

Evaluating the burgeoning literature is difficult for practising clinicians, service users and policy makers. Attempts to do so are fraught with problems owing to the use of multiple outcomes, lack of clinical relevance and paucity of information on clinically relevant variables such as side-effects. Given a patient with a diagnosis and a variety of concerns, the traditional randomised placebo-controlled ‘efficacy’ trial (RCT) gives little information on the appropriate treatment choice for that individual. A number of authorities, including the National Institute for Health and Clinical Excellence (NICE), recommend that patients are actively involved in treatment decisions, but do not opine on how this can realistically be achieved. Nevertheless, presenting information on treatment in an appropriate format has been demonstrated to improve adherence (Reference Halvorsen, Selmer and KristiansenHalvorsen 2007).
Little is known about how psychiatrists make prescribing decisions (Reference Hodgson, Bushe and HunterHodgson 2007), but the presentation of results in many reported clinical trials does not allow easy translation to the clinical situation. Simple questions posed by patients, such as the chances of a particular side-effect or whether a proposed treatment is better than one they have read about, are not readily answerable. We hope that the judicious use of the tables and concepts in this article may partially bridge this gap and aid evidence-based prescribing of antipsychotics.
The NNT and NNH
One way of presenting the results of trials in a readily understandable way is the number needed to treat (NNT). The NNT answers the question: ‘How many people would need to receive an intervention for one of them to benefit who would not have benefited had they all received a control intervention’. Analogous to the NNT, the number needed to harm (NNH) is an expression of the number of patients who would need to receive an intervention to cause one additional adverse event.
In randomised studies, patients are randomly allocated either to an experimental treatment or to a control intervention. The incidence of an event occurring owing to the experimental intervention is called the experimental event rate (EER) and the incidence of an event owing to the control intervention is the control event rate (CER).
The beneficial (or adverse) effects of the experimental intervention are usually measured by comparing the probabilities of events in the experimental and control group using the concept of absolute risk reduction (ARR). The absolute risk reduction is the difference between the control and experimental event rates (ARR = CER − EER). This number provides an idea of the clinical relevance of a given treatment but is problematic as it is a dimensionless and abstract number.
The NNT is the reciprocal of the absolute risk reduction: NNT = 1/(CER − EER) in relation to therapeutic events. Likewise, the NNH is the inverse of the absolute difference in adverse event rates between the experimental and control arms. By convention, the NNT and NNH are always rounded up to the nearest whole figure. Confidence intervals can be calculated for both the NNT and the NNH (Reference AltmanAltman 1998).
This may sound complicated but in practice the calculation of the NNT is easy. The only information required is a proportion for the variable of interest in both the control and the experimental groups. Box 1 summarises the elements involved and Box 2 shows an NNT calculation using data from the Clinical Antipsychotic Trials of Intervention Effectiveness – CATIE (Reference Lieberman, Stroup and McEvoyLieberman 2005).
BOX 1 The formulae
Experimental event rate (EER) The incidence of an event occurring owing to the experimental intervention
Control event rate (CER) The incidence of an event occurring owing to the owing to the control intervention
Absolute risk reduction (ARR) The difference between the control and experimental event rates:
The number needed to treat
BOX 2 Example NNT calculation from CATIE
In CATIE, 192 out of 257 patients discontinued the control intervention perphenazine, which gives
and 210 out of 330 patients discontinued the experimental intervention olanzapine, giving
The ARR is the CER minus the EER or in this case
Hence,
which by convention is rounded up to 10.
(Data from Reference Lieberman, Stroup and McEvoyLieberman 2005)
The clinical utility of presenting data as the NNT and NNH has been advocated by the Cochrane Collaboration and the evidence-based resource Bandolier (www.medicine.ox.ac.uk/bandolier). A number of websites provide NNT calculators (e.g. www.ebem.org/nntcalculator.html; www.nntonline.net/ebm/visualrx/try.asp).
Balancing the NNT with the NNH
Reference StrausStraus (2002) has proposed the likelihood of being helped or harmed (LHH) as a valid way of presenting risks and benefits. The LHH is the ratio of the NNT to the NNH, or the absolute risk reduction (ARR) divided by the absolute risk increase (ARI) for the relevant outcomes: LHH = ARR/ARI. However, this is confusing when different adverse events have to be included in the balance. For example, let us assume that a novel antipsychotic has an NNT of three for preventing hospital admission and an NNH of three for causing extrapyramidal side-effects. This would give a ratio of 1, suggesting a reasonable risk/benefit ratio. To the treating clinician a drug's effectiveness in reducing hospital admissions may far outweigh the risk of extrapyramidal side-effects. However, a patient may view the risk of extrapyramidal side-effects as unacceptable.
Interpreting and using the NNT
Statistical and methodological considerations
The number needed to treat should be interpreted in the appropriate clinical context. It cannot be applied to continuous variables. For example, if change in weight is an outcome of interest and the results are expressed as a mean change in weight, analysis is not possible. However, if they are presented as a categorical variable such as the proportion of participants whose weight increased by more than 7%, then the NNT can be calculated. Likewise, a mean drop in Positive and Negative Syndrome Scale (PANSS) score cannot be used, but if the data can be converted to give the proportion of patients achieving remission with response being defined as, for example, a 20% improvement in PANSS score then the NNT can be calculated.
The more effective an intervention, the smaller the NNT. When there is a placebo effect the lowest possible NNT is 2. Generally in medicine, an NNT of 8 or less can be considered useful for adjunctive treatment but monotherapies for acute conditions should have an NNT of 2–5.
The clinical relevance of an NNT is not solely a function of the actual value but is also dependent on the illness being treated. For life-threatening conditions, a higher NNT may be acceptable in the absence of an alternative treatment. For a serious condition, a low NNH (a high chance of harm) may be more acceptable than for a more benign condition.
The NNT is a measure of effect size. It is independent of the P -value, although confidence intervals can be calculated with a similar level of probability (e.g. 95%). Therefore, a significant P-value does not necessarily result in a clinically or statistically significant NNT. The NNT helps the clinician judge the clinical relevance of a statistically significant result. If confidence intervals are not quoted, caution should be exercised in assuming that one treatment is superior to another. If confidence intervals overlap between two treatments then there is no significant statistical difference between these treatments. As the NNT is a measure of effect size, it provides a common currency that allows comparisons between interventions from different medical specialties (Table 1).
TabLE 1 Numbers needed to treat (NNT) for medical interventions
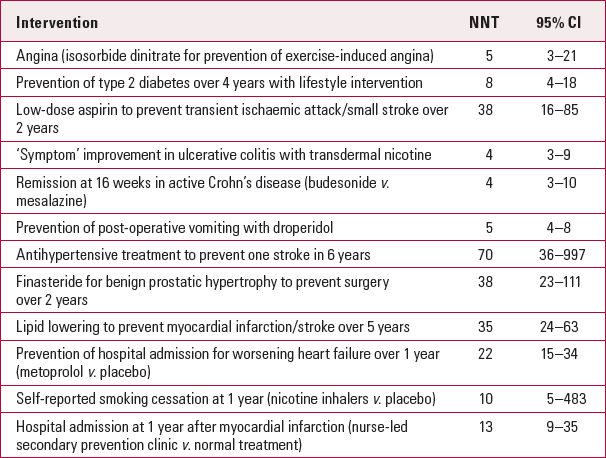
| Intervention | NNT | 95% CI |
|---|---|---|
| Angina (isosorbide dinitrate for prevention of exercise-induced angina) | 5 | 3–21 |
| Prevention of type 2 diabetes over 4 years with lifestyle intervention | 8 | 4–18 |
| Low-dose aspirin to prevent transient ischaemic attack/small stroke over 2 years | 38 | 16–85 |
| ‘Symptom’ improvement in ulcerative colitis with transdermal nicotine | 4 | 3–9 |
| Remission at 16 weeks in active Crohn's disease (budesonide v. mesalazine) | 4 | 3–10 |
| Prevention of post-operative vomiting with droperidol | 5 | 4–8 |
| Antihypertensive treatment to prevent one stroke in 6 years | 70 | 36–997 |
| Finasteride for benign prostatic hypertrophy to prevent surgery over 2 years | 38 | 23–111 |
| Lipid lowering to prevent myocardial infarction/stroke over 5 years | 35 | 24–63 |
| Prevention of hospital admission for worsening heart failure over 1 year (metoprolol v. placebo) | 22 | 15–34 |
| Self-reported smoking cessation at 1 year (nicotine inhalers v. placebo) | 10 | 5–483 |
| Hospital admission at 1 year after myocardial infarction (nurse-led secondary prevention clinic v. normal treatment) | 13 | 9–35 |
Clinical interpretation
Before the findings in any clinical trial can be applied to an individual patient, the clinician must decide whether the study results can be generalised to that patient. Box 3 illustrates some of the points to be considered. For example, if the patient has presented with a first episode of psychosis, the clinician may be better informed using the NNT and NNH from a trial for first-episode schizophrenia rather than a study for chronic schizophrenia. Most efficacy trials of new drugs exclude patients with physical health problems or substance misuse. In reality, many patients have these comorbid conditions and the clinician needs to tailor the treatment around these issues. Essentially, the clinician must decide whether or not the patient's problems are so different from those of participants in the trial that the results are not applicable. However, ‘off-label’ prescribing is common in psychiatry (Reference Hodgson and BelgamwarHodgson 2006a; Reference Taylor, Shajahan and LawrieTaylor 2008), suggesting that experienced clinicians are willing to experiment when there is limited trial evidence to guide treatment choices, and often off-label prescribing foreshadows later positive RCTs (Reference Hodgson and BelgamwarHodgson 2006a).
BOX 3 Do these results apply to my patient?
-
• Does my patient have a condition similar to those in the trial?
-
• Is my patient similar to patients in the trial (it is not appropriate to apply all the exclusion/inclusion criteria)?
-
• Are there any contraindications to the proposed medication for this particular individual?
-
• Can the results be converted to NNT and NNH?
-
• Are there any baseline considerations?
-
• How will the patient and I rank particular side-effects?
-
• What do the patient and I want from treatment?
Other baseline considerations that should influence treatment choice include formulary restrictions and resources such as access to blood testing and electrocardiography (Reference Hodgson, Belgamwar and KrishnaHodgson 2006b). In addition to research evidence, doctors will be guided by their clinical experience and personal preference (Reference Bleakley, Olofinjana and TaylorBleakley 2007; Reference Taylor and BrownTaylor 2007). Patients and their carers may also have views.
Practical applications of NNT and NNH in psychiatry
We will examine applications of NNT and NNH in psychiatry by focusing on large-scale and long-term pragmatic trials. Medication trials that have not been sponsored by the pharmaceutical industry are preferable, as published positive results seem often to be associated with the vested interest of the sponsor (Reference Als-Nielsen, Chen and GluudAls-Nielsen 2003), although this is not invariably the case (Reference Heres, Davis and MainoHeres 2006). Concentrating predominantly on non-industry-sponsored trials dramatically reduces the numbers of studies available for analysis and does not avoid all potential bias (Reference CoyneCoyne 2006).
Trials of antipsychotics
Much of the available clinical information on antipsychotic treatment comes from company-sponsored efficacy trials carried out for registration purposes. Since the publication of the NICE appraisal of the use of atypical antipsychotics (National Institute for Clinical Excellence 2002) they have been included as a first-line treatment for schizophrenia in the UK. A systematic review accompanying the appraisal indicated that, although the atypicals are a heterogeneous group of compounds, there was little evidence to suggest differential efficacy at the time of the review, other than for clozapine. Also, there were few head-to-head studies and outcomes were often based on rating scales and did not readily help clinicians in their prescribing choice. Another meta-analysis (Reference Geddes, Freemantle and HarrisonGeddes 2000) had concluded that any difference in efficacy between typical and atypical antipsychotics arose only when the dose of the typical antipsychotic was high. However, a later meta-analysis of the efficacy studies (Reference Davis, Chen and GlickDavis 2003) indicated that there were differences in terms of efficacy between different atypicals and between some atypical and typical agents that could not be explained by high doses of the typicals. This meta-analysis was noteworthy for both its size and its independence of the pharmaceutical industry.
CATIE
In the 18-month CATIE study (Reference Lieberman, Stroup and McEvoyLieberman 2005), 1493 patients with chronic schizophrenia were randomised to receive either the typical antipsychotic perphenazine, or one of the atypicals (in phase 1, olanzapine, quetiapine, risperidone or, after its licensing in 2002, ziprasidone). The study was sponsored by the US National Institute of Mental Health. It tried to simulate real-world prescribing, albeit within the structure of an RCT, by excluding as few patients as possible and by using pragmatic outcomes such as rate of discontinuation of medication. The discontinuation rate is regarded as a proxy for effectiveness (Reference Stroup, McEvoy and SwartzStroup 2003; Reference Hodgson, Belgamwar and Al-tawarahHodgson 2005). CATIE included assessment of symptoms, cognitive function and medication-related side-effects. Aspects of CATIE have been reviewed and commented on previously in Advances (Reference CooksonCookson 2008; Reference OwensOwens 2008). In interpreting CATIE, it should be noted that participants were not randomised to perphenazine if they had extrapyramidal side-effect markers such as tardive dyskinesia.
In phase 1 of CATIE the most ‘effective’ anti-psychotic (and the only one significantly different from perphenazine) was olanzapine. However, olanzapine was associated with a greater side-effect burden (notably, weight gain and metabolic effects) than the comparator antipsychotics. These differences in effectiveness and side-effect burden mean that CATIE is an appropriate study to explore further.
The initial results revealed an overall composite discontinuation rate (the primary outcome) of 74% over 12 months, with patients who were taking olanzapine least likely to discontinue their medication (64%), leading to the conclusion that it was the most effective of the antipsychotics in the study. However, olanzapine was associated with the highest discontinuation rate for intolerable side-effects (18%) and risperidone with the lowest (10%). Table 2 shows NNTs and NNHs for the antipsychotics in CATIE. Olanzapine is used as the comparator, as in the original publication. We can see that the NNT for avoiding discontinuation for any reason was 11 for olanzapine in comparison with risperidone, 10 in comparison with perphenazine, 7 with ziprasidone and 6 with quetiapine. Participants taking perphenazine, risperidone, quetiapine and ziprasidone were more likely to stop their medication owing to lack of efficacy than those taking olanzapine. However, patients taking olanzapine are more likely to stop their medication owing to side-effects than those taking risperidone.
TabLE 2 Numbers needed to treat (NNT) and numbers needed to harm (NNH) from CATIE
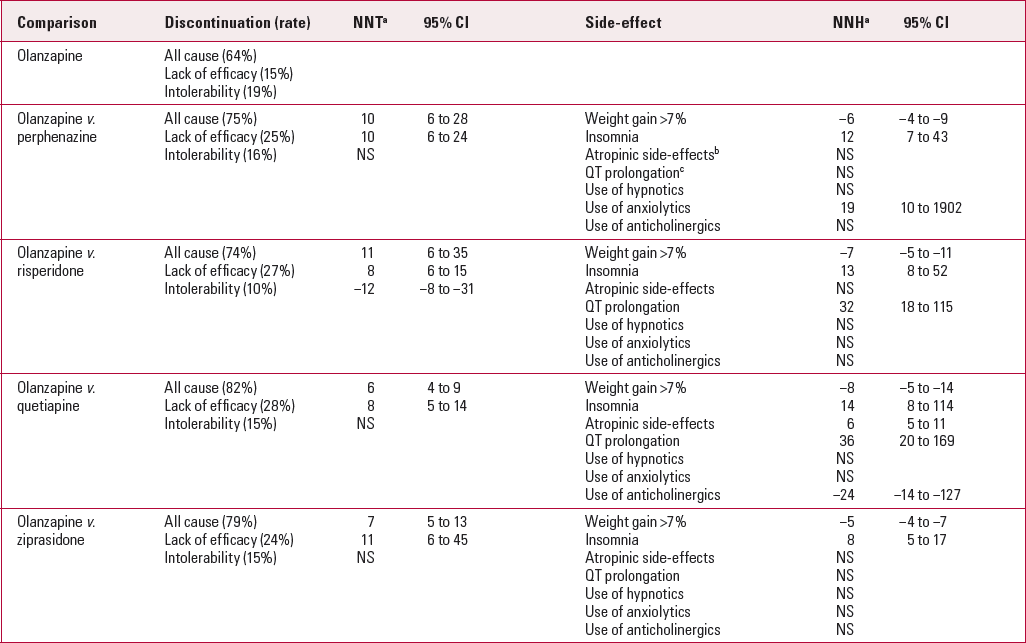
| Comparison | Discontinuation (rate) | NNT a | 95% CI | Side-effect | NNH a | 95% CI |
|---|---|---|---|---|---|---|
| Olanzapine | All cause (64%) | |||||
| Lack of efficacy (15%) | ||||||
| Intolerability (19%) | ||||||
| Olanzapine v. perphenazine | All cause (75%) | 10 | 6 to 28 | Weight gain >7% | −6 | −4 to −9 |
| Lack of efficacy (25%) | 10 | 6 to 24 | Insomnia | 12 | 7 to 43 | |
| Intolerability (16%) | NS | Atropinic side-effectsb | NS | |||
| QT prolongationc | NS | |||||
| Use of hypnotics | NS | |||||
| Use of anxiolytics | 19 | 10 to 1902 | ||||
| Use of anticholinergics | NS | |||||
| Olanzapine v. risperidone | All cause (74%) | 11 | 6 to 35 | Weight gain >7% | −7 | −5 to −11 |
| Lack of efficacy (27%) | 8 | 6 to 15 | Insomnia | 13 | 8 to 52 | |
| Intolerability (10%) | −12 | −8 to −31 | Atropinic side-effects | NS | ||
| QT prolongation | 32 | 18 to 115 | ||||
| Use of hypnotics | NS | |||||
| Use of anxiolytics | NS | |||||
| Use of anticholinergics | NS | |||||
| Olanzapine v. quetiapine | All cause (82%) | 6 | 4 to 9 | Weight gain >7% | −8 | −5 to −14 |
| Lack of efficacy (28%) | 8 | 5 to 14 | Insomnia | 14 | 8 to 114 | |
| Intolerability (15%) | NS | Atropinic side-effects | 6 | 5 to 11 | ||
| QT prolongation | 36 | 20 to 169 | ||||
| Use of hypnotics | NS | |||||
| Use of anxiolytics | NS | |||||
| Use of anticholinergics | −24 | −14 to −127 | ||||
| Olanzapine v. ziprasidone | All cause (79%) | 7 | 5 to 13 | Weight gain >7% | −5 | −4 to −7 |
| Lack of efficacy (24%) | 11 | 6 to 45 | Insomnia | 8 | 5 to 17 | |
| Intolerability (15%) | NS | Atropinic side-effects | NS | |||
| QT prolongation | NS | |||||
| Use of hypnotics | NS | |||||
| Use of anxiolytics | NS | |||||
| Use of anticholinergics | NS |
If weight gain is analysed using the criterion of an increase in baseline weight of more than 7%, then the NNHs for olanzapine against risperidone, quetiapine, perphenazine and ziprasidone are 7, 8, 6 and 5 respectively (Table 2). The minus signs in the table indicate an adverse outcome. Thus, a clinician choosing between risperidone and olanzapine would see from Table 2 that for every 11 patients who were prescribed olanzapine, one more would continue on that medication than if risperidone were prescribed. However, this would be balanced by the knowledge that for every 7 patients prescribed olanzapine rather than risperidone, one more would gain greater than 7% of their baseline weight.
Table 2 shows some other NNHs of interest, including atropinic side-effects (urinary hesitancy, dry mouth and constipation), QT prolongation and the need for concomitant medication. There is little difference between olanzapine and ziprasidone except that patients taking ziprasidone experienced more insomnia (NNH = 8). Atropinic side-effects were more common with quetiapine than olanzapine (NNH = 6). Conversion of other potentially relevant issues such as extrapyramidal side-effects, symptom profile, or metabolic parameters other than weight gain cannot be done as the data are not presented in a binary format. Other pair-wise calculations for the CATIE study drugs can be made but few are significant. For example, when all-cause discontinuation is considered, both risperidone (NNT = 13, 95% CI 7–54) and perphenazine (NNT = 15, 95% CI 8–336) outperformed quetiapine. Perphenazine was more likely than quetiapine to lead to prescription of anticholinergic medication (NNH = 15) and more likely than olanzapine to require anxiolytics (NNH = 17). Quetiapine was less likely than the other drugs to require concomitant antidepressants.
Prolongation of the QT interval occurred more frequently with risperidone and quetiapine than with olanzapine, ziprasidone and perphenazine. The difference was small, with an NNH in the 30s, and the clinical relevance is uncertain. However, for patients at high risk of QT prolongation this result may be clinically significant.
CUtLASS
The UK Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS) was a 1-year, open-label, randomised study of 227 people with schizophrenia (Reference Jones, Barnes and DaviesJones 2006). Patients were allocated to receive either a typical antipsychotic or a newer (atypical) drug, and the clinician was able to choose which drug the patient received from a list. After 1 year, 54% of patients were still on a typical antipsychotic and 63% on an atypical. This difference was not statistically significant. There were no significant differences in other relevant outcomes, including quality of life. Given the lack of significant differences, it is not meaningful to calculate NNTs or NNHs, as all treatments were effectively equal. However, the list of typicals included sulpiride, and this was the drug most often chosen (49%). Olanzapine was the most popular drug on the atypicals list (46%). Note that when it was first marketed in the 1970s, sulpiride was regarded as an atypical antipsychotic (Reference OwensOwens 2008).
First-episode psychosis
Two pragmatic trials have studied antipsychotic continuation in first-episode schizophreniform psychosis: the Comparison of Atypicals in First Episode of Psychosis (CAFE) study and the European First Episode Schizophrenia Trial (EUFEST).
CAFE
This was a double-blind study investigating the use of quetiapine (100–800 mg/day), risperidone (0.5–4 mg/day) or olanzapine (2.5–20 mg/day). It was funded by AstraZeneca Pharmaceuticals, the manufacturer of quetiapine (Reference McEvoy, Lieberman and PerkinsMcEvoy 2007). The research was coordinated by the University of North Carolina. The discontinuation rate over 50 weeks was around 70% for all the study drugs. So for this outcome, NNT calculations would reveal no differences between the drugs.
EUFEST
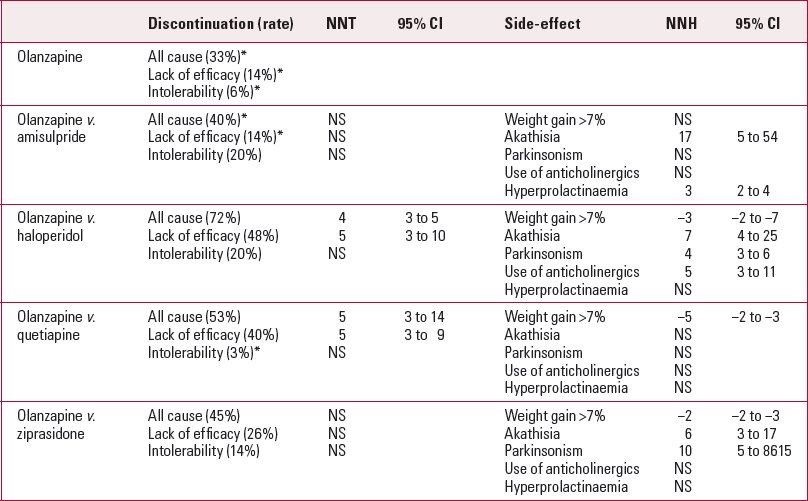
EUFEST (Reference Kahn, Fleischhacker and BoterKahn 2008) was a 1-year, multicentre, pragmatic, open-label, randomised comparison of low-dose haloperidol (maximum dose 1–4 mg/ day) with four atypical antipsychotics: amisulpride (200–800 mg/day), olanzapine (5–20 mg/day), quetiapine (200–750 mg/day) and ziprasidone (40–160 mg/day). The trial was sponsored by three companies (Sanofi-Aventis (amisulpride), AstraZeneca (quetiapine) and Pfizer (ziprasidone)) but these companies were said not to be involved in study design or data analysis. The primary outcome was treatment discontinuation and the study population (n = 489) comprised patients with first-episode schizophrenia (defined as having no more than 2 years of psychotic symptoms). The combined discontinuation rate was 47%; rates for the individual drugs varied from 33% for olanzapine to 72% for haloperidol. On this primary outcome all the atypicals in the trial were better than haloperidol. There were differences in adverse event rates for extrapyramidal side-effects, hyperprolactinaemia and weight gain. An increase in baseline weight of more than 7% just failed to reach significance (P = 0.053) but absolute weight change did (P < 0.0001). As the latter is a continuous variable it cannot be used to calculate NNHs, so to facilitate comparison we have used the >7% figures (Table 3). There were no statistically significant differences for many adverse events, such as glucose, lipid and electrocardiogram changes.
TabLE 3 Numbers needed to treat (NNT) and numbers needed to harm (NNH) from EUFEST
| Discontinuation (rate) | NNT | 95% CI | Side-effect | NNH | 95% CI | |
|---|---|---|---|---|---|---|
| Olanzapine | All cause (33%)* | |||||
| Lack of efficacy (14%)* | ||||||
| Intolerability (6%)* | ||||||
| Olanzapine v. amisulpride | All cause (40%)* | NS | Weight gain >7% | NS | ||
| Lack of efficacy (14%)* | NS | Akathisia | 17 | 5 to 54 | ||
| Intolerability (20%) | NS | Parkinsonism | NS | |||
| Use of anticholinergics | NS | |||||
| Hyperprolactinaemia | 3 | 2 to 4 | ||||
| Olanzapine v. haloperidol | All cause (72%) | 4 | 3 to 5 | Weight gain >7% | −3 | −2 to −7 |
| Lack of efficacy (48%) | 5 | 3 to 10 | Akathisia | 7 | 4 to 25 | |
| Intolerability (20%) | NS | Parkinsonism | 4 | 3 to 6 | ||
| Use of anticholinergics | 5 | 3 to 11 | ||||
| Hyperprolactinaemia | NS | |||||
| Olanzapine v. quetiapine | All cause (53%) | 5 | 3 to 14 | Weight gain >7% | −5 | −2 to −3 |
| Lack of efficacy (40%) | 5 | 3 to 9 | Akathisia | NS | ||
| Intolerability (3%)* | NS | Parkinsonism | NS | |||
| Use of anticholinergics | NS | |||||
| Hyperprolactinaemia | NS | |||||
| Olanzapine v. ziprasidone | All cause (45%) | NS | Weight gain >7% | −2 | −2 to −3 | |
| Lack of efficacy (26%) | NS | Akathisia | 6 | 3 to 17 | ||
| Intolerability (14%) | NS | Parkinsonism | 10 | 5 to 8615 | ||
| Use of anticholinergics | NS | |||||
| Hyperprolactinaemia | NS |
On some secondary measures, such as change in PANSS score (Reference Kay, Fiszbein and OplerKay 1987), there were no differences between any of the antipsychotics, but on the Clinical Global Impression Scale (Reference GuyGuy 1976) and the Global Assessment of Functioning Scale (Reference Endicott, Spitzer and FleisssEndicott 1976) the atypical antipsychotics outperformed haloperidol.
From Table 3 we can see that only four patients have to be treated with olanzapine rather than haloperidol to prevent one discontinuation. However, compared with haloperidol, olanzapine is associated with weight gain, with an NNH of 3.
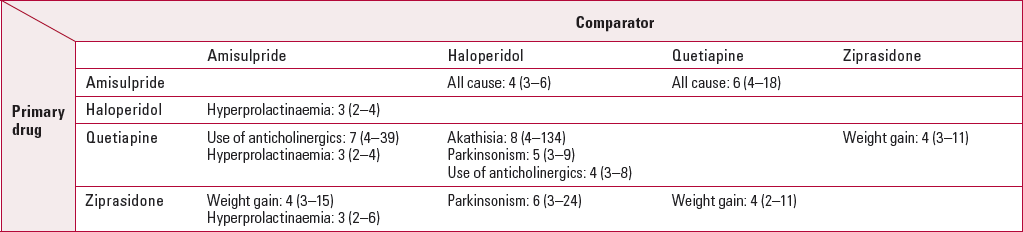
There are other differences between the study drugs (Table 4). For example, amisulpride has a higher continuation rate than haloperidol (NNT = 4) and quetiapine (NNT = 6) but is associated with a higher risk of hyperprolactinaemia (blood serum prolactin levels >0.38 U/l in males or >0.53 U/l in females) than these drugs (NNT = 3). The clinical significance of the hyperprolactinaemia is unclear. Sexual side-effects were common in all groups.
TabLE 4 Other significant differences a between EUFEST study drugs b
| Comparator | |||||
|---|---|---|---|---|---|
| Primary drug | Amisulpride | Haloperidol | Quetiapine | Ziprasidone | |
| Amisulpride | All cause: 4 (3–6) | All cause: 6 (4–18) | |||
| Haloperidol | Hyperprolactinaemia: 3 (2–4) | ||||
| Quetiapine | Use of anticholinergics: 7 (4–39) | Akathisia: 8 (4–134) | Weight gain: 4 (3–11) | ||
| Hyperprolactinaemia: 3 (2–4) | Parkinsonism: 5 (3–9) | ||||
| Use of anticholinergics: 4 (3–8) | |||||
| Ziprasidone | Weight gain: 4 (3–15) | Parkinsonism: 6 (3–24) | Weight gain: 4 (2–11) | ||
| Hyperprolactinaemia: 3 (2–6) | |||||
In EUFEST there is little difference between effectiveness for olanzapine and amisulpride but the side-effect profile is different, use of anticholinergics (NNH = 9) and hyperprolactinaemia (NNH = 3) being associated more with amisulpride.
Prospective observational studies
Reference Tiihonen, Walhbeck and LönnqvistTiihonen et al(2006) reported on an observational study of a cohort of 2230 consecutive patients with first-episode schizophrenia in Finland who were followed up over 7 years. Statistical analysis included the use of propensity scoring to minimise the effect of non-randomisation in the study. The main outcomes were medication discontinuation and hospital admission, which facilitates comparison with CATIE and EUFEST. There were differences in the medications studied, with a wider range of atypicals in CATIE and EUFEST and more typicals in the Finnish study. However, comparisons can be made for olanzapine, perphenazine and risperidone. The methodology used national databases so there was no information on adverse events other than cause of death.
Table 5 shows the NNTs for treatment discontinuation. The results shown must be interpreted with caution as confidence intervals cannot be calculated from the information provided in the original paper. Overall, it can be seen that depot perphenazine is the most effective treatment for the small group of patients receiving it. Clozapine is the most effective of the oral antipsychotics, followed by olanzapine, risperidone, perphenazine and haloperidol. These results are in keeping with other large observational studies (Reference Hodgson, Belgamwar and Al-tawarahHodgson 2005; Reference Haro and Salvador-CarullaHaro 2006). For olanzapine against risperidone, NNT = 8 in the Tiihonen study and NNT = 11 in CATIE. When oral perphenazine is considered against olanzapine and risperidone, it performs less well in the Tiihonen study than in CATIE, with NNTs of −2 and −4 respectively.
TabLE 5 Numbers needed to treat (NNT) for all-cause discontinuation for the Tiihonen study a
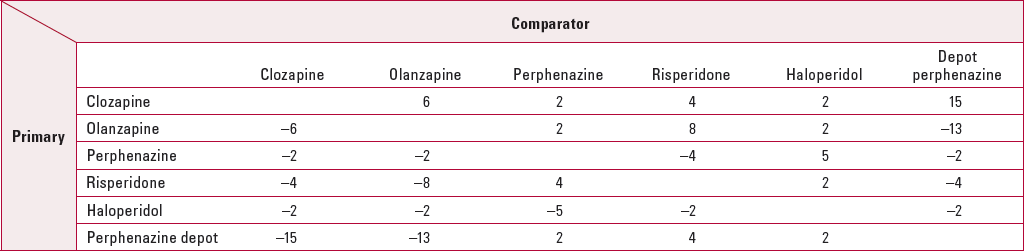
| Comparator | |||||||
|---|---|---|---|---|---|---|---|
| Primary | Clozapine | Olanzapine | Perphenazine | Risperidone | Haloperidol | Depot perphenazine | |
| Clozapine | 6 | 2 | 4 | 2 | 15 | ||
| Olanzapine | −6 | 2 | 8 | 2 | −13 | ||
| Perphenazine | −2 | −2 | −4 | 5 | −2 | ||
| Risperidone | −4 | −8 | 4 | 2 | −4 | ||
| Haloperidol | −2 | −2 | −5 | −2 | −2 | ||
| Perphenazine depot | −15 | −13 | 2 | 4 | 2 | ||
Tiihonen et al also reported outcomes for non-medicated patients. Although the majority of participants were on antipsychotic medication, mortality among those not taking antipsychotics was 10 times higher than among those who were. Overall, 9 patients taking antipsychotics died, compared with 75 not taking antipsychotics; the figures for suicide were 1 and 26 respectively. The NNT with antipsychotics to prevent one death was 34 (95% CI 27–47) and to prevent one suicide it was 90 (95% CI 64–150).
Clozapine
Clozapine was included in phase 2 of CATIE and in CUtLASS (Reference Lewis, Barnes and DaviesLewis 2006), and it was shown to be effective. In CATIE phase 2, clozapine was superior to risperidone (NNT = 4, 95% CI 2–15) and quetiapine (NNT = 3, 95% CI 2–6), but not to olanzapine, for all-cause discontinuation over 18 months. In CUtLASS, the presentation of the results precludes NNT analysis. Clozapine was also the most effective oral medication in Reference Tiihonen, Walhbeck and LönnqvistTiihonen et al(2006) (Table 5).
Discussion
Although there are methodological differences between the CATIE, EUFEST and Tiihonen studies in study design and length of follow-up, there are broad similarities in the differential effectiveness and adverse event rates for the antipsychotics studied. These similarities and differences are more apparent using NNT and NNH analysis than if the data are left in the original format. The range of NNTs presented so far is similar to ranges in other psychotropic treatment studies, including placebo-controlled trials. By comparison, psychological treatments such as family interventions in schizophrenia to prevent hospital admission have an NNT of about 8 (Reference Pharoah, Mari and RathbonePharoah 2006).
It is also clear that all drugs have attendant side-effects which may outweigh their benefit in certain circumstances. By using NNTs and NNHs, the data can be presented in a more meaningful way, which should facilitate discussions about medication choice with patients. Combining adverse event rates (NNH) with NNT provides an index that further extends this discussion, so that a patient and their treating clinician can balance treatment choices and also take into account factors that are relevant to the individual patient.
There are limitations to using NNTs. First, the data must be presented so that the relevant calculations can be made. This was particularly noticeable in CATIE, where much of the data presentation precluded analysis. Second, NNT analyses are only as good as the trial from which they are derived. Poor trials will result in misleading NNTs. There is also the impact of studies that do not demonstrate differences, and we have included two trials that did not significantly differentiate between trial drugs (CAFE and CUtLASS) to illustrate this point. Clinicians will still need to use their critical appraisal skills to determine which studies are most relevant to their patients and to assess the quality of the study in question. By presenting a number of studies we have also demonstrated that using NNT and NNH in isolation may be misleading.
Overall, clinicians need to be able to integrate evidence with clinical experience to optimise treatment in collaboration with their patients. These techniques may also be valuable tools to formulary committees, service commissioners and other funding bodies. Of course, these bodies may be interested in different outcomes from those that are important to clinicians and patients. Ultimately, although NNT and NNH help quantify clinical trial data in an understandable manner, they cannot account for an individual's personal weighting of risk v. benefit.
MCQs
Select the single best option for each question stem
-
1 The NNT:
-
a is a measure of effect size
-
b can only be calculated from continuous data
-
c cannot be calculated with confidence intervals
-
d is the reciprocal of NNH
-
e should always be more than 3 in a placebo-controlled trial.
-
-
2 A large NNT:
-
a indicates that the treatment is effective
-
b may be acceptable for a mild, self-limiting illness
-
c is often seen in immunisation programmes
-
d will be strongly associated with a large NNH
-
e with wide confidence intervals is likely to be clinically significant.
-
-
3 If 51.5% of patients taking placebo relapse in the year after their first episode of schizophrenia compared with 16.2% receiving an antipsychotic (P = 0.01), then:
-
a the NNT for the antipsychotic is 2
-
b the NNT for the antipsychotic is 3
-
c the NNT for placebo is −11
-
d the NNT is 1/(the sum of the relapse rates)
-
e the antipsychotic is ineffective in preventing relapse in patients with schizophrenia.
-
-
4 If 70% of patients on drug X achieve remission of their depressive symptoms compared with 50% on drug Y, and 60% of patients on drug X develop frequent headaches compared with 30% on drug Y, then:
-
a the NNT for drug X is 10
-
b the NNT for drug X is 5
-
c the NNH for frequent headaches with drug X is 3
-
d the NNH for frequent headaches with drug X is 6
-
e drug X is the better drug.
-
-
5 The Reference Tiihonen, Walhbeck and LönnqvistTiihonen (2006) study shows that:
-
a patients taking antipsychotics are likely to die by suicide
-
b clozapine has the lowest discontinuation rate of the oral antipsychotics
-
c patients taking antipsychotics are far more likely to die than those not taking antipsychotics
-
d all antipsychotics are equally effective at preventing hospital admission
-
e NNTs can be calculated from observational studies.
-
MCQ answers
| 1 | a | 2 | c | 3 | b | 4 | b | 5 | b |






eLetters
No eLetters have been published for this article.