



Introduction
Crop wild relatives (CWRs) play a key role in breeding by providing beneficial trait characteristics for improvement of related crops. However, the inter- and intraspecific diversity in CWRs is in decline due to global threats such as ecosystem degradation and climate change (Ford-Lloyd et al., Reference Ford-Lloyd, Schmidt, Armstrong, Barazani, Engels, Hadas, Hammer, Kell, Kang, Khoshbakht, Li, Long, Lu, Ma, Nguyen, Qiu, Ge, Wei, Zhang and Maxted2011). Given the importance of CWRs for agriculture, different ex situ conservation strategies have been developed (Heywood et al., Reference Heywood, Casas, Ford-Lloyd, Kell and Maxted2007). Seed banking has the advantage over other ex situ methods in that it allows long-term storage of plant material at a reasonable cost and that it can include a larger part of the gene pool (Li and Pritchard, Reference Li and Pritchard2009). Nevertheless, many CWR ex situ seed banks are underused because of the absence of genetic diversity information (Schoen and Brown, Reference Schoen and Brown2001; Dempewolf et al., Reference Dempewolf, Baute, Anderson, Kilian, Smith and Guarino2017). Moreover, the lack of genetic diversity assessments in ex situ seed banks may result in the loss of genetic diversity when germplasm is regenerated, because the subset of seeds used for regeneration might not sufficiently reflect the total diversity in the collection (Schoen and Brown, Reference Schoen and Brown2001; Fu, 2017). If the genetic diversity in an ex situ seed bank is known, genetic resources conservation can be optimized by delineating core collections. Core collections are subsets of accessions that incorporate the maximal amount of genetic diversity present in the original collection (Brown, Reference Brown1989). Genetic diversity in ex situ collections can be maximized by either maximizing allelic richness or genetic distance. A distant subset of widely-adapted accessions is desired by plant breeders, while subsets that include rare alleles are more interesting for taxonomists and geneticists (Marita et al., Reference Marita, Rodriguez and Nienhuis2000).
Dessert and cooking bananas (Musa spp.) belong to the most prominent tropical and subtropical food commodities in the world (FAO, 2019). The genetic contribution of the CWR Musa balbisiana Colla to banana cultivars has been associated with a higher tolerance to banana weevil infestation and drought (Stover and Simmonds, Reference Stover and Simmonds1987; Thomas et al., Reference Thomas, Turner and Eamus1998; Ocan et al., Reference Ocan, Mukasa, Rubaihayo, Tinzaara and Blomme2008; Kissel et al., Reference Kissel, Van Asten, Swennen, Lorenzen and Carpentier2015). M. balbisiana has a natural geographic range that reaches from India to South China (Perrier et al., Reference Perrier, De Langhe, Donohue, Lentfer, Vrydaghs, Bakry, Carreel, Hippolyte, Horry, Jenny, Lebot, Risterucci, Tomekpe, Doutrelepont, Ball, Manwaring, de Maret and Denham2011) with its centre of origin most likely situated in the northern Indo-Burma region (Janssens et al., Reference Janssens, Vandelook, De Langhe, Verstraete, Smets, Vandenhouwe and Swennen2016). In addition, feral M. balbisiana populations are found far outside its natural range (Perrier et al., Reference Perrier, De Langhe, Donohue, Lentfer, Vrydaghs, Bakry, Carreel, Hippolyte, Horry, Jenny, Lebot, Risterucci, Tomekpe, Doutrelepont, Ball, Manwaring, de Maret and Denham2011). M. balbisiana seeds can be stored after desiccation without losing their viability, making them suitable for ex situ seed bank conservation (Stotzky et al., Reference Stotzky, Cox and Goos1962).
Here, we quantified genetic diversity in seven M. balbisiana ex situ seed collections that were separately collected from three natural populations, two feral populations and two ex situ field collections (online Supplementary Table S1 and Fig. S1). Our research questions were: (i) how genetically diverse are these M. balbisiana seed collections and (ii) which core subsets of seeds maximize genetic distance, allelic richness, or both? Our study contributes to the delineation of a conservation strategy of M. balbisiana genetic resources, serving as an example for CWR seed conservation of dessert and cooking bananas.
Materials and methods
Sampling and genotyping
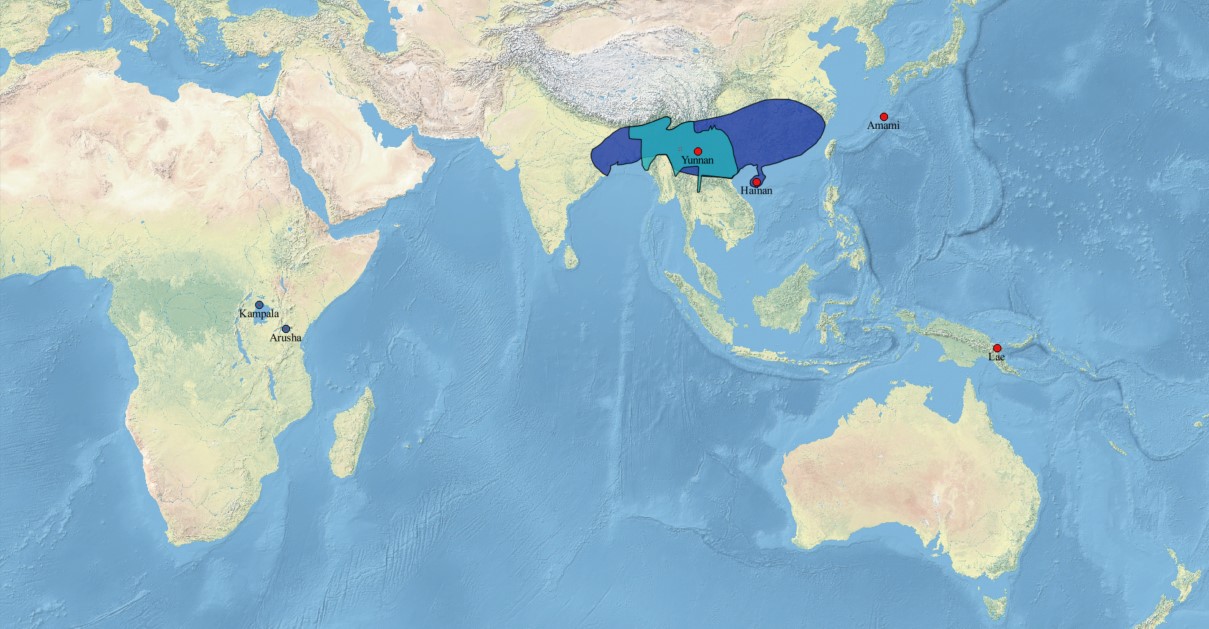
In total, 247 seeds belonging to seven ex situ seed collections available at the Bioversity International Musa Germplasm Transit Center (ITC) and Meise Botanic Garden were selected for this study (online Supplementary Table S1). Each seed collection was retrieved from one bunch of bananas, which is common practice in the collection of banana seeds. Three seed collections were obtained from two natural populations in Yunnan and one in Hainan (China). Two seed collections were retrieved from one feral population in Amami (Japan) and one in Lae (Papua New Guinea), while two other seed collections originated from two ex situ field collections at the IITA genebank facilities in Kampala (Uganda) and Arusha (Tanzania). An ex situ field collection consisted of M. balbisiana accessions originating from separate populations in separate regions that were brought together in one collection.
The seed embryo was isolated using embryo rescue and subsequently germinated on a culture medium, substantially increasing the germination rate compared to seeds that are sown in a greenhouse (Afele and De Langhe, Reference Afele and de Langhe1991). The leaves of the juvenile plants were dried on silica gel for DNA extraction. For the seed collection of Lae, DNA was directly taken from the embryo. DNA from the leaves and embryos was extracted using a modified cetyltrimethylammonium bromide protocol of Doyle and Doyle (Reference Doyle and Doyle1987). Eighteen polymorphic microsatellite markers (online Supplementary Table S2) were selected from previous studies on wild M. balbisiana accessions (Ge et al., Reference Ge, Liu, Wang, Schaal and Chiang2005; Wang et al., Reference Wang, Huang, Chen, Feng and Wu2011; Rotchanapreeda et al., Reference Rotchanapreeda, Wongniam, Swangpol, Chareonsap, Sukkaewmanee and Somana2016). The reverse primer of each marker was coupled to a universal primer sequence published by Schuelke (Reference Schuelke2000) and all primer combinations were arranged in four multiplexes using Multiplex Manager v1.2 (Holleley and Geerts, Reference Holleley and Geerts2009). Microsatellite regions were amplified using the Type-it Microsatellite PCR Kit (Qiagen, Venlo, the Netherlands), following a modified M13-like labelling protocol, which is described in detail in Vanden Abeele et al. (Reference Vanden Abeele, Hardy and Janssens2018). Afterwards, 1.5 µl of each polymerase chain reaction (PCR) amplicon was genotyped on an ABI 3730 sequencer (Applied Biosystems, Foster City, California, VS) with 12 µl of HiDi Formamide and 0.3 µl of the MapMarker 500 labelled with the DY-632 size standard (Eurogentec, Seraing, Belgium). The raw genetic data were scored with Geneious Pro v9.1.7 (Kearse et al., Reference Kearse, Moir, Wilson, Stones-havas, Cheung, Sturrock, Buxton, Cooper, Markowitz, Duran, Thierer, Ashton, Meintjes and Drummond2012). All microsatellite loci displayed distinct allelic patterns within each multiplex, validating the rearrangement of these markers into new multiplex PCRs.
Data analysis
Genetic diversity variables including the average number of alleles (N A), the average number of alleles with an allele frequency of at least 5% (N A≥5%), the number of private alleles (N priv) and observed (H O) and expected (H E) heterozygosity were calculated with the GenAlEx v6.5 plug-in in Microsoft Excel (Peakall and Smouse, Reference Peakall and Smouse2012). Genetic differentiation between seed collections was assessed based on Wright's F-statistics (F ST) and visualized by a principal coordinates analysis (PCoA) using the GenAlEx v6.5 plug-in in Microsoft Excel (Peakall and Smouse, Reference Peakall and Smouse2012). The significance of F ST values was tested with 999 permutations. Genetic clustering was examined using a Bayesian Markov Chain Monte Carlo (MCMC) clustering analysis implemented in STRUCTURE v2.3.4 (Pritchard et al., Reference Pritchard, Stephens and Donnelly2000). A series of independent runs with K values ranging from 1 to 10 was run in order to determine the best fitting number of clusters. Subsequently, the probability for each K was computed using the median of medians (MEDMEDK), the median of means (MEDMEAK), the maximum of medians (MAXMEDK) and the maximum of means (MAXMEAK) (Puechmaille, Reference Puechmaille2016) implemented in StructureSelector (Li and Liu, Reference Li and Liu2018). These statistics were demonstrated to be more robust for large differences in sampling size between populations that are included in the dataset (Puechmaille, Reference Puechmaille2016). The admixture model with correlated alleles was selected and the burn-in period length and the number of MCMC replicates were set to 150,000 and 200,000, respectively, as these estimates generated stable results for each value of K.
Core subset delineation
Five non-redundant accessions of M. balbisiana (i.e. core subsets) were selected using three different methods. First, the Maximization strategy (M-strategy) (Schoen and Brown, Reference Schoen and Brown1993), implemented in software CoreFinder (Cipriani et al., Reference Cipriani, Spadotto, Jurman, Di Gaspero, Crespan, Meneghetti, Frare, Vignani, Cresti, Morgante, Pezzotti, Pe, Policriti and Testolin2010), was used with an autogenerated random seed number and 99 permutations to delineate a core collection with the highest possible allelic richness. The M-strategy minimizes the sum of probabilities that alleles are not conserved in the core collection when a certain set of accessions is selected. At least one individual of every putative population is included in the final core collection (Schoen and Brown, Reference Schoen and Brown1993). Second, a maximum length subtree (MLST) (Perrier et al., Reference Perrier, Flori, Bonnot, Hamon, Seguin, Perrier and Glaszmann2003) was constructed using DARwin v6 software (Perrier and Jacquemoud-Collet, Reference Perrier and Jacquemoud-Collet2006) to select the genetically most distant individuals in our dataset. The MLST method required the reconstruction of a weighted neighbour-joining tree based on a dissimilarity matrix that was calculated for our dataset. The tree was subsequently pruned in a stepwise manner, each step removing one unit of each unit pair with the minimal length to the external edge. The number of individuals that remained present in the tree was set to be equal to the size of the subset that was determined with the M-strategy. Finally, the R package Corehunter III (De Beukelaer and Davenport, Reference De Beukelaer and Davenport2018), used in R v3.5.0 (R Core Team, 2018), was applied to maximize the Cavalli-Sforza and Edwards distance (CE distance) and the Shannon diversity index (SH index) through an advanced stochastic local search method. The CE distance is a Euclidean distance parameter that calculates distances between accessions as the square root of the differences between the allele frequencies of two individuals. The SH index reduces the redundancy of alleles in the collections by minimizing allele frequencies (Thachuk et al., Reference Thachuk, Crossa, Franco, Dreisigacker, Warburton and Davenport2009). A core collection that contained both a high number of alleles (high SH index) and genetically distant accessions (high CE distance) was constructed as well. The SH index and CE distance contributed in equal weight to the composition of this collection, resulting in a set of accessions that is interesting for both taxonomists, geneticists and plant breeders.
Results
Genetic diversity and differentiation
Eleven out of 18 amplified microsatellite loci were polymorphic in the ex situ seed collections. The seed collections from natural populations of M. balbisiana carried a higher average number of alleles than those gathered from feral populations (Table 1). Within the group of seed collections from natural populations, the average number of alleles was higher for the seeds of Yunnan (Yunnan-1 = 2.06 ± 0.25, Yunnan-2 = 2.06 ± 0.27) than for the seeds of Hainan (1.72 ± 0.23). Seed collections from natural populations also had a higher number of low-frequency alleles, while the seeds of feral populations had no polymorphic loci if rare alleles were not included (Table 1). Furthermore, the number of private alleles (N priv) was relatively low in all seed collections, but N priv was much higher (0.30 ± 0.14) in the ex situ field collection of Kampala than in the other seed collections. The highest heterozygosity levels were observed in the seeds of Yunnan, while the observed and expected heterozygosity were remarkably low in the seed collections of Amami (feral), Lae (feral) and Kampala (ex situ field collection) (Table 1).
Table 1. Genetic diversity parameters of seven seed collections of M. balbisiana based on 18 microsatellite markers

N A, average number of alleles per locus; N A⩾5%, average number of alleles per locus with an allele frequency higher than 5%; N priv., average number of unique alleles per locus; H O, observed heterozygosity; H E, expected heterozygosity; SE, standard error.
All F ST values were very high (>0.4), except for the F ST value between Yunnan-1 and Yunnan-2 (Table 2). The PCoA results showed a clear genetic clustering in the dataset. The Kampala seed collection was positioned in the top left corner of the PCoA graph (Fig. 1), clearly separated from all other collections. Three other clusters were recognized along the first principal axis: one cluster with all seeds from Arusha and Lae, a second cluster that combined the Yunnan seed collections, and a third cluster that contained the seeds of Hainan and Amami. The STRUCTURE analysis for the most optimal value of k (k = 6) delineated similar clusters compared to the PCoA results (Fig. 2). The seed collections from natural populations showed some admixture, especially between the collections of Yunnan, and encompassed three clusters that were not found in other seed collections. The feral populations and the ex situ field collections were clearly assigned to three clusters, combining the seeds of Amami and Hainan in one cluster and the seeds of Arusha and Lae in a second cluster. The third cluster exclusively consisted of seeds from Kampala (Fig. 2).

Fig. 1. The results of the PCoA displayed along the first two axes. Individuals of the same seed collection are shown in the same colour. The dark shade behind data points reflects overlapping data points.

Fig. 2. STRUCTURE bar plot with designation of the seed collection origin for the most optimal value of K (K = 6).
Table 2. F ST values calculated between all pairs of seed collections

Core subset delineation
The two core subsets that were constructed by methods that maximize allelic richness (i.e. the M-strategy and the SH-index) contained many seeds from the Yunnan and Kampala seed collections (Table 3). The core subset that was composed using the M-strategy contained 12 genotypes, but 80% of the allelic diversity in the dataset was found in only four seeds originating from Yunnan-1, Hainan, Kampala and Arusha (online Supplementary Fig. S2). The seed collection from the ex situ field collection in Arusha also contributed substantially to the core subset that maximized the Shannon diversity index (Table 3). The two distance-based core subsets (constructed by the CE distance and the MLST method) predominantly included seeds from natural populations in Yunnan and Hainan (Table 3, Fig. 3). When the allelic richness (SH index) and genetic distance (CE distance) were both optimized, the resulting core collection mainly consisted of seeds from natural populations and ex situ field collections.

Fig. 3. Maximal length subtree that was derived from a neighbour-joining tree with 200 bootstrap replicates. Each individual is named after its origin and sampling number.
Table 3. Number (N) and proportion (P) of seeds selected in core subsets to maximize genetic diversity using the Maximization strategy (M), the Cavalli-Sforza and Edwards distance (CE), the Shannon diversity index (SH) or a combination of CE and SH

Discussion
Genetic diversity assessment and sampling recommendations
This study assessed the genetic diversity in M. balbisiana seed collections retrieved from natural populations, feral populations and ex situ field collections. The genetic diversity in all seed collections (N A and H O) is low compared to that previously reported in wild M. balbisiana populations (Ge et al., Reference Ge, Liu, Wang, Schaal and Chiang2005; Jayaweera and Samarasinghe, Reference Jayaweera and Samarasinghe2016). However, some natural populations in China that were initially described as M. balbisiana populations were more recently assigned to another Musa species (i.e. Musa itinerans), which may partly explain the difference between our results and previously reported findings (Ge et al., Reference Ge, Liu, Wang, Schaal and Chiang2005). The lower genetic diversity in seed collections may additionally be explained by two factors. First, seeds from the same bunch of bananas have a common maternal ancestry. So each seed collection consists exclusively of half-siblings which are, by definition, genetically less diverse than M. balbisiana populations with more distantly related individuals assessed in previous studies. Second, pollen flow might be limited in M. balbisiana populations so that one bunch of bananas might only include alleles from a relatively small number of pollen donors. Consequently, the collection of a small number of seeds from different individuals within the same population might be necessary to efficiently conserve the genetic diversity in that population. Unfortunately, seeds of different individuals are in reality hard to find during one prospection, making it difficult to collect seeds from several individuals at once. Besides, M. balbisiana is a short-living species that clonally propagates via budding (Ge et al., Reference Ge, Liu, Wang, Schaal and Chiang2005), two life history traits that are believed to decrease the ability of populations to persist after seed harvest (Meissen et al., Reference Meissen, Galatowitsch and Cornett2015). Collecting seeds from multiple individuals might only be possible in large populations and is also preferentially spread through time in order to reduce the impact on population viability and allow for seed sampling from different individuals.
We observed a higher genetic diversity in the two seed collections from Yunnan than in the seed collections from other regions. Previously reported genetic analyses of natural M. balbisiana populations from China had the highest diversity in Yunnan as well (Ge et al., Reference Ge, Liu, Wang, Schaal and Chiang2005; Wang et al., Reference Wang, Chiang, Roux, Hao and Ge2007), confirming the high value of populations in its region of origin for conservation. Regional genetic diversity assessments of M. balbisiana only found moderate levels of genetic differentiation between populations (Ge et al., Reference Ge, Liu, Wang, Schaal and Chiang2005; Wang et al., Reference Wang, Chiang, Roux, Hao and Ge2007; Jayaweera and Samarasinghe, Reference Jayaweera and Samarasinghe2016), which is in accordance with the low genetic differentiation that we observed between the two seed collections from Yunnan. Hence, the collection of seeds from several populations within the same region might not strongly increase the total genetic diversity in the ex situ seed bank. In contrast, the high genetic differentiation that was observed between seed collections from different regions rather suggests that gathering seeds from regions that are part of a wide geographical range should result in a higher increase in genetic diversity in the ex situ seed bank. These findings align with Rivière and Müller (Reference Rivière and Müller2017) who provided evidence for common intraspecific sampling gaps in ex situ seed collections and argued that a more extensive sampling of the diversity across multiple biogeographic regions is required to fill these gaps. Prioritized sampling locations for seeds of M. balbisiana are especially located in its natural distribution area, and more specifically in its region of origin. The conservation of seeds from regions that are absent in the ex situ seed bank, such as the northeastern part of India and the northern regions of Laos, Vietnam and Myanmar should be of prior concern. In addition, land use changes have reduced the number of M. balbisiana populations in Papua New Guinea and in northern China during the last few decades, urging the conservation of the M. balbisiana genepool (Ge et al., Reference Ge, Liu, Wang, Schaal and Chiang2005; Wang et al., Reference Wang, Chiang, Roux, Hao and Ge2007).
Ex situ seed bank curation
The delineation of genetically diverse subsets substantially increases the manageability of collections, but the composition of a subset varies depending on whether a high genetic distance or high allelic richness is preferred. Distance-based methods only select seeds from Yunnan or Hainan, indicating that these collections are especially interesting for plant breeders. However, methods that capture the highest allelic diversity include more seeds from the ex situ field collections, which makes these collections more important for taxonomists and conservation biologists (Marita et al., Reference Marita, Rodriguez and Nienhuis2000; Thachuk et al., Reference Thachuk, Crossa, Franco, Dreisigacker, Warburton and Davenport2009). The high number of private alleles in the collection of Kampala suggests that these seeds contain a different part of the gene pool of M. balbisiana. However, the seeds in the ex situ field collections are open-pollinated and it cannot be excluded that certain unique alleles in the seeds from these field collections are introgressed from another Musa species, such as M. acuminata, which occurs in the proximity of M. balbisiana accessions. Furthermore, the seed collections from feral populations capture very low amounts of genetic variation, suggesting that the presence of these collections in the ex situ seed bank is only of secondary importance. However, these seed collections may serve as safety backups for alleles that are also conserved in other seed collections (van Hintum and Visser, Reference van Hintum and Visser1995; Milner et al., Reference Milner, Jost, Taketa, Mazón, Himmelbach, Oppermann, Weise, Knüpffer, Basterrechea, König, Schüler, Sharma, Pasam, Rutten, Guo, Xu, Zhang, Herren, Müller, Krattinger, Keller, Jiang, González, Zhao, Habekuß, Färber, Ordon, Lange, Börner, Graner, Reif, Scholz, Mascher and Stein2019).
In order to maintain a viable seed collection, it is necessary to regenerate a subset of seeds after a certain period of time. The regeneration of seeds can result in the loss of genetic diversity if the reared seeds do not properly cover the diversity in the entire seed collection or if the size of the regenerated sample pool is not large enough. Our results suggest that the number of seeds that must be used for regeneration to maintain the genetic diversity in a seed collection must be substantially larger for the seed collections from natural populations than for the collections from feral populations. Ex situ seed banking of M. balbisiana seed collections becomes much more efficient when these differences in genetic diversity between seed collections are taken into account.
The results of this study indicate that the seed collections from natural populations, feral populations and ex situ field collections of M. balbisiana are three complementary sources of genetic diversity. The seed collections from natural populations, preferably sampled within the centre of origin of the species, include high levels of genetic diversity, and conservation and collection efforts should primarily focus on these regions. We also recommend collecting a relatively small number of seeds from multiple M. balbisiana individuals within one population to efficiently conserve genetic diversity in the target population. The seed collections from ex situ field collections add unique genetic variation to the ex situ seed bank. These collections are also easily accessible and their storage in an ex situ seed bank additionally safeguards the diversity present in the ex situ field collections. The seed collections from ex situ field collections are interesting for genetic or taxonomic research, while our results suggest that the contribution of these seed collections to plant breeding might be limited if plant material from natural populations is available. The seed collections from feral populations provide safety backups for genetic resources in the seed collections from natural populations. A small number of seeds is probably sufficient to conserve the genetic diversity in feral populations. Nevertheless, the number of seed collections available for this study was limited to seven and the collection and characterization of additional seed material is needed to validate our results.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S1479262119000376.
Acknowledgements
We acknowledge Matthew Turner for providing the seed collection of Amami and Nicolas Roux for facilitating the transfer of seed collections for genetic analyses. Special thanks also go to the colleagues Kevin Longin and Tom Vanderstraeten at the International Transit Center who performed embryo rescue on the banana seeds and reared all embryos and to Wim Baert and Alexia Semeraro from Meise Botanic Garden for their support in the lab. Finally, we owe many words of gratitude to Professor Olivier Hardy and his research team at the University of Brussels (ULB) for their hospitality and for sharing their experiences in the analysis of microsatellite markers. Part of this work was funded by the Genebank CGIAR Research Program, the CGIAR Research Program on Roots, Tubers and Bananas (RTB) with a contribution of the Belgium government (DGD) through the PhenSeeData project, by Research Foundation - Flanders (FWO) (No. G0D9318N) and by the National Natural Science Foundation of China (No. 31261140366). In addition, this work was supported by the project ‘BBTV mitigation: Community management in Nigeria, and screening wild banana progenitors for resistance (2015–2020)’, funded by the Bill and Melinda Gates foundation. The funding agencies were not involved in the design of the study, in the provision or analysis of the data or in the writing and submission of the manuscript.










 Open access
Open access