


Crimean-Congo haemorrhagic fever virus (CCHFV) is prevalent in Africa, Asia, Russia and the Balkans. It is a tick-borne virus transmitted by ticks belonging to the Hyalomma genus. The virus is a member of Nairovirus genus and the family Bunyaviridae. The single-stranded, negative-sense, segmented RNA genome of CCHFV consists of small (S), medium (M), and large (L) segments of about 1672, 5360 and 12 200 base pairs (bp), respectively [Reference Elliott1]. The L segment encodes the viral polymerase, the M segment encodes the precursor polyprotein for glycoproteins, Gn and Gc and the S segment encodes the viral nucleocapsid protein (NP). Phylogenetic studies, based on the S, M and L segments, have grouped CCHFV into seven groups or genotypes. The tree topologies based on S and L gene sequence data are more likely to correspond with geographical location, with genotype 1 found predominantly in Asia and the Middle East, genotype 2 in Asia, genotype 3 in Africa, genotypes 4 and 6 in Europe and genotypes 5 and 7 in Africa [Reference Deyde2, Reference Mild3]. In some locations, more than one genotype has been identified. Incongruencies in the groupings have been identified based on M gene data and are likely to be the result of reassortment [Reference Deyde2, Reference Mild3]. Several phylogenetic studies have been performed to determine the genetic diversity of the S segment, but antigenic diversity and serological cross-reactivity between geographically distinct isolates has not been well investigated.
Ticks belonging to the Hyalomma genus are present in southern and southwestern Europe. During the last decade CCHFV emerged and re-emerged in several Balkan countries, Turkey, southwestern regions of the Russian Federation, and the Ukraine [Reference Maltezou4]. Re-emergence of the virus in Central Africa has also been described [Reference Grard5]. The re-emergence of CCHFV in 1999, in southwestern regions of the Russian Federation after a 27-year absence and the recent emergence and escalation of numbers of human cases in the Balkans has raised concerns that this virus could spread to currently non-endemic regions of Europe [Reference Maltezou4]. The reasons for re-emergence are probably multi-factorial. Global warming with changes in weather patterns that influence tick populations, increased animal movement as a result of livestock trade as well as human activities such as changes in farming practices and land development contribute to emergence [Reference Maltezou4, Reference Maltezou and Papa6]. Warmer temperatures are conducive to increased tick activity and populations, which may impact on higher risk of transmission. Monitoring possible spread of the virus will require development of safe, standardized reagents and assays that can contribute to increasing laboratory capacity with facilities to perform diagnosis and surveillance. Currently, for most enzyme-linked immunosorbent assays (ELISAs), preparation of reagents is dependent on culturing the virus in cell cultures or from infected mouse brain preparations which require biosafety level-4 (BSL-4) facilities. Recombinant antigens have been developed and evaluated by ELISAs using limited numbers of clinical samples. Results indicate that recombinant antigens have potential as safe reagents but their usefulness for early detection of antibody needs to be confirmed.
The global usefulness of recombinant antigens prepared using specific viral proteins as opposed to native antigens may limit the applicability of these antigens if there are antigenically diverse strains. To date CCHFV is considered to comprise of one antigenically similar species based on native antigens. Considering the genetic diversity of the genotypes there is potential for lack of cross-reactivity to specific proteins from diverse strains which could influence reagent preparation for epidemiological surveys.
To identify the isolate with the least nucleotide and amino-acid homology compared to a South African isolate, the open-reading frame (ORF) encoding the NP of 45 CCHFV isolates were retrieved from GenBank (GenBank accession numbers available on request). Data were available for isolates from Bulgaria, China, Democratic Republic of Congo, Greece, Iran, Mauritania, Nigeria, Oman, Pakistan, Russia, South Africa, Sudan, Tajikistan, Turkey, Uganda, Uzbekistan, Yugoslavia and Kosovo. All available sequence data in GenBank for the complete ORF of the NP gene were included in the study. Sequence alignments were generated with multiple alignment using fast-Fourier transform (MAFFT) software, version 6 (http://mafft.cbrc.jp). The phylogenetic analysis was performed using Molecular Evolutionary Genetics Analysis (MEGA) v. 4 and the bootstrap neighbour joining method with 1000 replicates. Sequence divergence was determined using MEGA to calculate the average p distances within and between groups. Seven genotypes were identified (Fig. 1). Genotype 1 comprised of isolates identified in Asia and the Middle East, genotype 2 comprised of isolates from Asia, genotype 3 comprised of isolates from South Africa and West Africa, genotype 4 was from Europe and Turkey, genotype 5 from the Democratic Republic of the Congo, Africa, genotype 6 was a single isolate (designated AP92) from Greece and genotype 7 included isolates from West Africa. Genotypes 1, 2, and 4 comprised of isolates from similar geographical locations, whereas African isolates were identified in more than one genotype (3, 5, 7).
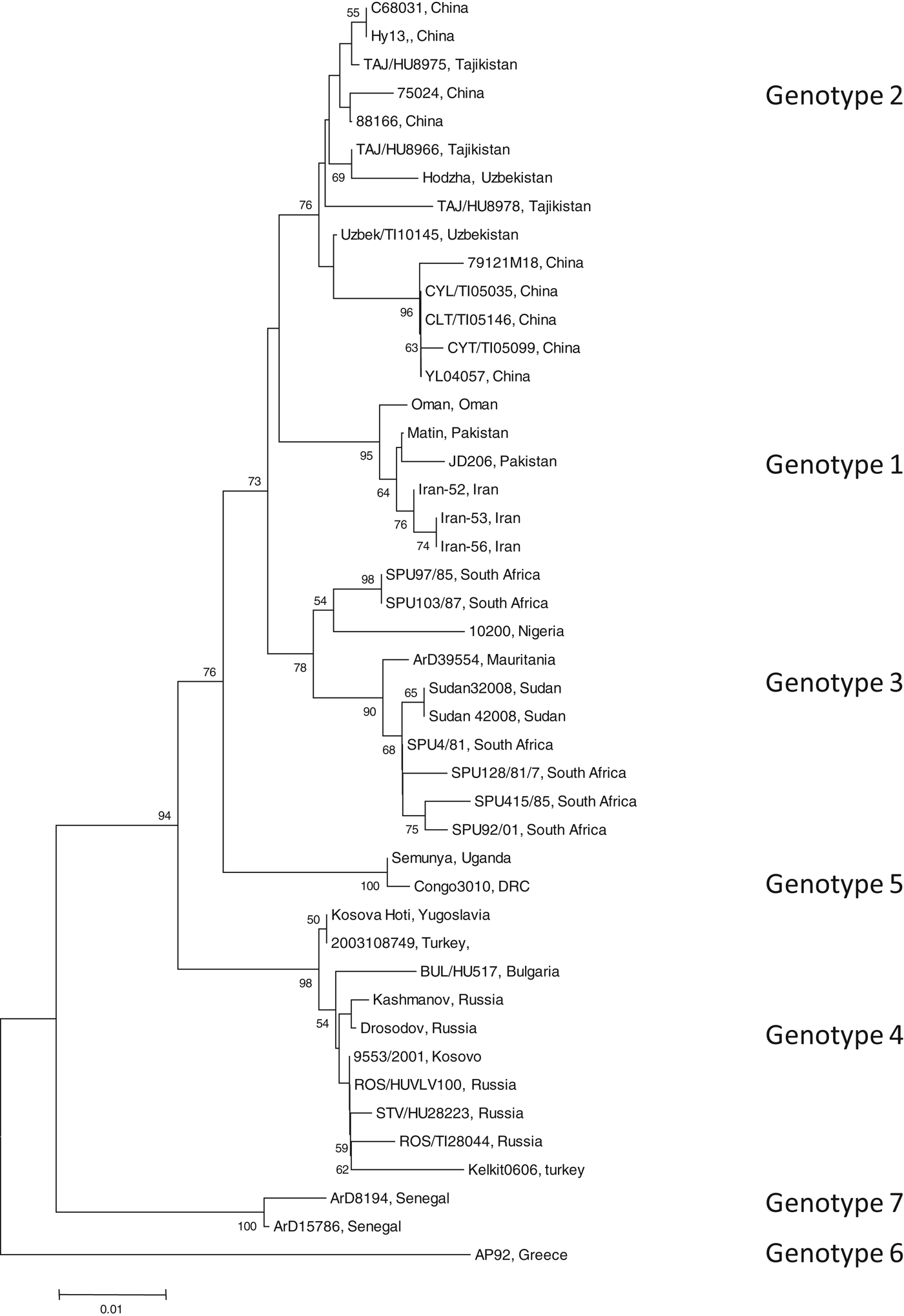
Fig. 1. Phylogenetic tree using nucleotide sequence data for the complete open reading frame encoding the nucleoprotein. There were a total of 1443 positions in the final dataset. Node values below 50 were omitted. Horizontal distances are proportional to nucleotide differences. Isolates are labelled according to name of the strain and country of isolation and genotyped using nomenclature previously described [Reference Mild3]. Isolates SPU415/85 and AP92 for which sequence data were used to prepare recombinant antigens are highlighted. Phylogenetic analyses were conducted using MEGA v. 4.
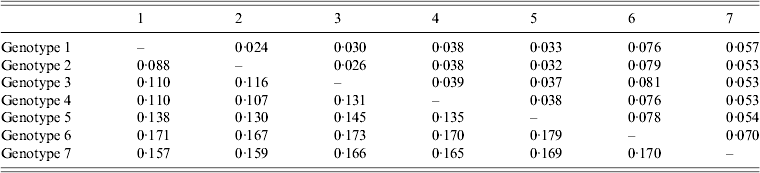
The average genetic p distance is the proportion of nucleotide or predicted amino-acid sites at which the two sequences differ. Within each genotype, nucleotide distances ranged from 1·2% to 4·5% and predicted amino-acid distances from 0·2% to 1·2%. Table 1 summarizes the p distance between the genotypes. Genotypes 1 and 2 were the most similar with homology of 91·2%, and, excluding genotype 6, genotypes 5 and 7 were the most diverse, exhibiting maximum homology of 83·1%. Excluding the diversity between genotypes 5 and 7, singly, AP92 (genotype 6) was consistently more diverse (homology ranging from 82·1% to 83·3%). The amino-acid diversity between genotypes was significantly lower than the nucleotide diversity between the different genotypes. Amino-acid diversity between groups excluding strain AP92 ranged from 2·4% to 5·7%. The nucleotide homology between genotypes 5 and 7 (83·1%) translated to an amino-acid homology of 94·6%. The Greek isolate showed the least homology with other isolates with similarity ranging from 91·3% to 93·0%. In agreement with previous studies, despite the relatively high percentage of nucleotide changes, there were fewer changes in the predicted protein with pairwise nucleotide differences ranging from 0·4% to 21% and deduced amino-acid differences ranging from 0% to 8·7% indicating that many of the nucleotide differences were silent, resulting in synonymous amino-acid codons [Reference Deyde2, Reference Mild3, Reference Burt and Swanepoel7]. This could explain the antigenic similarity of the nucleocapsid region observed between different isolates worldwide. The broad geographical distribution of strains is probably related to the distribution and dispersal of the vectors of the virus. Based on the sequence data from the NP gene, isolates circulating in Africa clustered in one of three genotypes, while isolates circulating in Eastern Europe and Turkey, and the Middle East and Asia and Greece grouped in geographically defined genotypes.
Table 1. Mean distance between genotypes based on nucleotide sequence data (bottom left) and predicted amino acids (top right). The number of base or amino-acid differences per site from averaging over all sequence pairs between groups is shown
The sequence for the gene encoding the CCHFV NP of isolate AP92 (retrieved from GenBank, accession no. DQ211 638) was analysed using GenScript rare codon analysis tool software (GenScript, USA). The gene was codon-optimized for expression in a bacterial system using GenScript's OptimumGene™ gene design tools to optimize codon usage, guanine cytosine (GC) content and to eliminate polyadenylation sites, splicing sites, killer motifs and RNA secondary structure. The optimized gene was synthesized by GenScript and supplied in pUC57 with BamHI restriction sites engineered at the 5′ and 3′ ends. The codon-optimized gene was rescued from pUC57 plasmid using restriction enzyme digestion and subsequently subcloned into pCOLD-TF cold-shock expression vector (TakaraBio, France) to generate pColdTF-opAP92. Glycerol stock of E. coli cells expressing recombinant CCHF NP using plasmid, pColdTF-opCCHFVNP, prepared in a previous study and incorporating a codon-optimized gene for a South African isolate (415/85), was included in this study [Reference Samudzi8]. The recombinant NP had previously been shown to detect IgG antibody against CCHFV in South African survivors. Similarly, a glycerol stock of cultures of pCold-TF plasmid lacking an inserted gene was used as control. The construct, pColdTF-opAP92, was used to transform E. coli OverExpress C43 (DE3) competent cells (Lucigen, USA). Positive transformants were identified by PCR amplification of colonies using CCHFV-specific primers and confirmed using restriction enzyme digestions. A positive transformant and glycerol stocks of the cultures of pCOLD-TF plasmid control and pColdTF-opCCHFVNP were initially propagated in 5 ml Luria–Bertani (LB) broth containing 100 μg/ml ampicillin and incubated overnight at 37°C. Recombinant protein expression was induced as described previously [Reference Samudzi8]. The recombinant CCHFV NP fusion proteins designated recNP 415 protein and recNP AP92 protein, and recTF protein were all purified from the soluble fraction using Protino Ni-TED resin according to the manufacturers' instructions for purification under native conditions (Machery-Nagel, Germany). Expression and purification of the proteins were confirmed by SDS–PAGE.
In-house ELISA using recNP 415 was performed as described previously [Reference Samudzi8]. Checkerboard titrations of each reagent were used to optimize the ELISA protocol using recNP AP92. Unless stated otherwise, all volumes were 100 μl/well, plates were incubated for 1 h at 37°C and washed 3 × 15 s with 0·1% Tween-20 in phosphate-buffered saline (PBS), pH 7·0. The diluent was 2% skimmed milk/PBS. A 96-well microtitre Polysorb plate was coated overnight at 4°C with recombinant NP diluted at 1:1000 and recombinant pColdTF antigen diluted 1:4000 in PBS. The plates were washed, and human serum samples diluted 1:100 in diluent were added to each well in duplicate. The plates were incubated, washed and anti-human IgG horseradish peroxidase (HRP; Zymed Laboratories, USA) diluted 1:6000 was added to each well. After further incubation and washing, positive reactors were visualized using the substrate 2,2′-azino-di-ethyl-benzothiazoline-sulfonic acid peroxidase substrate (ABTS; Kirkegaard & Perry Laboratories, USA). The plates were incubated at room temperature (22–25°C) in the dark for 30 min and the optical density (OD) values were read at 405 nm. Subsequent to the overnight coating, the ELISA takes 3·5 h to complete. The net OD value (OD on CCHF NP minus OD on pColdTF) for each test and negative serum was determined. A panel of negative sera from six patients with no history of CCHFV infection was used to determine cut-off values in the ELISA. Known positive serum samples from 14 convalescent patients who survived CCHFV infection in South Africa were collected and screened for the presence of IgG antibodies in an indirect ELISA. Ethical approval was obtained for the study (ETOVS no. 152/06) and informed consent was obtained from all patients that provided samples for the study. The patients had previously been confirmed as CCHF positive by the Centre for Emerging and Zoonotic Diseases, National Institute for Communicable Diseases, Johannesburg using virus isolation, RT–PCR or detection of IgM antibody against CCHFV during acute illness. The serum samples used in this study were confirmed IgG positive in a related study using immunofluorescent antibody tests supplied by EuroImmun (Germany).
The optimized genes encoding the CCHFV NP of SPU415/85 and AP92 comprise 1443 bp, excluding the start and stop codons, encoding for a protein of 481 amino-acid residues. For AP92 gene, codon optimization increased the codon adaptation index (CAI) from 0·62 to 0·84. CAI is a measure of codon usage bias of a gene and is given a score from 0 to 1 determined from the CAI values of the individual codons. Improving the CAI to a score closer to 1 may contribute to increasing expression levels. OverExpress C43 cells transformed with pCOLD-TF-opCCHFVNP and pCOLD-TF-opCCHFAP92 expressed high levels of 6 × His-tagged NP proteins with a 48 kDa-associated chaperone protein, Trigger Factor, after a 24-h incubation period. Solubility studies indicated presence of protein in both soluble and insoluble phases hence recombinant CCHFV NP fusion proteins, designated recNP 415, recNP AP92 and recTF proteins were purified from the soluble phase using a native purification method. In a previous study recNP 415 was purified from the insoluble phase using a denaturing method [Reference Samudzi8]. The protein was found to be unstable and it was hoped that using the soluble fraction would improve stability. The functional activity of the soluble protein fraction for detection of IgG antibody against CCHFV was confirmed by testing serum samples from survivors. The soluble protein fraction was able to detect IgG antibody as effectively as the protein previously purified from the insoluble fraction; however, it was not significantly more stable over time.
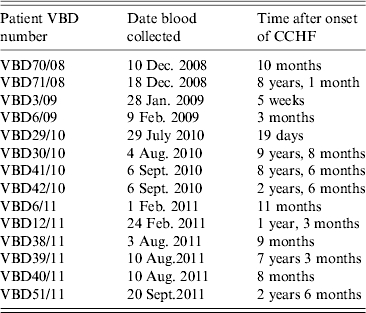
Serum samples were collected from 14 patients at various intervals after disease, from day 19 to 9 years and 8 months, as shown in Table 2. Each serum sample was tested in duplicate. A panel of six negative sera were used to determine the cut-off value and establish a level of non-specific background. Sera were reacted against mock antigen to determine background levels. The cut-off was calculated using net OD values determined from the mean net OD of the negative panel plus 2 standard deviations. A cut-off of 0·12 was calculated for recNP 415 and 0·20 for recNP AP92. Figure 2 illustrates the mean net OD values plus minimum and maximum values. All sera tested reacted with the recNP 415 protein (Fig. 2 a). Similar results were obtained using the recNP AP92 protein, with 13/14 sera reacting positively (Fig. 2 b).
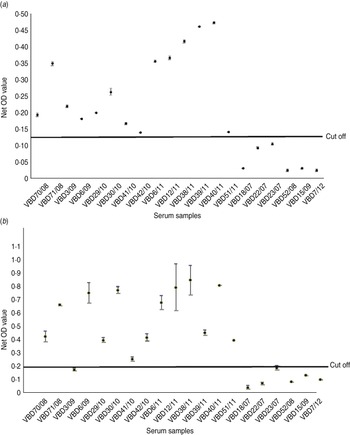
Fig. 2. Detection of anti-CCHFV IgG by ELISA using recombinant antigens. The maximum and minimum net optical density (OD) values are shown for each serum sample and the cut-off value is shown. Negative reactors are depicted below the cut-off line. (a) recNP 415; (b) recNP AP92.
Table 2. Serum samples from CCHF survivors in South Africa. Serum samples and time after illness that samples were collected
To develop standardized reagents that can be used worldwide, global diversity of CCHFV isolates, based on nucleic acid, protein and antigenic level must be investigated. There is high rate of genetic variation in CCHFV and although the general pattern of genetic diversity observed in the S segment appears to be related to the geographical distribution of the virus (genotypes 1, 2, 4), there are similar subtypes found in distant geographical locations (genotype 3) [Reference Deyde2, Reference Mild3]. The use of native antigens requires BSL-4 facilities for culturing and handling the virus. This subsequently limits the number of laboratories worldwide that can prepare reagents for diagnostic and epidemiological tools. The use of recombinant antigens provides a safe alternative. The NP is considered the most abundant protein in many Bunyavirus infections and has previously been used for preparation of recombinant antigen for detection of antibody against CCHFV [Reference Samudzi8–Reference Dowall10]. A genetic analysis of isolates using complete sequence data from GenBank, identified South African isolate 415/85 and a single isolate from Greece, AP92, as having the highest nucleotide and amino-acid diversity (8·7%) and hence were selected to compare serological cross-reactivity. Amino-acid sequences for the NP of South African strain SPU415/85 and Greek strain AP92 were aligned for comparison and a total of 41 amino-acid changes were identified occurring throughout the NP. The majority of changes involved amino acids with similar properties with respect to polarity and hydrophobicity that are unlikely to influence the secondary structure of the protein. More significantly there were only two amino-acid changes involving proline at amino acids 47 and 73. In previous studies truncation of recombinant NP antigens suggested that the central region of the NP and the region from amino acids 123 to 396 include a highly antigenic region of the NP with application in development of antibody detection assays [Reference Burt11–Reference Saijo13].
ELISA is a sensitive assay that has been developed for CCHFV serosurveillance and also diagnostic applications [Reference Samudzi8–Reference Dowall10, Reference Marriot12–Reference Shepherd, Swanepoel and Gill17]. In a previous study recombinant NP derived from AP92 strain was able to detect IgG antibody in 7/8 human sera collected from haemorrhagic fever patients in Albania, Pakistan and Saudi Arabia and from serosurveys in Greece and Senegal [Reference Marriot12]. In this study, two in-house ELISAs were developed using genetically diverse genes to express recombinant CCHFV NP. A panel of positive samples from South African CCHFV survivors were shown to react against both recombinant NP antigens. One serum from a patient collected 5 weeks after onset of illness was consistently negative by ELISA. The detection of antibody in the remaining samples suggests that it is unlikely to be due to antigenic differences and more likely related to sensitivity of the ELISA for low antibody titre samples. Use of a larger panel of sera, particularly collected from early stages of infection, is required to determine the sensitivity of the antigen for diagnostic application. Application of the antigen in an ELISA for detection of IgM antibody has not been evaluated but would be important for diagnostic application. Addressing the issue of protein stability would be paramount in considering commercial availability of the ELISA. Cross-reactivity was assessed to determine the usefulness and application of recombinant antigens in different laboratories worldwide. Although the assays described in this study will require further validation with larger numbers of samples than is available in our laboratory the results suggest that there is sufficient serological cross-reactivity between genetically and geographically distinct CCHFV isolates to allow recombinant NP to detect antibody in sera from geographically distinct locations. Hence a recombinant antigen expressed from a South African NP gene is likely to have application in other laboratories worldwide.
ACKNOWLEDGEMENTS
The authors acknowledge the Polio Research Foundation for funding the project and the National Health Laboratory Services, Universitas, and Faculty of Health Sciences, University of the Free State where the study was performed.
DECLARATION OF INTEREST
None.




