, Pons, Poulain, Qin, Sicheritz-Ponten, Tims, Torrents, Ugarte, Zoetendal, Wang, Guarner, Pedersen, de Vos, Brunak, Dore, Antolin, Artiguenave, Blottiere, Almeida, Brechot, Cara, Chervaux, Cultrone, Delorme, Denariaz, Dervyn, Foerstner, Friss, van de Guchte, Guedon, Haimet, Huber, van Hylckama-Vlieg, Jamet, Juste, Kaci, Knol, Lakhdari, Layec, Le Roux, Maguin, Merieux, Melo Minardi, M'rini, Muller, Oozeer, Parkhill, Renault, Rescigno, Sanchez, Sunagawa, Torrejon, Turner, Vandemeulebrouck, Varela, Winogradsky, Zeller, Weissenbach, Ehrlich and Bork2011; Karlsson et al. Reference Karlsson, Tremaroli, Nookaew, Bergstrom, Behre, Fagerberg, Nielsen and Backhed2013). Curiously, comparatively little attention has focused on the question of whether metagenomics can be used to discover, detect and characterize pathogens in samples from diseased individuals.
VIRUS DISCOVERY
Not surprisingly, given the difficulty or impossibility of culturing most viruses, virologists were the first to explore the potential of open-ended, shotgun sequencing to identify and detect human-associated viruses. The genomes of DNA viruses can be recovered through shotgun sequencing of DNA directly extracted from a sample. To detect RNA viruses, RNA extracted from a sample has to be converted to cDNA (Batty et al. Reference Batty, Wong, Trebes, Argoud, Attar, Buck, Ip, Golubchik, Cule, Bowden, Manganis, Klenerman, Barnes, Walker, Wyllie, Wilson, Dingle, Peto, Crook and Piazza2013). In such cases, one is searching for a viral genome in the midst of a sample-derived metatranscriptome.
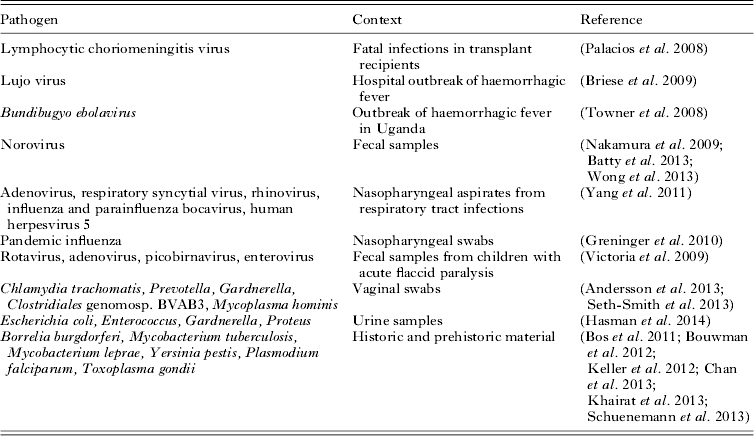
Diagnostic viral metagenomics has now been used to detect an unknown pathogen in a number of high-profile cases or outbreaks of disease (Table 1). In an early example, sequencing of cDNA from three transplant recipients with fatal infections yielded 14 sequences resembling segments of the genome of lymphocytic choriomeningitis virus (Palacios et al. Reference Palacios, Druce, Du, Tran, Birch, Briese, Conlan, Quan, Hui, Marshall, Simons, Egholm, Paddock, Shieh, Goldsmith, Zaki, Catton and Lipkin2008). Similarly diagnostic metagenomics uncovered a novel arenavirus responsible for a hospital outbreak of haemorrhagic fever in South Africa (Briese et al. Reference Briese, Paweska, McMullan, Hutchison, Street, Palacios, Khristova, Weyer, Swanepoel, Egholm, Nichol and Lipkin2009) and identified a novel species of Ebola virus (Bundibugyo ebolavirus) from Uganda (Towner et al. Reference Towner, Sealy, Khristova, Albarino, Conlan, Reeder, Quan, Lipkin, Downing, Tappero, Okware, Lutwama, Bakamutumaho, Kayiwa, Comer, Rollin, Ksiazek and Nichol2008).
Table 1. Examples of diagnostic metagenomics
Metagenomics has now been used widely in virus discovery (Capobianchi et al. Reference Capobianchi, Giombini and Rozera2013; Smits and Osterhaus, Reference Smits and Osterhaus2013), revealing the existence of numerous ‘orphan viruses’ that have not been associated with any disease and thereby establishing the existence of a normal human virus microbiome or ‘virome’ (Li and Delwart, Reference Li and Delwart2011; Lecuit and Eloit, Reference Lecuit and Eloit2013). Diagnostic metagenomics has also been used for the detection of established viral pathogens (e.g. influenza and norovirus) in clinical samples (Nakamura et al. Reference Nakamura, Yang, Sakon, Ueda, Tougan, Yamashita, Goto, Takahashi, Yasunaga, Ikuta, Mizutani, Okamoto, Tagami, Morita, Maeda, Kawai, Hayashizaki, Nagai, Horii, Iida and Nakaya2009).
DETECTION OF BACTERIAL PATHOGENS
In a pioneering 2008 metagenomics survey of fecal samples from a single individual, Nakamura and colleagues showed the feasibility of detecting bacterial pathogens such as Campylobacter through unbiased sequencing of DNA extracted from stool samples (Nakamura et al. Reference Nakamura, Maeda, Miron, Yoh, Izutsu, Kataoka, Honda, Yasunaga, Nakaya, Kawai, Hayashizaki, Horii and Iida2008). In this case, 156 Campylobacter sequences were found in a sample taken during a bout of illness, but were absent from a convalescent sample from the same individual.
More recently, Loman, myself and others, in collaboration with clinical microbiologists from Hamburg, explored the potential of diagnostic metagenomics on stool samples collected during the outbreak of Shiga-toxigenic E. coli O104:H4 that struck Germany in May–June 2011 (Loman et al. Reference Loman, Constantinidou, Christner, Rohde, Chan, Quick, Weir, Quince, Smith, Betley, Aepfelbacher and Pallen2013). Contrary to expectations, we obtained deep coverage of the outbreak strain genome from several stool metagenomes, even using a benchtop-sequencing platform (the Illumina MiSeq). We subsequently sequenced a larger set of stool metagenomes on a higher-throughput instrument (the HiSeq2500) and in some cases obtained much deeper coverage of a pathogen genome. We also recovered genome-level coverage of other pathogens (Campylobacter jejuni, Clostridium difficile, Salmonella enterica) that had been detected by routine microbiological investigation in several STEC-negative samples.
In this study, we established proof-of-principle that metagenomics could be used not only to detect, but also to characterize, bacterial pathogens within a sample. For example, we were able to obtain typing data and phylogenetic profiles for strains of use in epidemiology and population genetics directly from the metagenomes without culture. Another study has recently confirmed the utility of metagenomics in recovering single-nucleotide polymorphisms from different strains of gut bacteria (Schloissnig et al. Reference Schloissnig, Arumugam, Sunagawa, Mitreva, Tap, Zhu, Waller, Mende, Kultima, Martin, Kota, Sunyaev, Weinstock and Bork2013).
In our study, we also demonstrated the open-endedness of the approach by detecting organisms that we did not expect to find. In one sample positive for C. difficile, we also discovered an abundance of sequences from Campylobacter concisus, which has been described as an emerging diarrhoeal pathogen. In several STEC-positive samples, we discovered sequences from C. difficile. These findings not only illustrate the ability of diagnostic metagenomics to find ‘unknown unknowns’ but also question the assumption that a single headline pathogen causes disease on its own: are the second-line pathogens in these samples merely colonizing the altered microenvironment caused by disease or are they also contributing to pathology?
In the initial phase of our study, we aligned reads from the metagenomes against the known outbreak strain genome. This might be seen as ‘cheating’, as we were in effect using a crib sheet to answer the question of whether the strain was present or not. In a subsequent phase, we used the MetaPhlAn pipeline to identify reads that were likely to originate from pathogens (Segata et al. Reference Segata, Waldron, Ballarini, Narasimhan, Jousson and Huttenhower2012). It was this more open-ended approach that allowed us to discover the unknown unknowns. In the final phase of the project, we devised an entirely de novo approach to identify the outbreak strain and recover its genome. We started with two simplifying assumptions: that strain-specific reads from the outbreak strain would be present in at least half of the samples and that they would be absent from the stool metagenomes of healthy individuals. Applying these criteria to the collection of fecal metagenomes, we obtained a strain-specific set of reads. By clustering these reads with other reads that showed the same patterns of abundance in different samples, we were able to obtain the entire outbreak strain genome.
Several recent studies have shown the ability of diagnostic metagenomics to make a diagnosis of infection long after the individual is dead. In 2011, a study of the genome of the Tyrolean ice mummy Ötzi revealed sequences from the bacterium Borrelia burgdorferi, identifying the first known case of Lyme disease (Keller et al. Reference Keller, Graefen, Ball, Matzas, Boisguerin, Maixner, Leidinger, Backes, Khairat, Forster, Stade, Franke, Mayer, Spangler, McLaughlin, Shah, Lee, Harkins, Sartori, Moreno-Estrada, Henn, Sikora, Semino, Chiaroni, Rootsi, Myres, Cabrera, Underhill, Bustamante, Vigl, Samadelli, Cipollini, Haas, Katus, O'Connor, Carlson, Meder, Blin, Meese, Pusch and Zink2012). Krause and colleagues subsequently recovered a genome from the leprosy bacillus, Mycobacterium leprae, from the metagenome obtained from a historical dental sample (Schuenemann et al. Reference Schuenemann, Singh, Mendum, Krause-Kyora, Jager, Bos, Herbig, Economou, Benjak, Busso, Nebel, Boldsen, Kjellstrom, Wu, Stewart, Taylor, Bauer, Lee, Wu, Minnikin, Besra, Tucker, Roffey, Sow, Cole, Nieselt and Krause2013). Around the same time, we described the first example of post-mortem metagenomic diagnosis of tuberculosis, from mummified lung tissue from a young woman who died in 1797 (Chan et al. Reference Chan, Sergeant, Lee, Minnikin, Besra, Pap, Spigelman, Donoghue and Pallen2013). We found evidence of mixed infection with two distinct genotypes of Mycobacterium tuberculosis, both related to strains circulating in Europe and North America.
Recently, diagnostic metagenomics has also been applied to urine samples, allowing the recovery of uropathogen genome sequences directly from clinical material without culture, while also providing taxonomic, epidemiological and phylogenetic data (Hasman et al. Reference Hasman, Saputra, Sicheritz-Ponten, Lund, Svendsen, Frimodt-Moller and Aarestrup2014). In low-biomass samples, it may still be possible to obtain enough pathogen DNA for sequencing thanks to target-independent whole-genome amplification. A recent metagenomics study using this approach on a vaginal swab reported recovery not just of a chlamydial genome, but also high genome-wide coverage for Prevotella melaninogenica, Gardnerella vaginalis and Mycoplasma hominis (Andersson et al. Reference Andersson, Klein, Lilliebridge and Giffard2013).
RELEVANCE TO PARASITOLOGY
Diagnostic metagenomics has so far seen little use in the detection of parasitic infection, although there is no reason to expect that parasitology will not soon catch up with other branches of microbiology in this regard. In a single unreplicated study, it has been claimed that Plasmodium and Toxoplasma sequences can be found in the metagenomes of Egyptian mummies (Khairat et al. Reference Khairat, Ball, Chang, Bianucci, Nerlich, Trautmann, Ismail, Shanab, Karim, Gad and Pusch2013). However, no sequence data were presented, so it is hard to assess the veracity of this claim. Shotgun metagenomics of cDNA has been used to analyse the taxa associated with Lutzomyia longipalpis, the vector of visceral leishmaniasis (McCarthy et al. Reference McCarthy, Diambra and Rivera Pomar2011). Potential future applications of metagenomics in parasitology include recovery of genomic-epidemiological data (Griffing et al. Reference Griffing, Mixson-Hayden, Sridaran, Alam, McCollum, Cabezas, Marquino Quezada, Barnwell, De Oliveira, Lucas, Arrospide, Escalante, Bacon and Udhayakumar2011; Khaireh et al. Reference Khaireh, Assefa, Guessod, Basco, Khaireh, Pascual, Briolant, Bouh, Farah, Ali, Abdi, Aden, Abdillahi, Ayeh, Darar, Koeck, Rogier, Pradines and Bogreau2013) and determining the influence of parasites on the microbial ecology of the gut. It remains to be seen how much parasite DNA can be detected in samples without amplification or capture: this might present a challenge for bloodstream infections. However, in samples such as feces, where parasite biomass is likely to be relatively high, this will be much less of an issue. Also of interest might be the recovery of parasite sequences from historical material; museum samples might provide insights into extinct parasite lineages.
FUTURE DIRECTIONS
Diagnostic metagenomics brings the promise of an open-ended, assumption-free one-size-fits-all workflow that could be applied to any specimen to detect any kind of pathogen (viruses, bacteria, fungi and parasites). Nonetheless, the sceptic will dismiss diagnostic metagenomics as ‘a sledgehammer to crack a nut’ in that it is orders of magnitude too expensive for routine use and requires significant expert technical and bioinformatics input. Plus, why bother when there are easier methods? However, with likely future improvements in the ease, throughput and cost-effectiveness of sequencing, twinned with commoditization of laboratory and informatics workflows, one can foresee a tipping point when a unified automated metagenomics-based workflow will start to compete with the plethora of methods currently in use in the diagnostic laboratory, while also delivering additional useful information (e.g. on genomic epidemiology, antimicrobial resistance, virulence). In the case of an outbreak of life-threatening infection with an unknown cause, we may already have reached that point.
However, we are faced with a number of challenges before diagnostic metagenomics can be used routinely. One is the broad range of abundances of different taxa within a specimen. Metagenomics does appear to be able to deliver whole-genome coverage of the most abundant organisms, but it is probably not safe to assume that the pathogenic species in a sample are always the most abundant. In addition, assembly of a metagenome presents considerable computational challenges, although there have been recent methodological improvements in this area.
A number of potential approaches to simplifying the problem can be envisaged. It is possible to capture or enrich for cells or sequences from a pathogen of interest. This approach has been applied to clinical samples to obtain genomes from Chlamydia genomes without culture (Seth-Smith et al. Reference Seth-Smith, Harris, Skilton, Radebe, Golparian, Shipitsyna, Duy, Scott, Cutcliffe, O'Neill, Parmar, Pitt, Baker, Ison, Marsh, Jalal, Lewis, Unemo, Clarke, Parkhill and Thomson2013) and has been used to recover leprosy, tuberculosis and plague genomes from historical and ancient material (Bos et al. Reference Bos, Schuenemann, Golding, Burbano, Waglechner, Coombes, McPhee, DeWitte, Meyer, Schmedes, Wood, Earn, Herring, Bauer, Poinar and Krause2011; Bouwman et al. Reference Bouwman, Kennedy, Muller, Stephens, Holst, Caffell, Roberts and Brown2012; Schuenemann et al. Reference Schuenemann, Singh, Mendum, Krause-Kyora, Jager, Bos, Herbig, Economou, Benjak, Busso, Nebel, Boldsen, Kjellstrom, Wu, Stewart, Taylor, Bauer, Lee, Wu, Minnikin, Besra, Tucker, Roffey, Sow, Cole, Nieselt and Krause2013). If used alone, this risks compromising the open-endedness of diagnostic metagenomics, but such approaches could be used as adjuncts to an open-ended approach.
It is possible to simplify the analysis of the metagenome by sorting cells or DNA fragments into sub-metagenomic bins: for example, a recent study reported use of cell sorting to recover the genome of an unculturable bacterium from a hospital sink biofilm (McLean et al. Reference McLean, Lombardo, Badger, Edlund, Novotny, Yee-Greenbaum, Vyahhi, Hall, Yang, Dupont, Ziegler, Chitsaz, Allen, Yooseph, Tesler, Pevzner, Friedman, Nealson, Venter and Lasken2013). Another attractive option would be to normalize a sample, so that all taxa and their sequences appear equally abundant. Experimental approaches to a similar problem for cDNA libraries have proven successful using nuclease cleavage of abundant re-annealed DNA duplexes (Christodoulou et al. Reference Christodoulou, Gorham, Herman and Seidman2011) and this might also work for metagenomes. The imminent launch of cheap and accessible long-read high-throughput nanopore sequencing also promises to improve the prospects for recovering pathogen genomes from metagenomes (Maitra et al. Reference Maitra, Kim and Dunbar2012). If these technologies live up to the hype, it will soon be possible to pipette a sample into a highly portable USB-stick-sequencer and see sequences stream off on to a computer in real time, which could transform field work and make inroads into the diagnostic laboratory.
In conclusion, it is clear that diagnostic metagenomics works in a research setting and already has a role to play in identifying the causes of unknown illnesses and outbreaks. A number of hurdles need to be overcome before it can be integrated into routine practice. Nonetheless, it is possible to envisage a tipping point, sometime in the next decade or two, when the old Gram–Koch paradigm gives way to a diagnostic metagenomics approach to the detection and characterization of pathogens in clinical samples fit for the 21st century.
ACKNOWLEDGEMENTS
I thank the British Society for Parasitology for the invitation to speak at their 2013 Autumn Symposium.


 Open access
Open access