



Introduction
Aging is a biological process that causes the organism's quality of life to diminish over time due to time-dependent cellular and functional degradation. Age-related conditions as a group pose a serious socioeconomic burden on the world and a substantial healthcare concern. Finding treatment approaches that slow the deterioration of numerous age-related pathological disorders is therefore crucial (Ref. Reference Aman1).
The incidence of various diseases such as cancer, cardiovascular diseases, neurodegenerative diseases (NDs) and metabolic dysfunction rises with aging (Ref. Reference Menzies2). Above-mentioned illnesses can be brought on by dysregulation of cellular autophagy. Autophagy is a quite preserved catabolic cellular process that expresses lysosomal degradation and recycling of nucleic acids, proteins, lipids and other intracellular components. Autophagy mechanism is associated with homoeostasis, differentiation, development and survival processes, as well as defence against pathogens and conservation of cellular energy. Decreased autophagic activity in aging affects a variety of cellular and molecular processes and results in the accumulation of damaged macromolecules and organelles. Therefore, it has been suggested that maintenance of regular autophagic activity is associated with longevity (Ref. Reference Ruckenstuhl3).
Autophagy functions as both a tumour suppressor and promoter (through protective autophagy) in cancer. Furthermore, it plays a crucial role in preventing cardiac aging and maintaining neuronal homoeostasis. Early regulation of the formation of the mammalian heart, proper maintenance of cardiac structure and function, and the beginning and progression of cardiovascular disorders are all regulated by autophagic activity (Ref. Reference Gatica4). In addition to protein aggregation, NDs have dysregulated autophagy along with mitochondrial dysfunction and reduced lysosomal activity. The prevalence of NDs like Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD) increases with aging and neuronal autophagy dysregulation (Ref. Reference Tan5). Moreover, in metabolic illnesses including obesity and diabetes, autophagy is suppressed or increased (Ref. Reference Jaishy6). Studies have demonstrated the significance of the signalling pathways PI3K/AKT/mTOR, AMP-activated protein kinase (AMPK), MAPK, SIRT1, and FoXO in the control of autophagy, cell aging, and metabolic diseases.
There are many synthetic compounds that have been discovered that modify autophagy and are good candidates for the therapy of cancer, but they come with unfavourable side effects. As a result, several phytochemicals have drawn a lot of interest for their potential to be used as autophagy modulators with minimum adverse effects (Ref. Reference Rahman7). Indeed, naturally occurring compounds including, alkaloids, terpenoids, flavonoids, phenylpropanoids, lignans, phenolic acids, stilbenes, tannins, senevol glycosides, steroids, lectins have been found to be powerful autophagy inducers or inhibitors, opening up new treatment regimens.
In the present review new aspects of autophagy and speculations on how they might be connected to both aging and the onset and progression of age-related diseases are discussed. We also aim to summarise the natural compounds that can be used as autophagy modulators. Eventually, developing practical applications that support long-term health will be made easier with a better understanding of the specific interactions between autophagy and the risk of age-related diseases across organisms.
Autophagy and aging
Three different types of autophagy have been described; macroautophagy; is the basic mechanism of autophagy and occurs when larger particles and organelles are taken into the autophagosome and fused with lysosomes, microautophagy; small particles are taken into the lysosome with the indentation in the membrane and degraded, chaperone-mediated autophagy (CMA); the recognition and transport of cytosolic proteins with a certain peptide sequence by Hsp73, which is complex with molecular chaperones, to lysosomes and this type of degradation pathway does not require vesicular traffic (Refs Reference Majeski8, Reference Kroemer9). On the other hand, selective forms of macroautophagy and microautophagy in which the degradation process occurs due to either the binding of HSC70 to the hydrophobic residues exposed in misfolded or aggregated proteins exists, namely chaperone-assisted selective autophagy (CASA) or the sequence-mediated targeting of proteins by HSC70, namely endosomal microautophagy (eMI) (Refs Reference Tekirdag and Cuervo10, Reference Vitale11).
The morphology of autophagy was first described in mammalian cells in the 1950s, and 31 autophagy-related (ATG) genes involved in the basic mechanism in autophagy were identified (Refs Reference Huang12, Reference Klionsky13). Most yeast genes encoding autophagy proteins are orthologous to the Caenorhabditis elegans genome (ATG2, ATG3, ATG4, ATG5, ATG7, ATG9, ATG10, ATG12, ATG18 genes), but there are two homologues of ATG4, ATG8 and ATG16. The ATG proteins encoded by these genes act at different stages of autophagosome biogenesis such as initiation of autophagy (unc-51/ATG1), vesicle nucleation (bec-1/ATG6, vps-34/VPS34), protein conjugation system (ATG-7/M7.5/ATG7, lgg-1/ATG8, lgg-3/ATG12), uptake and vesicle transformation (ATG-18/F41E6.13/ATG18) (Ref. Reference Kovacs14).
Macroautophagy can be induced in the presence of various stress factors such as hypoxia, starvation, presence of damaged protein or organelles. This type of autophagy involves 5-steps including initiation, nucleation, elongation, fusion and degradation. In the initiation stage, inhibition of mTOR, the master regulator of autophagy, leads to activation of autophagy. The nucleation stage involving the ULK1 complex (ULK1/2, ATG13 and FIP200) and the class III PI3K complex (Beclin 1, ATG14L, p150) participates in phagophore formation. The phagophore membrane is closed via the ATG12 conjugation system (ATG12, ATG5 and ATG16L) and the LC3-II conjugation system in the elongation stage. This structure, called the autophagosome, contains the autophagic cargo. Finally, in the degradation stage, the autophagosome fuses with the lysosomal membrane and the autophagic cargo is sequestered by lysosomal hydrolases (Refs Reference Karampa15, Reference Hu and Reggiori16, Reference Sanchez-Mirasierra17). In macroautophagy and microautophagy (also called endosomal microautophagy in mammals), cargo transport to the lysosome is vesicle-mediated, whereas in CMA it occurs through specific receptors (Refs Reference Scrivo18, Reference McCarty19). In CASA, which is one of the selective macroautophagy types, polyubiquitinated aggregate proteins are transmitted to the autophagosome by helper chaperone Bcl2-related athanogen 3 (Bag3) and cytosolic chaperone heat shock cognate 70 kDa protein (HSC70) without binding to a pentapeptide amino acid motif (KFERQ)-like motif (Ref. Reference Scrivo18). CMA is a variant of autophagy defined only for proteins carrying a specific targeting motif in the KFERQ amino acid sequence. The KFERQ-like motif is recognised by the cytosolic chaperone HSC70 and translocates to the substrate lysosome by the lysosome-associated membrane protein type A (LAMP2A) receptor. CASA is distinct from CMA, by being ubiquitin-independent (Fig. 1) (Refs Reference Massey, Zhang and Cuervo20, Reference Arndt21).
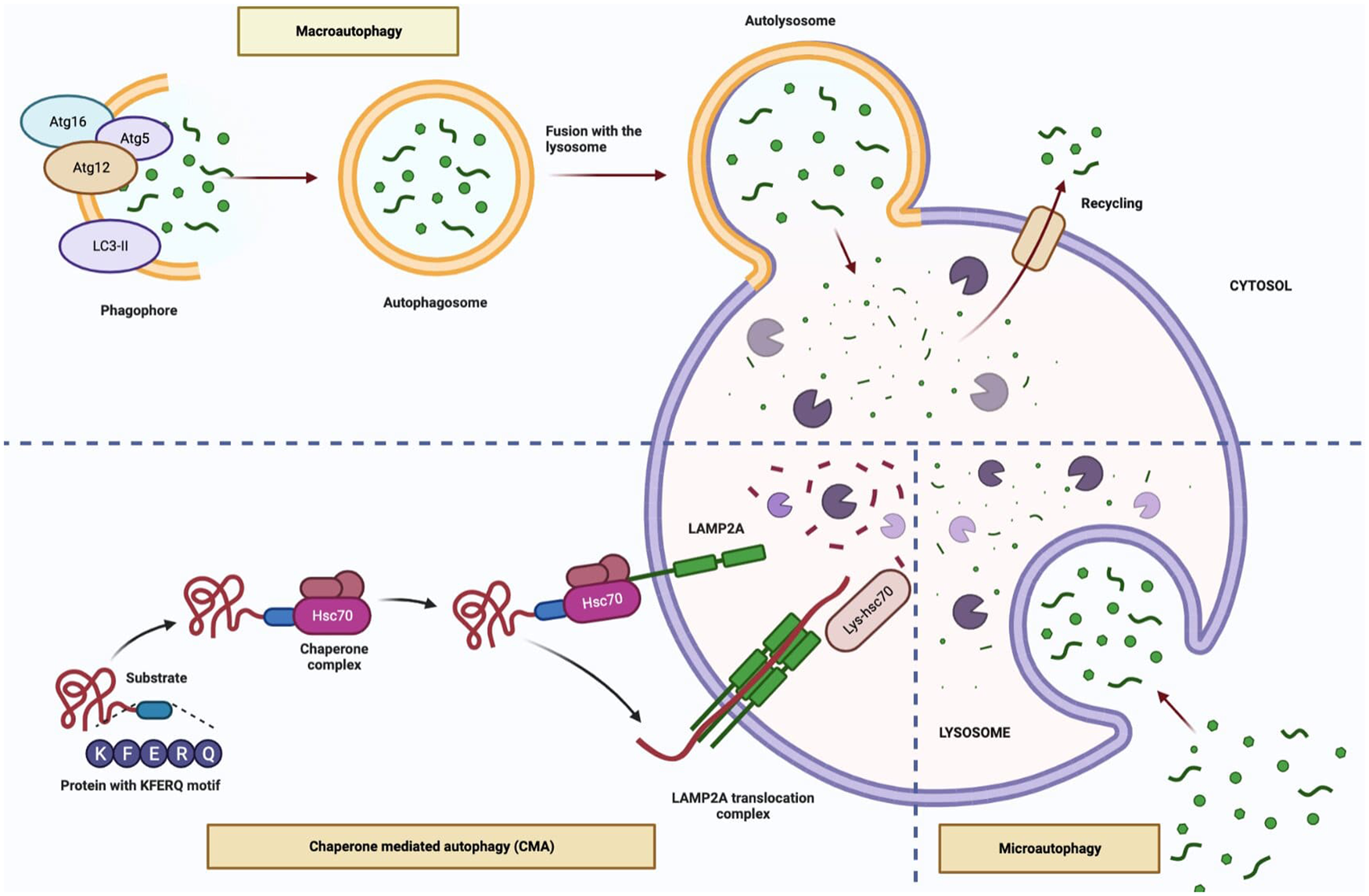
Figure 1. The types of autophagy (Figure created with Biorender.com).
The processes of autophagy are related to aging in terms of biochemical regulation of cells. It has been reported that the decrease in gene expression of ATG5, ATG7, and Beclin-1 with age in humans is a result of decreased stimulation of macroautophagy (Ref. Reference Lipinski22). Activation of macroautophagy as a result of disruption in the Beclin-1-BCL2 complex prolongs the lifespan of mice (Ref. Reference Fernández23). While the impairment of macroautophagy in cells such as neurons, melanocytes, and fibroblasts increases aging (Ref. Reference Moreno-Blas24), chemical regulation of macroautophagy in tendon stem cells decreases senescence (Ref. Reference Nie25). Although the mechanism of microautophagy is well elucidated, the information on the relationship between microautophagy and aging is quite insufficient. One of the limited studies has shown that the accumulation of protein and lipid peroxidation products via microautophagy increases with aging (Ref. Reference Cannizzo26). The accumulation and aggregation of proteins known as CMA substrates are associated with the pathogenesis of aging and its accompanying diseases. Studies have shown that an increase in LAMP2A levels improves the reduction in CMA associated with aging (Ref. Reference Valdor27). Moreover, in previous preclinical studies, impaired CASA was shown to cause Z disc disintegration and progressive muscle weakness in flies, mice, and human, revealing the importance of chaperone-assisted degradation for the preservation of cellular structures of muscle tissues, thereby shedding light on aging (Ref. Reference Arndt21).
Hallmarks of aging
Hallmarks of aging are mainly loss of proteostasis, mitochondrial dysfunction, disordered nutrient perception, genomic instability, telomere attrition, cellular senescence, cellular stemness.
The role of autophagy in the loss of proteostasis
During the aging process, a decrease in the proteostasis activity of many cells and tissues is observed. Loss of proteostasis, a key feature of aging, is characterised by the appearance of misfolded or aggregated proteins. On the other hand, autophagy is capable of detecting proteotoxic stress and developing an appropriate response. Autophagy, which plays an important role in removing misfolded or unfolded protein aggregates, is impaired with age. With this degradation, protein aggregates accumulate and cause proteotoxicity (Ref. Reference Di Domenico28).
Autophagy in mitochondrial dysfunction
The reduced mitophagy (known as clearing damaged mitochondria) associated with aging in mitochondria also leads to decreased energy production. Autophagy plays a key role in cleaning damaged mitochondria. Dysregulation in the autophagy mechanism causes mitochondria accumulation and thus oxidative stress (Ref. Reference Um and Yun29).
Autophagy and cellular stemness
Autophagy plays a major role in the ability of adult stem cells to self-renew and differentiate into specialised cells. The decrease in autophagic activity with age results in a decrease in stem cells (Ref. Reference Boya, Codogno and Rodriguez-Muela30).
Autophagy and senescence
Oxidative stress, DNA damage, telomere shortening lead to cellular senescence. While autophagy over-stimulation induces cell death, its deficiency can trigger cellular senescence (Ref. Reference Rajendran31).
Autophagy and genomic instability
UV light, toxic chemicals, agents such as reactive oxygen species (ROS), mutations in nuclear and mitochondrial genes cause DNA damage. Weakening of the DNA repair mechanism and accumulating DNA damage trigger cellular senescence (Ref. Reference Hewitt and Korolchuk32).
Autophagy and telomere attrition
Damage to telomeres or a decrease in their activity is directly related to the aging process (Ref. Reference Lopez-Otín33). An increase in cytoplasmic vacuoles and autophagy-related ATG5, ATG12 and LC3 proteins was observed in telomere-defected cells (Ref. Reference Cheon34).
Molecular mechanisms of autophagy
Many signalling pathways have been identified to be responsible for regulating longevity and aging. Studies have shown that PI3K/AKT/mTOR, AMP-activated protein kinase (AMPK), MAPK, SIRT1, FoXO, signalling pathways are important in the regulation of autophagy as well as cell aging and metabolic disorders.
The PI3K/AKT/mTOR axis induced by growth factors is an essential signalling pathway in cancer cell growth in nutrient-rich conditions. In nutrient deficient conditions, inhibition of this pathway promotes autophagy (Ref. Reference Fairlie, Tran and Lee35). TOR is an evolutionarily conserved serine/threonine kinase mainly localised in cytoplasm that regulates various cellular functions, including cell metabolism, survival, cytoskeleton, and autophagy (Ref. Reference Ge36). mTOR is encoded by a single gene and there are two types of mTOR complexes as the protein product, functionally and structurally: mTOR complex 1 (mTORC1) sensitive to rapamycin and mTOR complex 2 (mTORC2) insensitive to rapamycin (Ref. Reference Bhaskar and Hay37). mTORC1 consists of mTOR, raptor (regulatory-associated protein of mTOR), mLST8 (mammalian lethal with sec-13 protein 8), PRAS40 (proline-rich AKT substrate 40 kDa), Deptor (the DEP domain containing mTOR-interacting protein), and TTI1/TEL2 (Ref. Reference Papadopoli38). mTORC1 is responsible for negative regulation of cell growth, differentiation and autophagy. Phosphorylation of mTORC1 inactivates ULK/ATG1, which promotes autophagy, increases anabolism, and promotes cell growth. Rapamycin is a well-known inhibitor of mTOR and also an inducer of macroautophagy. The mTORC2 consists of mTOR, mLST8, rictor, mSIN1, protor, and Deptor subunits. mTORC2 is mainly involved in the insulin signalling pathway and indirectly regulates autophagy (Ref. Reference Wang39). The activity of mTORC1 is regulated by the PI3K/AKT/mTOR signalling pathway as well as by the AMPK and Ras-dependent MAPK pathways (Ref. Reference Zada40). It was shown that regulation of autophagy dependent on the AMPK-mTOR signalling pathway can delay cardiomyocyte senescence (Ref. Reference Chen41). It was also determined that overactivation of mTOR leads to impaired autophagy and results in premature aging (Ref. Reference Pan42). ULK1, one of the important autophagy proteins, is a serine/threonine kinase and cellular autophagy is triggered by ULK1 binding to ATG13, FIP200 and ATG101 proteins to form a complex (Ref. Reference Zachari and Ganley43). Under normal energy conditions, mTORC1 phosphorylates ULK1 and AMPK cannot activate ULK1. Cell autophagy is inhibited because it cannot form complexes with inactive ULK1, ATG and FIP200. In the absence of nutrients in cells, AMPK is activated, mTOR is inhibited, and autophagy is activated (Ref. Reference Turco, Fracchiolla and Martens44). Consequently, the activated mTOR pathway inhibits the activation of ULK1 to induce senescence by inhibiting autophagy. When AMPK, the negative regulator of mTOR, is activated, it either activates ULK1 or inhibits mTOR and can delay aging by initiating autophagy.
SIRT1 is the mammalian homologue of Sir2 (silent information regulator) protein, known as stress resistance and longevity factor in yeasts. SIRT1 is the most studied sirtuin of seven human sirtuins (Ref. Reference Kitada, Ogura and Koya45). SIRT1 is a class III protein deacetylase and uses nicotinamide adenine dinucleotide (NAD) as a cofactor to deacetylase lysine residues, thereby silencing genes, which can delay aging, reduce inflammation, improve energy metabolism. It was revealed that SIRT1 can form complexes with autophagy-related ATG5, ATG7, LC3 proteins and regulate autophagy by causing their direct deacetylation (Ref. Reference Lee46). Calorie restriction delays the onset of age-related diseases such as NDs, cardiovascular diseases, and diabetes, therefore it is the only strategy to extend lifespan in many organisms (Ref. Reference Colman47). Although the mechanism of extension of lifespan is unclear, it has also been determined that autophagy is increased during calorie restriction and energy deficiency (Ref. Reference Hansen48). SIRT1 level in humans decreases with age, therefore it causes cellular proteins misfolding and cell damage. SIRT1 and AMPK are positive regulators of each other. NAD expression is increased by AMPK and activates SIRT1 (Ref. Reference Liu49). Calorie restriction also induces activation of SIRT1 with an anti-aging effect. The FoXO family of transcription factors (FoXO1, FoXO3a, FoXO4 and FoXO6) is also an important regulator of cellular metabolism and proliferation (Ref. Reference Fahy50). Recent studies have shown that signalling pathways related to SIRT1 and FoXO3 have an important role in the regulation of autophagy. SIRT1-mediated activation of FoXO1 induces expression of autophagosomes, and Rab7 (GTPase) which is highly important in autophagosome formation (Ref. Reference Ao, Zou and Wu51). Overexpression of Rab7 also triggers autophagy, and activated FoXO3 protein increases the expression of autophagy-related proteins (LC3 and BNIP3) (Fig. 2) (Ref. Reference Salminen and Kaarniranta52).

Figure 2. The molecular signalling mechanisms of autophagy (Figure created with Biorender. com).
Autophagy and age-related diseases
Cellular components that have been implicated in the aging process have been linked to progressively impaired autophagy. In the elderly, dysfunctional autophagy may play a role in age-related disorders such as cancer, cardiovascular and neurological diseases and metabolic syndrome. Thus, restoring defective autophagy to its healthy state may aid in preventing age-related diseases and boosting longevity (Ref. Reference Cheon34).
Autophagy in cancer
Cancer occurs when the balance between cell proliferation and death is lost, and more cells multiply than die or differentiate (Ref. Reference Mak53). Unlike apoptosis, autophagy helps maintain homoeostasis by reversing intracellular molecules in the absence of nutrients or in cellular stress (Ref. Reference Anding and Baehrecke54). Basal autophagy works as a tumour suppressor mechanism during tumorigenesis; excessive autophagy works as a survival pathway in certain cancers (Ref. Reference Yu, Chen and Tooze55). Unlike normal cells, basal levels of autophagy are high in tumour cells, stimulated in hypoxic regions, and provide a constitutive survival advantage (Ref. Reference Ding, Chen and Yin56). Therefore, inhibition of autophagy may prevent the survival of tumour cells and may be a new target for cancer therapy (Ref. Reference Maycotte and Thorburn57) (Table 1).
Table 1. Studies on autophagy in cancer

Autophagy is used to provide components and energy to cells for basal survival in each cell (Ref. Reference Anding and Baehrecke54). Autophagy is controlled by class I PI3K/mTOR and AMPK signalling pathways. During nutrient availability (growth factors and amino acids), mTOR kinase, a target of rapamycin, suppresses autophagy. Mutations in the PI3K/AKT/mTOR pathway, in which autophagy is negatively regulated, may lead to oncogenic transformations by increasing signalling in the pathway (Ref. Reference Alers58).
Beclin-1, which has a central role in autophagy, can function as a tumour suppressor (Ref. Reference Liang59). Beclin-1 is monoallelic deleted in many cancers, such as breast, ovarian, brain, and prostate cancers, and low protein levels have been found in the tumours of these cancers. Studies in mice have shown that heterozygous disruption of beclin-1 increases permeability to spontaneous tumour development (such as lymphomas, lung carcinomas, hepatocellular carcinomas, and mammary precancerous lesions), whereas Beclin-1 restoration in MCF-7 cells reduces proliferation and tumorigenesis (Refs Reference Liang59, Reference Mathew, Karantza-Wadsworth and White60, Reference Miracco61). Bif-1 plays a role in inducing autophagosome formation in autophagy and interacts with Beclin-1 via UVRAG. UVRAG proteins have been observed to be abnormal or deleted in various types of cancer, such as colorectal and gastric cancer. It is also a tumour suppressor candidate because it reduces proliferation and tumour size in cases of excessive UVRAG expression (Ref. Reference Liang62). It has been reported that the MAP1-LC3 gene, which encodes LC3-II located in the inner and outer membrane of the autophagosome and functions in autophagy substrate selection and autophagosome biogenesis, is frequently deleted in the liver, breast, prostate, and ovarian cancers (Ref. Reference Salvi63). Thus, it can be considered that autophagy is both a tumour suppressor and promoter (via protective autophagy) mechanism, tumours with autophagy deficiency grow faster and this situation increases exponentially with defects in apoptosis.
Modulation of autophagy in cancer therapy is a promising potential strategy. Autophagy is also a protective mechanism in cancer cells during anticancer treatment. The common treatment strategy in cancer treatment is chemotherapy, but the development of chemoresistance (such as the protective autophagy mechanism) limits the success rate. Some autophagy regulators, such as the best-known mTOR inhibitors rapamycin and its derivatives (temsirolimus and everolimus), chloroquine (CQ), and hydroxychloroquine (HCQ), are also used in cancer treatment.
Rapamycin, an mTOR inhibitor, was developed to inhibit proliferation signals in the PI3K/AKT/mTOR pathway, which is important for cell growth and proliferation in multiple tumour types (Ref. Reference Huang64). Moreover, rapamycin may sensitise cancer cells to radiation therapy and chemotherapeutic agents such as adriamycin, cisplatin and hormonal therapies (Ref. Reference Rao, Buckner and Sarkaria65). In addition rapamycin is used in rare lung diseases, coronary stents (Ref. Reference Zaytseva66). Temsirolimus was the first mTOR inhibitor class drug to be shown to improve survival in patients with advanced renal cell carcinoma (Refs Reference Motzer67, Reference Jurczak68). Increased PI3K/AKT expression and activation are common in renal cell cancer and this drug is used in the first-line treatment of metastatic renal cell cancer with poor prognosis (Ref. Reference Zaytseva66). It is also used in the treatment of relapsed or refractory mantle cell lymphoma in the European Union. Everolimus is used in combination with exemestane in the treatment of advanced neuroendocrine tumours and breast cancer (Refs Reference Yao69, Reference Zhao70). In preclinical studies in bladder cancer and pancreatic adenocarcinoma, it has been shown that CQ and HCQ can suppress cancer cell growth by inhibiting autophagy. Several studies have demonstrated that these reagents promote apoptosis through suppression of autophagy and enhance the therapeutic effects of chemotherapy by inhibiting autophagy-mediated resistance seen in therapy (Refs Reference Zhao70, Reference Liang, El-Zein and Dave71). Lys05, a water-soluble analogue of HCQ, showed higher anticancer effects than HCQ in low doses of melanoma and colon cancer xenograft models (Ref. Reference Barnard72). 5-Fluorouracil (5-FU), which is used in solid cancers such as colorectal, breast, and pancreatic cancer, has limited efficacy in the treatment because it induces protective autophagy and creates chemoresistance (Refs Reference Malet-Martino, Jolimaitre and Martino73, Reference Liang74). Spautin-1, SAR405 are newly developed autophagy-related anticancer drugs (Ref. Reference Kunimasa75).
Autophagy in cardiovascular diseases
Autophagic activity is implicated in the regulation of mammalian heart development in the early stage, maintenance of cardiac structure and function under normal conditions, in the onset and progression of cardiovascular diseases including ischaemia/reperfusion (I/R) and heart failure (Ref. Reference Gatica4). In addition, autophagy is important in delaying cardiac aging (Ref. Reference Shirakabe77).
Activation of autophagy during myocardial ischaemia ensures the maintenance of energy substrates and removal of damaged mitochondria that may initiate apoptosis by causing oxidative stress (Ref. Reference Zhang78). Autophagy activation during ischaemia relies on AMPK mediated inhibition of the mTORC1 pathway (Ref. Reference Matsui79). Inhibition of autophagy during chronic ischaemia can cause cardiomyocyte death by activating apoptosis. Therefore, induction of autophagy during myocardial ischaemia may be protective.
During reperfusion, tissues exposed to ischaemia are reloaded with nutrients and oxygen. Reperfusion injury is defined as damage to tissues or organs during the re-bleeding period following the ischaemic period. Studies have shown that autophagy is a key regulator with a dual role for I/R and is activated during I/R (Ref. Reference Hao80). AMPK activation is involved in autophagy resulting from ischaemia, whereas activation of Beclin 1 is involved in autophagy during reperfusion. Autophagy plays different roles during ischaemia and reperfusion; while protective during ischaemia, it could be harmful during reperfusion (Ref. Reference Matsui79). (Table 2).
Table 2. Studies on autophagy in cardiovascular diseases

The heart's first response to various stresses, such as hypertension, aortic stenosis, is hypertrophy, and if this situation continues, heart failure may develop (Ref. Reference Dämmrich and Pfeifer81). Furthermore, age-related progressive loss of autophagic activity leads to the development of hypertrophy (Ref. Reference Shirakabe77). It remains unclear whether the altered autophagy observed in cardiac hypertrophy and heart failure is beneficial or harmful. β-adrenergic stimulation, which induces cardiac hypertrophy and heart failure, inhibits autophagy and stimulates apoptosis (Ref. Reference Hao80). The deficiency of ATG5 causes cardiac hypertrophy by inhibition of autophagy with increased ubiquitination levels, while overexpression of ATG5 increases autophagy levels with Beclin-1 function and extends lifespan (Ref. Reference Nakai82). In conclusion, basal autophagy in the heart under normal conditions is a homoeostatic mechanism for maintaining cardiac structure and function.
The prevalence of autophagic activation in cardiac diseases has led to studies in this area for therapeutic targets. In the conjectural applications of cardiovascular diseases, 8-methylchroman-7-ol derivatives and glycosylated anti-tumour ether lipids inhibit autophagy; Rubicon, which binds the class III PI3K/Vps34–Beclin 1 complex, and AP23573, a phosphorus-rapamycin analogue, are used in the regulation of autophagy (Ref. Reference Nemchenko83).
Autophagy in neurological aging
Autophagy is important for the maintenance of neuronal homoeostasis by mediating the clearance of unfolded or misfolded protein aggregates.
Age-related decrease in autophagy may lead to accumulation of intracellular toxic wastes (Ref. Reference Di Domenico28). This condition is characterised by aggregated and abnormally deposited proteins in NDs. Apart from protein aggregation, mitochondrial dysfunction and decreased lysosomal activity accompany dysregulated autophagy in NDs. Aging and neuronal autophagy dysregulation increase the incidence of NDs such as AD, PD, HD (Table 3). In particular, misfolded protein accumulation and disruption of the autophagy pathway due to mTOR are seen in these diseases. With aging, there may be decreases in the ubiquitin-proteasome system and autophagy-lysosomal pathway, which are associated with the clearance of misfolded proteins in the brain (Ref. Reference Hansen, Rubinsztein and Walker84). Measures to eliminate the disorders in the autophagy mechanism may contribute to the prevention of age-related diseases. Gene polymorphisms involved in autophagy mechanisms contribute to aging-related neurodegeneration. Plaques formed as a result of tau protein accumulation in AD, mutant α-synuclein proteins (Lewy bodies) in PD, and mutant Huntingtin protein (mHtt) accumulation in the cytoplasm are seen in HD. Autophagy plays an active role in the clearance of these proteins. There is evidence that overexpression of ATG5, which is involved in autophagy induction, activates autophagy and increases survival. However, it was shown that there was a decrease in the expression of the ATG7 and Beclin-1 genes. In this case, it can be concluded that the decrease in autophagy activation decreases with aging (Ref. Reference Lipinski22).
Table 3. Studies on autophagy in neurodegenerative diseases
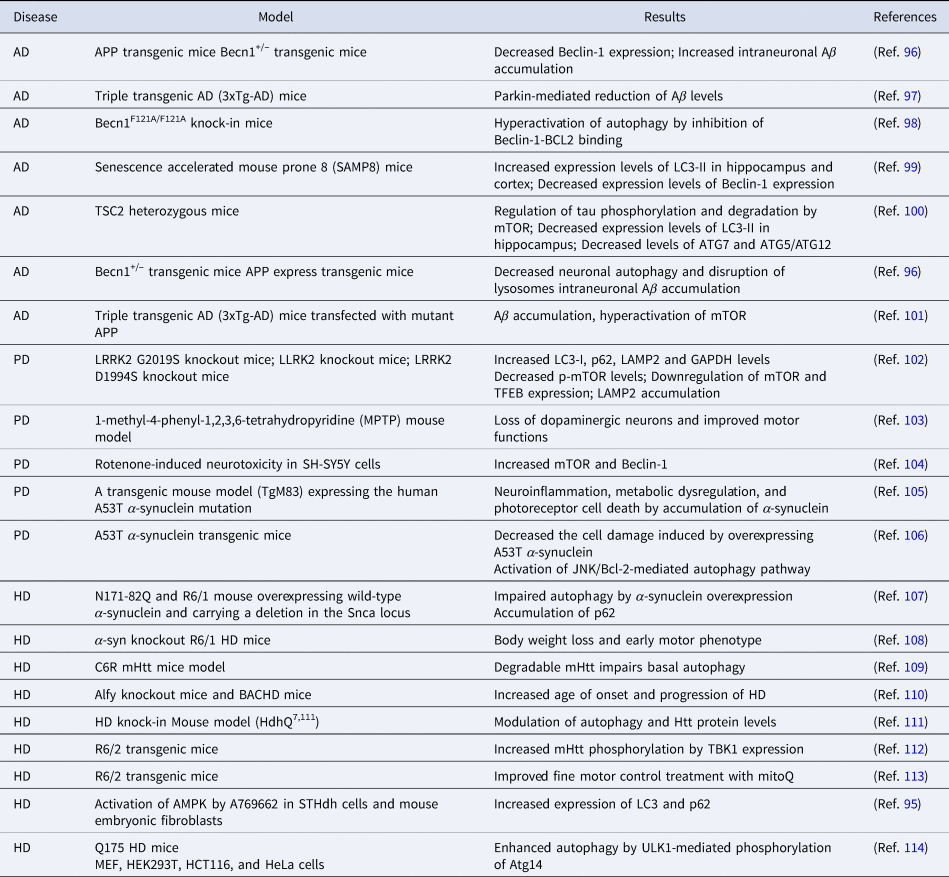
AD, Alzheimer's Disease; PD, Parkinson Disease; HD, Huntington's Disease.
Alzheimer's disease
AD is a ND characterised by the accumulation of extracellular β-amyloid (Aβ) plaques and intracellular neurofibrillary tangles containing hyperphosphorylated tau (Ref. Reference Tan5). Only 5% of AD is familial and results from mutations in presenilin 1, presenilin 2, and amyloid precursor protein (APP). Aβ and tau protein are substrates for autophagy. An abundance of autophagic structures containing these substrates is observed in AD. In addition, it has been shown that abnormal conformations of Aβ reduce lysosomal amplification, lead to synaptic defects and increase the course of the disease (Ref. Reference Holtzman, Morris and Goate85). Neurofibrillary tangles caused by mutant tau proteins can block CMA, resulting in decreased autophagy. Beclin-1, an autophagy-related protein, decreases with age in the brains of AD models, and increased levels of Beclin-1 ameliorate amyloid pathology.
Due to the morphological structure of nerve cells, the levels of clearance by autophagosomes may be different compared to other cells. The importance of autophagy in these cells is due to their post-mitotic nature. Autophagy is very important in cell survival as it can remove toxic aggregates in post-mitotic nerve cells. It has been found that autophagosome clearance is impaired in late-stage disease in neuron cells from patients with AD only. In some studies, it has been reported that the levels of some autophagy genes are increased in AD brains (Ref. Reference Lipinski22).
Parkinson disease
PD is a ND caused by selective loss of dopaminergic neurons and decreased dopamine content in the striatum (Ref. Reference Halliday, Lees and Stern86). This loss can be induced by mitochondrial toxins such as 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP) and the complex I inhibitor rotenone. As in AD, mutations that cause PD can directly cause disruption of autophagy. Some of these mutations are α-synuclein (SNCA or PARK1), also known as Lewy bodies, Parkin (PRKN or PARK2), ubiquitin carboxy-terminal hydrolase L1 (UCH-L1), PTEN-induced putative kinase 1 (PINK1), protein deglycase DJ-1 (PARK7) and leucine-rich repeat kinase 2 (LRRK2) (Ref. Reference Senkevich and Gan-Or87). Furthermore, many PD-related gene mutations can cause loss of function in the autophagy-lysosome pathway (ALP). Because these genes play a role in mitophagy, mutations in these genes cause insufficient mitochondrial quality control (Ref. Reference Pickrell and Youle88). Two major mutations in α-synuclein (A53T and A30P) are associated with PD (Ref. Reference Recchia89). Mutant α-synuclein causes disruption of CMA activity, inhibition of autophagosome formation and disruption of lysosomal degradation (Ref. Reference Winslow90). Increased expression of mTOR, one of the important autophagy pathways, has been reported after α-synuclein accumulation in patients with PD. Apart from aging and genetic mutations, PD may also develop due to dopaminergic neuron-specific toxins such as 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP) and rotenone.
Huntington's disease
HD is an autosomal dominant ND caused by long CAG trinucleotide expansion (over 37 repeats) in the Htt gene (Ref. Reference McColgan and Tabrizi91). In patients with HD, an abnormally long polyglutamine-encoding CAG trinucleotide expansion produces perinuclear cytoplasmic aggregates and intranuclear inclusions. This results in mutant Htt, an autophagic substrate, ingestion of defective autophagic substrate into autophagosomes. In addition, this expansion of polyglutamine causes misfolded proteins or accumulation of protein aggregates. Deletion of polyglutamine in the Htt gene has been shown to improve disease symptoms and increase autophagosome formation (Ref. Reference Jeong92). In addition, the decrease in Beclin-1 expression with aging may lead to the accumulation of mutant Htt (Ref. Reference Wu93). It has also been reported that the mTOR pathway plays a role in HD pathology. In a recent study, they identified the Homeodomain Interacting Protein Kinase 3 (HIPK3) gene as a negative modulator of autophagy and a positive regulator of mHtt expression levels in HD cells. It is considered that modulation of mHtt by HIPK3 can be a therapeutic target for HD (Ref. Reference Fu, Sun and Lu94). Stimulation of autophagy usually occurs by inhibition of mTOR. However, since mTOR is involved in many cellular processes, its inhibition can lead to unexpected effects. Walter et al. showed that AMPK activation causes increased expression of LC3-II and p62, and induced autophagy in an mTOR-independent manner. This resulted in decreased aggregates containing mHtt and cell viability (Ref. Reference Walter95).
Autophagy in obesity and diabetes
Obesity, which is associated with excessive calorie intake, is a chronic disease that occurs as a result of many genetic and environmental factors, affecting the metabolism and physiology of organs (Ref. Reference Zhang, Sowers and Ren115). Since triglycerides are stored in adipose tissue and metabolised by the liver, adipose tissue is at the centre of obesity and metabolic diseases (Ref. Reference Oga and Eseyin116). Autophagy, on the other hand, is a physiological process that removes damaged organelles, misfolded proteins, and lipids in case of excessive calorie intake. Autophagy is a highly sensitive mechanism to excessive calorie intake. Suppression or increase of autophagy is seen in metabolic diseases such as obesity and diabetes (Ref. Reference Jaishy6).
Insulin resistance, which develops due to the increase in calorie intake, suppresses the activity of the mTOR pathway; this initiates the autophagy process in adipose tissues. However, it has been determined that calorie restriction increases autophagy activity and, accordingly, insulin sensitivity in obese individuals. In addition, an increase in the LC3 level and the number of autophagosomes in the fat cells of obese patients was observed. In another study, it was determined that the mRNA expression level of the autophagy gene ATG5 was higher in patients with higher BMI and larger visceral adipose tissue (Ref. Reference Kovsan117). Obesity was shown to stimulate inflammation by causing hypothalamic resistance to insulin and leptin hormones through an inhibitor of the Nuclear Factor kappa B (NF-kB) pathway and induces hypothalamic dysfunction (Ref. Reference Zhang118). Therefore, hypothalamic dysfunction can be considered to play a role in the pathophysiology of obesity and diabetes. In studies, hypothalamic inhibition of autophagy via siRNA-mediated ATG7 suppression was achieved in mice fed a high-fat diet, resulting in increased energy consumption. In other words, feeding with HFD in mice caused disruption of lipolysis and insulin resistance. It has also been observed that hypothalamic autophagy is impaired in HFD-induced obesity (Ref. Reference Kaushik119).
Diabetes is a metabolic disease identified as chronic hyperglycaemia. There are two main clinical types, insulin-dependent (Type 1) and insulin-independent (Type 2) (Ref. Reference Gonzalez120). Type 1 diabetes mellitus (T1DM) is a disease that develops by autoimmune destruction of pancreatic beta cells. It is known that insulin reduces tau phosphorylation and autophagy by inhibiting the PI3K/AKT signalling pathway-mediated Glycogen synthase kinase-3 (GSK-3). Phosphorylation of tau protein is increased in T1DM with insulin deficiency (Ref. Reference Santos121). On the other hand, insulin resistance, hyperglycaemia and relative insulin deficiency determine the development of Type 2 diabetes mellitus (T2DM). The increase of adipose tissue in T2DM causes insulin resistance by activating the mTOR pathway. mTORC1 is activated by high amounts of glucose, fatty acids and amino acids. Moreover, insulin is a hormone that inhibits autophagy (Ref. Reference Brännmark122). Insulin resistance causes autophagy activation in adipose tissues of obese patients. In the previous studies, it was demonstrated that insulin resistance was induced and autophagy was inhibited by decreasing the expression levels of autophagy-related LC3, ULK2, ATG12 proteins in livers of mice fed a high-fat diet (Ref. Reference Ueno and Komatsu123). Studies on autophagy in obesity and diabetes are summarised in Table 4.
Table 4. Studies on autophagy in obesity and diabetes

Natural active compounds in longevity intervention
Age is the greatest risk factor for all major age-related pathologies. In recent years, it has been reported that the molecular mechanisms underlying aging are associated with cancer, cardiovascular diseases and neurodegeneration. It can also affect the same pathway, autophagy, as these diseases. Therefore, autophagy is gaining importance in the discovery of therapeutic interventions that promote healthy aging and increase longevity (Ref. Reference Niso-Santano142).
Natural products were widely used to treat different medical conditions before the development of modern pharmaceuticals. Today they still serve as an important pool for the identification of new drug precursors against various diseases including cancer and chronic diseases, which become widespread with increasing age. The structural complexity and diversity of natural products provide a valuable resource for future drug discovery.
In the following section, we summarise the autophagic mechanisms of natural compounds in cancer, cardiac diseases, neurological aging, diabetes and obesity.
Autophagic mechanisms of natural compounds in cancer
Natural compounds have been identified as potent inducers or inhibitors of autophagy to exhibit antitumour mechanisms, opening up new therapeutic regimens against cancer. Some studies suggest that autophagy exerts a tumour-suppressing role, while others suggest a tumour-promoting role. This complexity of autophagy in cancer biology may arise from different contexts such as stress types, tumour staging and cancer types. In vitro studies on different groups of natural compounds modulating autophagy and molecular target mechanisms were summarised in Table 5 and Figure 3.

Figure 3. Autophagy-related pathways regulated by natural compounds in cancer (Figure created with Biorender.com).
Table 5. In vitro studies on natural compounds modulating autophagy in various cancers signalling pathways

Alkaloids are a group of secondary metabolites that are extensively found in nature and have strong pharmacological activities. They can target the autophagy process in different cancer types including breast, lung, cervical, colon, liver and pancreatic cancer. Among the alkaloids, berbamine, cepharanthine, dauricine, daurisolin, liensinine inhibited the autophagy via different molecular target mechanism while matrine induced autophagy by p53/AMPK pathway (Refs Reference Fu143, Reference Tang144, Reference Wu145, Reference Zhou146, Reference Xie, He and Yao147). Natural cyclopeptide RA-XII suppresses protective autophagy through AMPK/mTOR/P70S6 K pathways in HepG2 cells (Ref. Reference Song148). Oridanin, concanavalin and ferulic acid induced autophagy and displayed an anticancer effect against cervical cancer (Refs Reference Cui149, Reference Roy150, Reference Gao151). Kaempferol and quercetin are natural flavonoids that are found in many fruits, vegetables, and medicinal plants. They mediated autophagy in gastric and pancreatic cancer cell lines via inhibition of HDAC/G9a and mTOR activity (Refs Reference Kim152, Reference Lan153). An indole derivative of 3,3′-diindolylmethane also plays a regulatory role in gastric cancer and inhibits miR-30e leading to ATG5 activation (Ref. Reference Ye154). Honokiol is a lignan isolated from the genus Magnolia L. (Magnoliaceae) that inhibits melanoma stem cells by targeting notch signalling and induced in autophagic cell death (Ref. Reference Kaushik155). Paeoniflorigenone regulated autophagy to induce anticancer bioactivities in human head and neck squamous cell carcinomas. This compound inactivated PI3K/AKT/mTOR/p70S6K in YD-10B cells (Ref. Reference Park156). β-lapachone and plumbagin, the active naphthoquinone, showed potent anticancer effects through autophagy induction (Refs Reference Park, Choi and Kwon157, Reference Li158). p-Coumaric acid, an ubiquitous plant phenolic acid, had cytotoxicity on neuroblastoma via generation of ROS that enhanced autophagy-induced mitochondria dysfunction (Ref. Reference Shailasree159). Elaiophylin, a natural autophagy inhibitor, exerted antitumour activity in multiple myeloma and ovarian cancer (Refs Reference Zhao70, Reference Wang160). Polyphenols are a large group of natural compounds that played an important role in modulating autophagy. They had significant antioxidant and anti-inflammatory properties as well as autophagic regulation in cancer cells. Curcumin is a natural polyphenol derived from rhizomes of Curcuma longa L. (Zingiberaceae) commonly known as turmeric. Numerous studies indicated that curcumin was able to modulate autophagy against various cancer including prostate, colon, oral, pancreatic cancers and brain tumours (Refs Reference Aoki161, Reference Teiten162, Reference Lee163, Reference Kim164, Reference Zhu and Bu165, Reference Liu166). Withaferin A had a steroidal lactone structure and inhibited lysosomal activity to block autophagic flux in breast cancer cells (Ref. Reference Muniraj167). Periplocin is a natural active steroid isolated from Periploca forrestii Schltr. (Apocynaceae). This compound promoted autophagy in pancreatic cancer cells via regulating the AMPK/mTOR pathway (Ref. Reference Zhang168). Resveratrol (3,5,4-trihydroxystilbene) is a stilbenoid found mainly in red grape, cranberry, mulberry, and peanut. It has gained more attention over the past two decades because of its ability to prevent and treat various cancers (Ref. Reference Tian169). A link between resveratrol and autophagy regulation by different moleculer mechanism was reported in breast, colon and ovarian cancers (Refs Reference Fu170, Reference Miki171, Reference Ferraresi172). Corilagin and punicalagin, members of the tannin group, induced autophagy in gastric cancer and glioblastoma cell lines, respectively (Refs Reference Xu173, Reference Wang174). Lucidone, a terpeneoid, inhibited autophagy via HMGB1/RAGE/PI3K/Akt signalling pathway in pancreatic cancer cells (Ref. Reference Chen175). Celastrol induced autophagy by targeting AR/miR-101 and served as epigenetic regulator in prostate cancer cell lines (Ref. Reference Guo176). Two natural triterpenoids, toosendanin and isotoosendanin, suppressed triple-negative breast cancer growth through inducing autophagy (Ref. Reference Zhang177).
Autophagic mechanisms of natural compounds in cardiac diseases
Despite recent advances in therapeutic regimens, cardiac disease is still a leading cause of morbidity and mortality and remains one of the greatest threats to public health. Dysregulation of autophagy in cardiomyocytes is associated with myocardial infarction, cardiac hypertrophy, heart failure and diabetic cardiomyopathy. In this context, autophagy appears to be an important therapeutic target and delays cardiac aging (Ref. Reference Wu178). Different groups of natural compounds including berberine, tanshinone IIA, oridanin, hesperidin, icariin, luteolin, nobiletin, puerarin, melatonin, hinokitiol, thymoquinone, gallic acid, spermidine, curcumin, allicin, resveratrol, ginsenoside Rg3, cucurbitacin B were reported to display cardioprotective effect in experimental animal models and in vitro studies via autophagy modulation (Refs Reference Huang179, Reference Zeng180, Reference Wang181, Reference Xu182, Reference Li183, Reference Hu184, Reference Hu185, Reference Wu186, Reference Liu187, Reference Chen188, Reference Xie189, Reference Xiao190, Reference Liu191, Reference Yan192, Reference Eisenberg193, Reference Yan194, Reference Liu195, Reference Ba196, Reference Gu197, Reference Xu198, Reference Sun199, Reference Xiao200). Autophagy regulating natural compounds in cardiac diseases and molecular target mechanisms were summarised in Table 6 and Figure 4.

Figure 4. Autophagy-related pathways regulated by natural compounds in cardiac disease (Figure created with Biorender.com).
Table 6. Autophagy regulating natural compounds in cardiac diseases

Autophagic mechanisms of natural compounds in neurological aging
Many studies indicated that natural compounds had therapeutic benefits in NDs via different mechanisms by targeting autophagy. Autophagy regulating natural compounds in neurological aging and molecular target mechanisms were given in Table 7 and Figure 5. Berberine, conophylline, curcumin, ginsenoside-Rg2, and celastrol improved cognitive impairment via autophagy induction in mice models of AD (Refs Reference Huang201, Reference Umezawa202, Reference Wang203, Reference Fan204, Reference Yang205). Caffeine is a well-known natural compound commonly used to increase alertness and energy. It prevented human prion protein-mediated neurotoxicity via autophagy induction (Ref. Reference Moon206). Dendrobine, an alkaloid, induced autophagy flux in hippocampus primary neurons of rats (Ref. Reference Li207). Abnormalities in neuron axonal transport-related proteins inhibit autophagosome maturation in AD. Curcumin increased autophagic flux by inducing interactions among autophagic axonal transport-related proteins and promoting lysosome-autophagosome fusion in N2a/APP695swe cells (Ref. Reference Liang208). Curcumin also showed a protective effect on amyloid-β-42 induced cytotoxicity in HT-22 cells (Ref. Reference Zhang209). β-asarone and cubeben inhibited amyloid-β, and PI3K-AKT, respectively via promoting autophagy in AD model with neuronal cells (Refs Reference Wang210, Reference Li, Song and Dong211). Wogonin, natural active flavonoid, increased β-amyloid clearance and inhibited mTOR in SH-SHY5Y cells (Ref. Reference Zhu and Wang212). Baicalein inhibited caspase-3 activity and exhibited beneficial effect in PD model with SH-SY5Y (Ref. Reference Kuang, Cao and Lu213). Calycosin, an phytoestrogen, showed protective effect against paraquat (PQ)-induced neurodegeneration by reducing pS6K and p4EBP1 levels (Ref. Reference Chaouhan214). Quercetin, a flavonoid known for its neuroprotective effects, acted as an autophagy enhancer and upregulated Beclin-1 in PD rat model (Ref. Reference El-Horany215). n-Butylidenephthalide induced autophagic down-regulation in motor neurons and prolonged the survival of amyotrophic lateral sclerosis (ALS) mice (Ref. Reference Hsueh216). Oleuropein is the active constituent of olive leaves and fruits and known as antioxidant, neuroprotective and autophagy-regulating properties. These polyphenolic compounds showed therapeutic benefits against PD in neuronal PC12 cells and TgCRND8 mouse model (Refs Reference Achour217, Reference Rigacci218). Resveratrol was reported to have neuroprotective potential in HD. It protected mutant SH-SY5Y cells from dopamine toxicity via rescuing ATG4-mediated mediated autophagosome formation (Ref. Reference Vidoni219). Also, resveratrol showed beneficial effects by inducing autophagy in both in vitro and in vivo AD and PD models (Refs Reference Lin220, Reference Deng and Mi221, Reference Guo222). D-limonene is a fragrance agent and belongs to the terpene group. D-limonene increased LC3-II and reduced p62 levels by inducing autophagy in SH-SY5Y cells (Ref. Reference Russo223). Cucurbitacin E is a tetracyclic triterpenoid isolated from Cucurbitaceae plants, decreased autophagic flux and neuronal death in a postmitotic cellular model of PD (Ref. Reference Arel-Dubeau224). Geraniol also diminished autophagy flux in an in vitro model of PD (Ref. Reference Rekha and Sivakamasundari225). Cannabidiol, a natural compound from Cannabis sativa L. (Cannabaceae) extended lifespan and improved neuronal health in C. elegans by induction of autophagy (Ref. Reference Wang226).

Figure 5. Autophagy-related pathways regulated by natural compounds in neurological aging (Figure created with Biorender.com).
Table 7. Autophagy regulating natural compounds in neurological aging

Autophagic mechanisms of natural compounds in obesity and diabetes
Autophagy has a modulating role in the process of adipocyte conversion. It is also an important signalling pathway for T2DM. Autophagy regulating natural compounds in obesity and diabetes and molecular target mechanisms were given in Table 8 and Figure 6. Berberine, an alkaloid, inhibited basal autophagy in adipocytes in mice fed a high-fat diet by decreasing Beclin-1 (Ref. Reference Deng227). Resveratrol increased AMPK and improved health and survival of high-calorie diet induced mice (Ref. Reference Baur228). Resveratrol activated SIRT1/FoXO1/Rab7 and ameliorated myocardial oxidative stress injury in diabetic mice (Ref. Reference Wang229). It alleviated I/R injury of diabetic myocardium by promoting autophagy (Ref. Reference Qu230). Apart from resveratrol, epigallocatechin-3-gallate, trehalose, dihydromyricetin, puerarin, melatonin, ferulic acid, arjunolic acid, and curcumin were reported to effective in experimental animal models and in vitro studies of diabetes via autophagy modulation (Refs Reference Yan231, Reference Xu232, Reference Shi233, Reference Xu234, Reference Zhang235, Reference Ma236, Reference Zhang237, Reference Yao238, Reference Zhang239).

Figure 6. Autophagy-related pathways regulated by natural compounds in obesity and diabetes (Figure created with Biorender.com).
Table 8. Autophagy regulating natural compounds in obesity and diabetes

Conclusion and future perspectives
Aging, an irreversible biological process, is an independent risk factor for cancer, neurodegeneration, cardiovascular diseases, obesity and diabetes. Under both healthy and pathological circumstances, autophagy is a crucial mechanism. Studies have revealed that the dysregulation of autophagy plays a role in the pathogenesis of illnesses associated with aging and have proposed potential treatments involving the control of the autophagy system. It is thought that a number of bioactive substances generated from medicinal plants have the ability to regulate autophagy by concentrating on autophagic pathways. The targeted control of autophagy is thought to offer a method of treating age-related disorders, hence regulation of the autophagic process is increasingly seen as an intriguing drug development technique. The regulating function of natural substances on autophagy to delay or treat age-related illnesses in vitro and in animal models was the main focus of this review. However, there is broad agreement about the effectiveness of natural pleiotropic substances capable of enhancing or reestablishing deficient autophagy in aggregate-prone proteins. According to literature data, different groups of phytochemicals have attracted attention as promising autophagy modulators. Curcumin and resveratrol stand out in terms of effectiveness and popularity and these two compounds can be identified as potent autophagy regulating compounds in cancer, cardiovascular and NDs, and metabolic dysfunction. Keep in mind that autophagy can be a two-way process. To successfully combat age-related illnesses, significant improvements in our understanding of their modes of action, pharmacokinetics, and nonspecific effects are required.















 Open access
Open access