


Introduction
Ortleppascaris sinensis, a new nematode species, is described from specimens found in the stomach and intestine of the captive Chinese alligator Alligator sinensis Fauvel 1879 (Crocodilian: Alligatoridae) in China (Zhao et al., Reference Zhao, Wang, Tu, Zhou, Wu and Li2015, Reference Zhao, Wang, Tu, Zhou and Wu2016). Recently, mitochondrial (mt) genomics have been widely used for species-specific identification and differentiation of many zoonotic nematodes (Xie et al., Reference Xie, Zhang, Wang, Lan, Li, Chen, Fu, Nie, Yan, Gu, Wang, Peng and Yang2011b). Moreover, mitochondrial genomes contain useful genetic markers for studying the genetic structure and systematics of nematodes, as well as clinical diagnosis and epidemiology of parasitic disease, due to mutation rates that are proposed to be more rapid than those of nuclear genes (Hu et al., Reference Hu, Chilton and Gasser2004). To date, over 100 complete mitochondrial genome sequences have been published for class Chromadorea nematodes and 20 for class Enoplea, although about 15,000 species of nematodes have been recorded (https://www.ncbi.nlm.nih.gov/genomes/OrganelleResource.cgi?opt=organelle&taxid=6231). However, complete information on the mt genome of nematodes in the genus Ortleppascaris was not previously available.
Here, we report the complete nucleotide sequence of the mt genome from O. sinensis for the first time, and compare the sequence and genome organization with the sequences from related nematodes in the same order, Ascaridida. Additionally, the phylogenetic relationships of the genus Ortleppascaris within the order Ascaridida were investigated by the construction of phylogenetic trees (Bayesian inference (BI), maximum parsimony (MP) and maximum likelihood (ML)) using the sequence of protein-coding genes (PCG) dataset. More mitogenomes from related species will further enhance our understanding of molecular evolution and phylogenetic relationships within Ascaridida.
Materials and methods
Parasite collection and DNA extraction
Adult nematodes were obtained from the stomach and intestine of the Chinese alligator A. sinensis in the National Nature Reserve of China Alligator (Chinese Crocodile Lake) in Anhui Province, China. Nematodes were washed in physiological saline, identified to species O. sinensis based on morphological and molecular characterization (Zhao et al., Reference Zhao, Wang, Tu, Zhou and Wu2016), fixed in 75% ethanol and stored at −20°C until use. Total genomic DNA was isolated from individual nematodes using a TIANamp Genomic DNA Kit (Tiangen, Beijing, China) according to the manufacturer's protocol.
Primer design, PCR amplification and sequencing
To sequence the complete mitogenome of O. sinensis, short segments of cox1, cox1–rrnL and rrnL–nad3 were amplified by polymerase chain reaction (PCR) using a known specific degenerate primer (Hu et al., Reference Hu, Chilton and Gasser2002). After obtaining the mitogenome segments, we designed species-specific primers for the remaining parts, based on the conserved sequences in Ascaridida nematodes (see supplementary table S1).
PCR amplification reactions were performed in 50-μl volumes: 25 μl Premix Taq (LA Taq Version 2.0; Takara, Japan), 1.0 μl of each primer (forward and reverse; 20 μm), 2.0 μl template DNA and 21 μl sterile double-distilled water (ddH2O). PCRs were performed with the following cycling parameters: 1 min at 94°C; followed by 35 cycles of 94°C for 30 s and 55–60°C for 5.0 min; and a final extension at 72°C for 10 min. PCR products were electrophoresed, excised and purified with a column DNA gel extraction kit (Sangon Biotech, Shanghai, China), and cloned into the pMD™19-T vector (Takara). The positive clones were sequenced directionally (ABI 3730; Applied Biosystems, Foster City, California, USA). To verify the number of variable-length AT-repeats in the largest non-coding region, DNA extracted from ten individual nematodes was used as a template for PCR.
Sequence analysis and annotation
The nucleotide sequences obtained were compared using BLAST against known sequences in GenBank, and then assembled with SeqMan software (DNAStar version 5.0; http://www.dnastar.com/). Protein-coding genes (PCGs) were determined by DNAStar version 5.0, implemented at the National Center for Biotechnology Information (NCBI) website with the invertebrate mitochondrial genetic code, as well as aligning their similarity against the published mitochondrial sequences of Ascaridida nematodes using Clustal X 2.0 and MEGA 6.0 (Tamura et al., Reference Tamura, Stecher, Peterson, Filipski and Kumar2013). The majority of tRNA genes and their secondary structures were identified using tRNAscan-SE software V1.21 (Mohandas et al., Reference Mohandas, Jabbar, Podolska, Zhu, Littlewood, Jex and Gasser2014). Two large non-protein-coding regions were candidates for the rRNA genes (rrnL and rrnS) by comparison with sequence similarity to genes in Ascaridida nematodes and previously reported conserved areas (Hu et al., Reference Hu, Chilton and Gasser2002; Xie et al., Reference Xie, Zhang, Wang, Lan, Li, Chen, Fu, Nie, Yan, Gu, Wang, Peng and Yang2011b). The secondary structure of non-coding regions (containing a putative control region) was folded using Mfold Server (Zuker, Reference Zuker2003). When multiple secondary structures were possible, the most stable one (lowest free energy (−ΔG)) was preferred. The base composition, codon usage, relative synonymous codon usage (RSCU) values and nucleotide substitution were analysed with Mega 6.0. Nucleotide composition skewness was calculated as AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C), respectively.
Phylogenetic analyses
For the phylogenetic analysis, 17 nematode mitochondrial genome sequences of Ascaridida available from GenBank were used, in addition to the complete mtDNA sequences of O. sinensis determined in this study (see supplementary table S2); the classification status of the nematodes listed in supplementary table S2 was according to the GenBank. To infer the phylogenetic relationships of genus Ortleppascaris in the Ascaridida, we constructed phylogenetic trees based on the 12 protein-coding genes. Phylogenetic analyses were conducted using BI, MP and ML methods using Wuchereria bancrofti (NC_016186) as the outgroup (McNulty et al., Reference McNulty, Mullin, Vaughan, Tkach, Weil and Fischer2012). The GTR + G model was selected as the most appropriate model by MODELTEST v.3.7 (Abascal et al., Reference Abascal, Zardoya and Posada2005) based on the Akaike information criterion (AIC). MP trees were implemented with a gap option as ‘missing data’ using PAUP* 4.0b10 (Swofford, Reference Swofford2002). The ML tree was reconstructed using PhyML 3.1 software (Guindon et al., Reference Guindon, Dufayard, Lefort, Anisimova, Hordijk and Gascuel2010) to assess the amount of phylogenetic signal in the dataset. As for BI analysis, the MrBayes 3.2 software was used to construct the tree (Ronquist et al., Reference Ronquist, Teslenko, van der Mark, Ayres, Darling, Hohna, Larget, Liu, Suchard and Huelsenbeck2012). The first 25% of the trees were discarded and the remaining trees were used to calculate Bayesian posterior probabilities (PP). Statistical tests for the alternative tree topology based on previously proposed hypotheses were performed using the likelihood-based Shimodaira–Hasegawa test implemented in BI, MP and ML trees.
Results and discussion
Genome structure, organization and composition
The complete mitogenome of O. sinensis was 13,828 bp in length, and its sequence has been deposited in GenBank under the accession number KU950438. The mitogenome encodes 12 PCGs (cox1–3, nad1–6, nad4L, atp6, cytb), 2 ribosomal RNAs (rrnS and rrnL), 22 tRNAs (trnA, trnR, trnN, trnD, trnC, trnQ, trnE, trnG, trnH, trnI, trnL1 CUN, trnL2 UUR, trnK, trnM, trnF, trnP, trnT, trnW, trnY, trnV, trnS1 AGN and trnS2 UCN) , a non-coding region (NCR), an AT-rich region and lacks atp8. These genes are all located on the J-strand and are transcribed in the same direction, like the genomes of other nematodes of Chromadorea (fig. 1) (Lavrov & Brown Reference Lavrov and Brown2001; Waku et al., Reference Waku, Segawa, Yonezawa, Akiyoshi, Ishige, Ueda, Ogawa, Sasaki, Ando, Kohno and Sasaki2016). The mitochondrial arrangement of O. sinensis was fully in accordance with the mode of GA7 (gene arrangement, GA) described by Yatawara et al. (Reference Yatawara, Wickramasinghe, Rajapakse and Agatsuma2010). Eleven intergenic spacers totalling 50 bp, and five overlapping regions totalling 5 bp were scattered throughout the whole genome (table 1). Compared to the mitochondrial arrangement of Ascaridida nematodes, that of O. sinensis was similar to those of the Ascaridae, Toxocaridae and Anisakidae, but different from those of Cucullanidae and Ascaridiidae (fig. 2) (He et al., Reference He, Jones, Armstrong, Lamberti and Moens2005; Li et al., Reference Li, Lin, Song, Wu and Zhu2008; Xie et al., Reference Xie, Zhang, Niu, Wang, Wang, Lan, Deng, Fu, Nie, Yan, Yang, Hao, Gu, Wang, Peng and Yang2011a). Compared to the mitochondrial arrangement of Ascaridia columbae and Ascaridia galli in the Ascaridiidae, there were large differences: cox1 and rrnS were translocated, and tRNAs, such as trnD, trnE, trnS2, trnF and trnC, were severely rearranged, translocated and/or inverted. Moreover, the position of the AT-rich region of O. sinensis was between the genes trnS2 and trnN, and the position of the NCR of O. sinensis was between nad4 and cox1. This is the same as reported for the nematodes of the families Ascaridae, Toxocaridae and Anisakidae (Li et al., Reference Li, Lin, Song, Wu and Zhu2008; Xie et al., Reference Xie, Zhang, Wang, Lan, Li, Chen, Fu, Nie, Yan, Gu, Wang, Peng and Yang2011b), whereas the AT-rich region was located between genes trnA and trnP in hookworms (Ancyclostoma caninum, A. duodenale, Bunostomum phlebotomum and Necator americanus). Such differences and similarities in the mitogenome of different nematodes suggests that structure and composition of the mitochondrial genome can be used to analyse phyletic evolution and population genetics for nematode species (J.K. Park et al., Reference Park, Sultana, Lee, Kang, Kim, Min, Eom and Nadler2011; Mohandas et al., Reference Mohandas, Jabbar, Podolska, Zhu, Littlewood, Jex and Gasser2014).
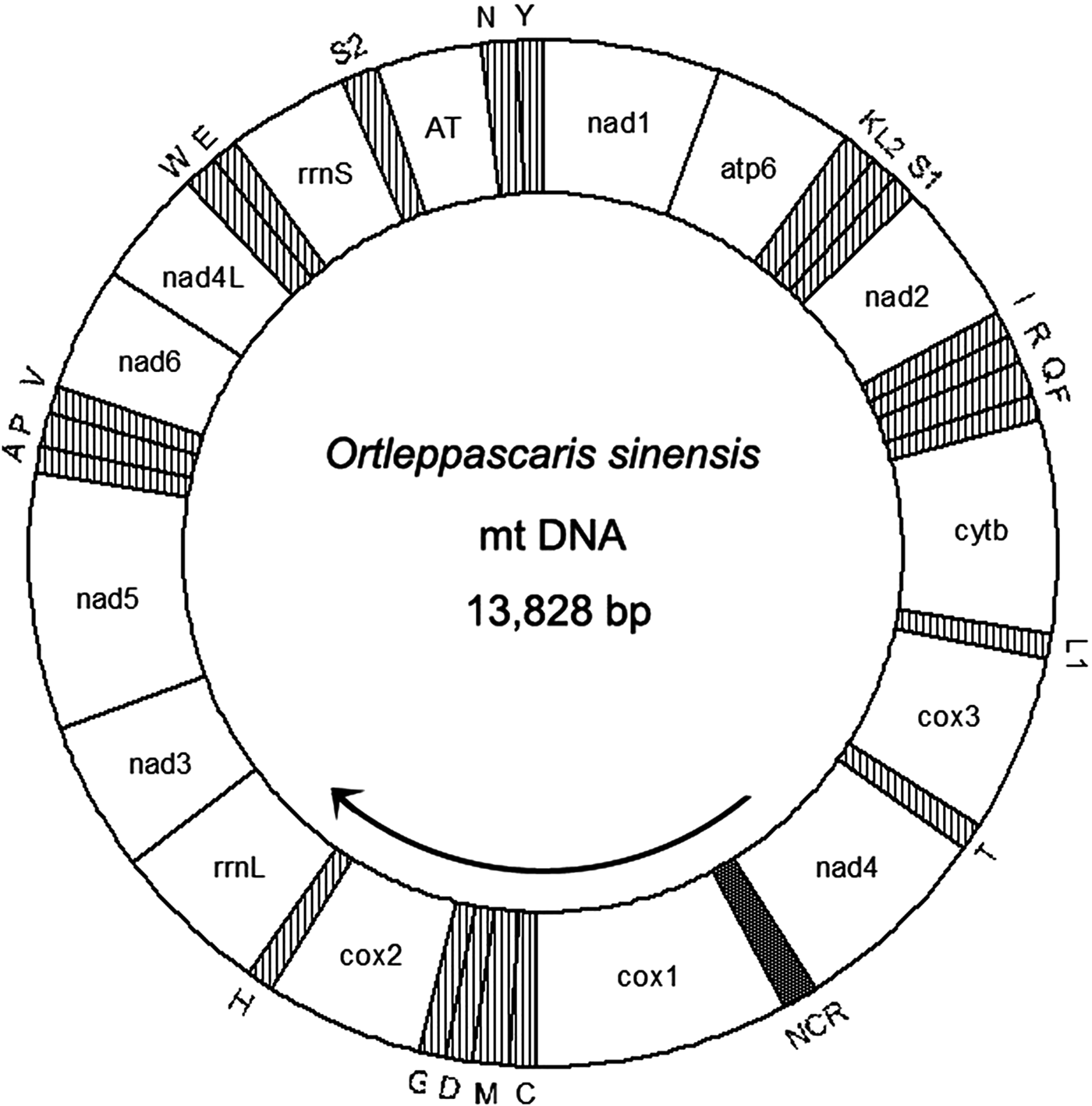
Fig. 1. Circular representation of the mitochondrial genome of O. sinensis. Protein-coding and ribosomal genes are shown with standard abbreviations. Genes for tRNAs are abbreviated by the single-letter amino acid code, with S1 = AGN, S2 = UCN, L1 = CUN, and L2 = UUR. AT: AT-rich region; NCR: non-coding region. Genes are not to scale and the tRNA genes are indicated by hatched areas.
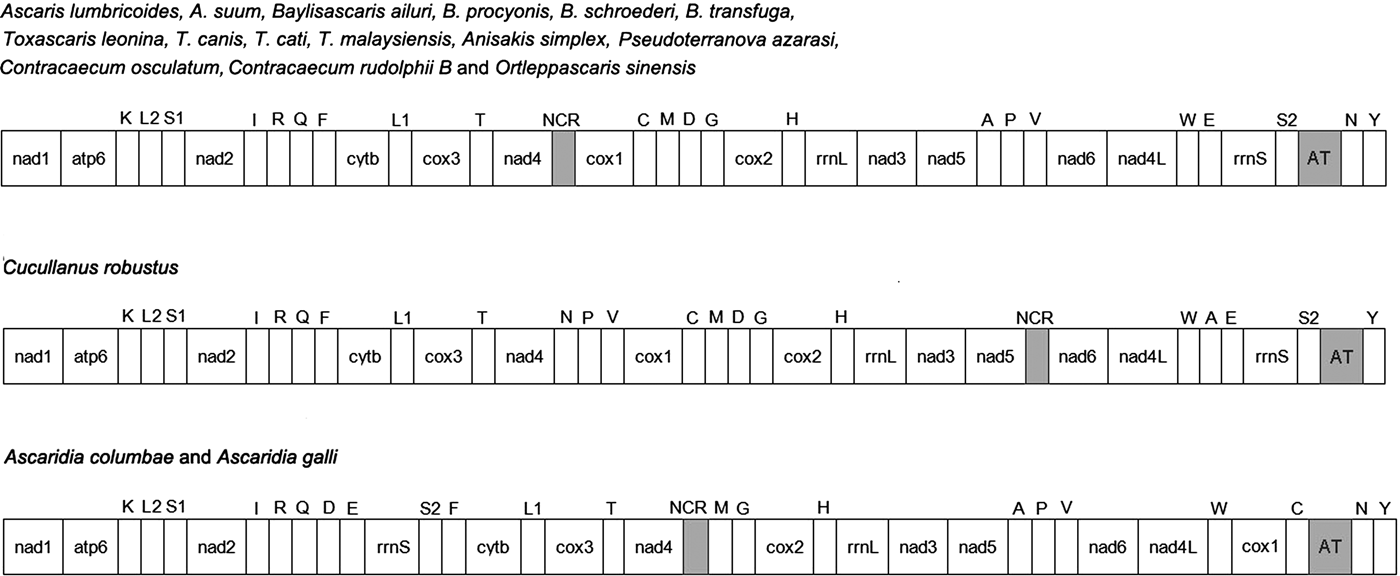
Fig. 2. Mitochondrial genome organization of 18 Ascaridida nematodes. Protein-coding and ribosomal genes are shown with standard abbreviations. Genes for tRNAs are abbreviated by the single-letter amino acid code, with S1 = AGN, S2 = UCN, L1 = CUN, and L2 = UUR. AT: AT-rich region; NCR: non-coding region. Genes are not to scale.
Table 1. Organization of the O. sinensis mitochondrial genome.
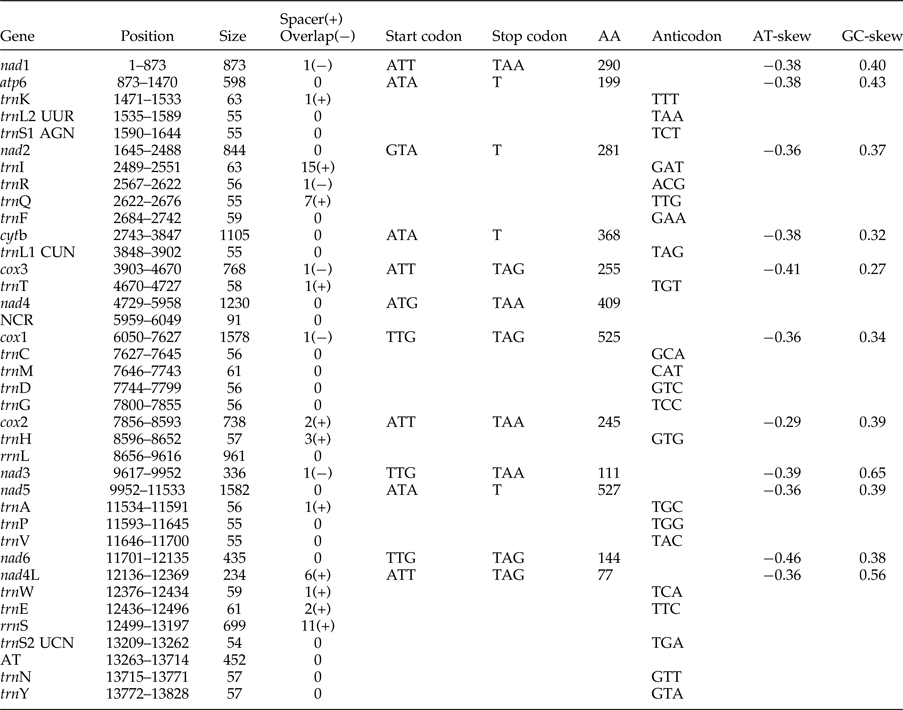
AT: AT-rich region. NCR: Non-coding region.
Base content
Like other Ascaridida nematodes, the nucleotide composition for the O. sinensis mitochondrial DNA was also biased toward A and T (A = 26.1%, T = 48.0%, C = 8.4%, and G = 17.6%) (see supplementary table S3). We can see from the table that the AT content of O. sinensis (74.0%) was the highest among the 18 Ascaridida nematodes, though its mtDNA length was the shortest apart from that of Contracaecum osculatum. In each Ascaridida nematode, the nucleotide composition of these genes (PCGs, rRNA, tRNA and AT-rich region) were biased toward A and T; the AT content of the AT-rich region was the highest, followed by that of rRNA; and the AT content was lowest in PCGs compared to the other genes. In general, Ascaridida nematodes have an AT-specific bias and rejection of C in nucleotide composition (Kim et al., Reference Kim, Eom and Park2006; Li et al., Reference Li, Lin, Song, Wu and Zhu2008).
A comparative analysis of AT% vs. AT-skew and GC% vs. GC-skew across complete mitogenomes of 18 Ascaridida nematodes is shown in fig. 3. The average AT-skew of the mitogenome of Ascaridida nematodes was −0.36, ranging from −0.28 in A. columbae to −0.41 in A. galli, whereas the AT-skew in the mitogenome of O. sinensis was −0.30. The average GC-skew of the mitogenome of Ascaridida nematodes was 0.41, ranging from 0.32 in Anisakis simplex to 0.49 in A. galli, whereas the GC-skew in the O. sinensis mitogenome was 0.35. The mitochondrial genomes of Ascaridida nematodes usually have negative AT-skews and positive GC-skews, like the mitochondrial genomes of metazoans (Lavrov & Brown Reference Lavrov and Brown2001). Although the exact mechanism causing the AT bias in mtDNA remains unknown, such a situation could be the result of inversions of the AT-rich regions and replication origin (Oliveira et al., Reference Oliveira, Haukka and Kaguni2015).
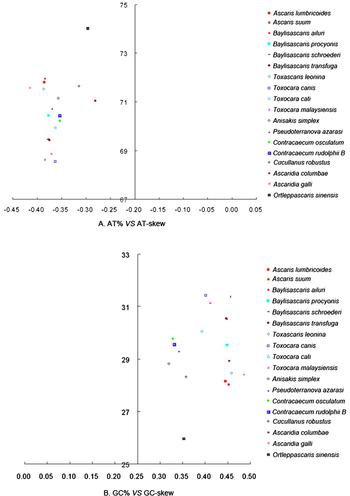
Fig. 3. AT% vs. AT-skew and GC% vs. GC-skew of the mitochondrial genomes of 18 Ascaridida nematodes. Values are calculated for full-length mitochondrial genomes. The x-axis shows the level of nucleotide skew and the y-axis indicates the nucleotide percentages.
Protein-coding genes and codon usage
The overall length of the 12 PCGs in the O. sinensis mitogenome was 10,319 bp, accounting for 74.62% of the total length of the mitogenome, encoding 3419 amino acids. Among the 12 PCGs of O. sinensis, nad5 is the longest (1582 bp) and nad4L is the shortest (234 bp). However, among the 12 PCGs of the 18 Ascaridida nematodes, nad3 (333 bp or 336 bp) and nad6 (1230 bp or 1236 bp) are the most conserved genes, and cytb (1101 bp, 1102 bp, 1104 bp, 1105 bp, 1107 bp, 1095 bp, 1098 bp or 1099 bp) is the most variable gene in length; the AT content of nad4L and nad6 is relatively high, but cox1, cox2 and cytb kept a relatively low level (see supplementary table S4). This nucleotide bias is bound to have an effect on the codon use of mtDNAs and the kinds of amino acids.
In O. sinensis all the PCGs utilized typical ATN, GTA and TTG start codons (see supplementary table S5). In detail, four genes (nad1, cox3, cox2 and nad4L) started with codon ATT, three genes (atp6, cytb and nad5) with ATA, three genes (cox1, nad3 and nad6) with TTG , one gene (nad4) with ATG and one gene (nad2) with GTA. Although almost all the PCGs utilized the conventional stop codons TAA (four times) or TAG (four times), the remaining genes (atp6, cytb, nad5 and nad2) used incomplete stop codons, such as a single T. In Ascaridida nematodes, the most commonly used start codons were TTG and ATT, but GTA, GTT, GTG, ATA, ATG and TTT were also used, of which TTT was only used as a start codon by nad6 genes in A. galli. The stop codons most commonly used were the complete termination codons TAA or TAG, but incomplete termination codons (T or TA) were also found. The presence of an incomplete termination codon is commonly observed in metazoan mitochondrial genomes and these incomplete termination codons would likely be completed via post-transcriptional polyadenylation (Kim et al., Reference Kim, Eom and Park2006; Li et al., Reference Li, Lin, Song, Wu and Zhu2008; Oliveira et al., Reference Oliveira, Haukka and Kaguni2015; Grosemans et al., Reference Grosemans, Morris, Thomas, Rigaux, Moens and Derycke2016).
The codons of the 12 PCGs of O. sinensis are analysed in supplementary table S6, including the frequency of each codon and relative synonymous codon usage (RSCU). We found that three codon positions possess different nucleotide biases – mean U in the second codon position and U, A or G in the third codon position. Among them, UUU was the most common (468), CUC, CAC and CCG were used only once, yet CGC (R) and CCG (P) did not exist in the codons of the 12 PCGs for O. sinensis. There was also codon bias (a tendency to use codons with A, U and G) in the other nematodes of Ascaridida. The codons with higher frequency of use were UUU (F), AUU (I), UUA (L), UUG (L), GUU (V), UAU (Y), UCU (S), GGU (G), AAU (N) and AGU (S). In addition, codons with a lower frequency of use, and even a lack of some codons, were all seen in some nematodes of Ascaridida. For example, the codons CUC (L), CCC (P) and CGC (R) were not present in Ascaris lumbricoides, and the codons CUC (L), CCC (P), UGC (C) and CGC (R) were not present in Ascaris suum. The RSCU for the mitochondrial proteins in O. sinensis are shown in supplementary fig. S1. The highest frequency of RSCU was CGU (3.64), and the lowest frequency was CUC (0.01). The frequency of RSCU is significantly reduced if the third base position is replaced by C; for example, the frequency of the UUU codon encoding phenylalanine (F) for O. sinensis mtDNA is 13.69%, while that of the synonymous codon UUC was just only 0.38%. This phenomenon could be explained by synonymous codon usage bias. Generally codon bias was proposed to be highest in gene regions of functional significance and is believed to be important for maximizing translation efficiency (Duret & Mouchiroud, Reference Duret and Mouchiroud1999).
Excluding the start and stop codons, the amino acid frequencies are similar among the 18 nematodes of Ascaridida (see supplementary table S7). The most overused amino acids were Leu (145.92–167.60‰), Phe (129.95–151.04‰), Ser (102.76–113.20‰) and Val (89.21–117.73‰), and the amino acid with lowest use was Arg (9.102–10.93‰). The mitogenome of O. sinensis has a relatively lower value in the proportion of the amino acids Leu (147.70‰, average 151.25‰), Phe (140.68‰, average 143.15‰) and Val (89.21‰, average 103.54‰). Usually, proteins encoded by mitochondrial genomes are mostly membrane proteins, and Phe and Leu occur in hydrophobic side chains of such proteins (Li et al., Reference Li, Lin, Song, Wu and Zhu2008; Xie et al., Reference Xie, Zhang, Wang, Lan, Li, Chen, Fu, Nie, Yan, Gu, Wang, Peng and Yang2011b).
Transfer RNA genes
Twenty-two tRNA genes were identified in the mitochondrial genome of O. sinensis (table 1). The tRNA sequences ranged from 55 to 63 bases in length, and have non-canonical secondary structures (see supplementary fig. S2). The tRNA structures shared some features with those of other metazoans, including a 7-bp amino-acyl arm, a 4-bp dihydrouridine (DHU) arm, a 5-bp anticodon stem, a 7-base anticodon loop, a T always preceding an anticodon, as well as a purine always following an anticodon. Twenty tRNAs (trnA, trnR, trnN, trnD, trnC, trnQ, trnE, trnG, trnH, trnI, trnL1 CUN, trnL2 UUR, trnK, trnM, trnF, trnP, trnT, trnW, trnY and trnV) have a 4-bp DHU stem and a DHU loop of 4–11 bases, but lack the TΨC arm and instead have a ‘TV-replacement loop’ of 7–13 bases. The tRNA genes without TΨC arm for A. suum also had normal biological function (Okimoto et al., Reference Okimoto, Macfarlane, Clary and Wolstenholme1992). The trnS for O. sinensis possesses a secondary structure consisting of a DHU replacement loop of 7 bases (trnS1 AGN) or 4 bases (trnS2 UCN), a 3-bp TΨC arm, a TΨC loop of 6 bases (trnS1 AGN) or 7 bases (trnS2 UCN) and a variable loop of 4 bases (trnS1 AGN) or 5 bases (trnS2 UCN). Their tRNA secondary structures were similar to those of all other Secernentea nematodes studied to date (Kim et al., Reference Kim, Eom and Park2006; Li et al., Reference Li, Lin, Song, Wu and Zhu2008), but were distinct from those of Trichinella spiralis (Lavrov & Brown, Reference Lavrov and Brown2001). This unusual feature has been described not only in Ascaridida nematodes but also in other invertebrates, such as Pupa strigosa (opisthobranch gastropod), Laqueus rubellus (articulate brachiopod) and Katharina tunicata (chiton) (Kurabayashi & Ueshima, Reference Kurabayashi and Ueshima2000). The secondary structure and anticodon of tRNA genes in the mitochondrial genome of metazoans were relatively conservative, which can be used for the classification of the families or genera for analysis of system evolution. There were also a certain number of base mismatches in the amino-acyl arm, DHU arm, anticodon arm and TΨC arm, and give priority to G·U weak pairing. This phenomenon of base mismatch can be corrected by RNA editing (Fujishima & Kanai, Reference Fujishima and Kanai2014).
Ribosomal RNA genes
The rrnS and rrnL genes of O. sinensis were identified by sequence comparison with Ascaridida nematodes. The rrnS gene was located between trnE and trnS2 UCN, and rrnL was located between trnH and nad3. The two genes were separated from one another by the protein genes nad3, nad5, nad6 and nad4L (fig. 2). The size of the rrnS gene for O. sinensis was 699 bp, and the size of the rrnL gene was 961 bp. The sizes of these two genes for O. sinensis were similar to those of other nematodes (for rrnS, 677–703 bp; for rrnL, 955–961 bp) (see supplementary table S3) (Kim et al., Reference Kim, Eom and Park2006; Li et al., Reference Li, Lin, Song, Wu and Zhu2008). The AT contents of the rrnS (73.8%) and rrnL (77.9%) for O. sinensis were higher when compared with other Ascaridida nematodes (for rrnS, 66.4–73.9%; for rrnL, 70.1–78.9%), and only slightly lower than those of Cucullanus robustus (supplementary table S3).
Non-coding regions
In the O. sinensis mitochondrial genome, the longest non-coding region (AT-rich region) was located between genes trnS UCN and trnN, as in the mitochondrial genomes of nematodes A. lumbricoides, Baylisascaris ailuri, Toxascaris leonina, A. simplex, Pseudoterranova azarasi and C. osculatum, but differing from C. robustus and A. columbae (J.K. Park et al., Reference Park, Sultana, Lee, Kang, Kim, Min, Eom and Nadler2011; Y.C. Park et al., Reference Park, Kim and Park2011; Liu et al., Reference Liu, Shao, Li, Zhou, Li and Zhu2013, Reference Liu, Zhou, Zhao, Xiong, Liang and Zhu2014; Mohandas et al., Reference Mohandas, Jabbar, Podolska, Zhu, Littlewood, Jex and Gasser2014; Liu et al., Reference Liu, Liu, Zhu and Weng2015; Xie et al., Reference Xie, Zhang, Wang, Lan, Li, Chen, Fu, Nie, Yan, Gu, Wang, Peng and Yang2011b) (fig. 2). The length of the AT-rich region of O. sinensis was 452 bp, which was the shortest, apart from C. osculatum, compared with other Ascardida nematodes (239–1516 bp). The AT-rich region is the most variable portion of the genome, both in terms of length and nucleotide sequence, among the mitochondrial genomes of 18 Ascardida nematodes (see supplementary table S8). The AT content of the AT-rich region for O. sinensis was 88.9%, significantly greater than that of other Ascaridida nematodes (68.3–89.1%) apart from Contracaecum rudolphii B. Additionally, the AT-rich region of the O. sinensis mtDNA contained four regions with varying numbers of the dinucleotide TA repeats (n = 8, 10, 26, 33) within a total of 154 bp. Similar multiple TA repeats have been described in the AT-rich region of the mitochondrial genomes of other Ascaridida and Strongylida species (Li et al., Reference Li, Lin, Song, Wu and Zhu2008). The AT-rich region has been known to serve as an essential element involved in the initiation of replication and transcription of the mitogenome. Thus, it possibly functions as a control region (Janssen et al., Reference Janssen, Karssen, Verhaeven, Coyne and Bert2016).
The second-longest non-coding region (NCR) of O. sinensis mtDNA was located between genes nad4 and cox1, and is relatively shorter (91 bp, average 113 bp) compared with the other 17 Ascaridida nematodes; the NCR of A. galli (157 bp) is the longest in length, and A. suum (70 bp) is the shortest. However, the NCR of O. sinensis has a relatively higher value of AT content (76.9%, average 75.1%) (supplementary table S8). The NCR may function as splicing recognition sites during processing of the transcripts (He et al., Reference He, Jones, Armstrong, Lamberti and Moens2005).
Phylogenetic analysis
The phylogenetic relationships were constructed using BI, MP and ML analyses based on a concatenated mitochondrial sequence dataset of the 12 PCGs of O. sinensis and all the other 18 Ascaridida nematodes available (supplementary table S2). Three phylogenetic trees inferred by BI, MP and ML analyses had identical topologies (fig. 4). Almost all nodes were well supported by high bootstrap values and high Bayesian posterior probabilities, except for some node confidence values. All the nematodes of the order Ascaridida belong to three superfamilies (Ascaridoidea, Seuratoidea and Heterakoidea), six families (Anisakidae, Ascarididae, Toxocaridae, Heterocheilidae, Cucullanidae and Ascaridiidae) and nine genera (Anisakis, Pseudoterranova, Contracaecum, Baylisascaris, Toxocara, Ortleppascaris, Cucullanus and Ascaridia). Heterocheilidae and Anisakidae/Toxocaridae/Ascarididae have a closer bond than Cucullanidae and Ascaridiidae within Ascaridida. In addition, our analyses showed that Baylisascaris (four nematodes) and Ascaris (two nematodes) had a close affinity, and then clustered as a taxonomic group with Toxocara (four nematodes) within the family Ascarididae; Anisakis (one nematode) and Pseudoterranova (one nematode) had a close affinity, and then made a sister taxon with Contracaecum (two nematodes). The results are similar to both morphological classification and phylogenetic analysis based on molecular evidence (Xie et al., Reference Xie, Zhang, Wang, Lan, Li, Chen, Fu, Nie, Yan, Gu, Wang, Peng and Yang2011b).
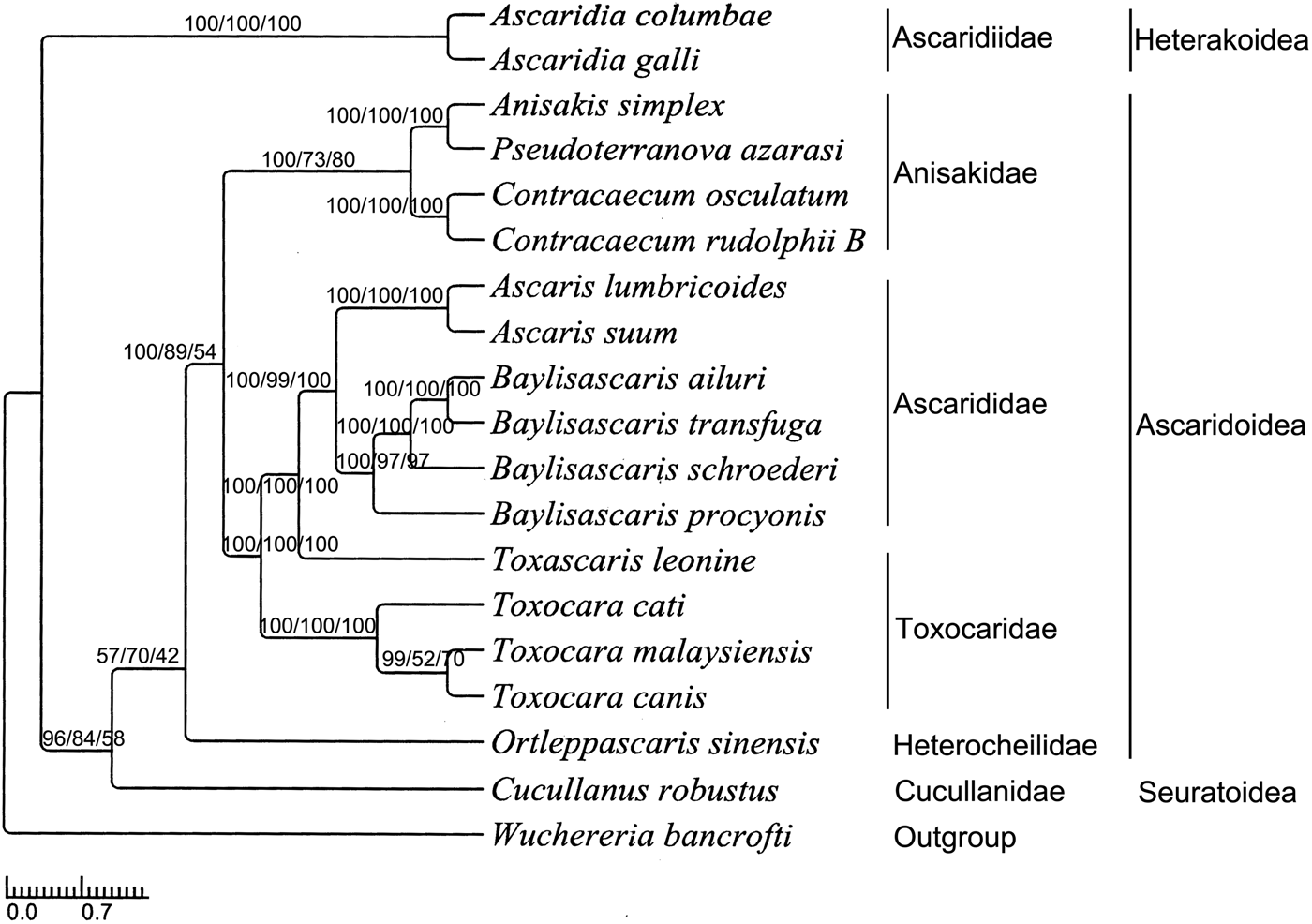
Fig. 4. Phylogenetic tree of 18 Ascaridida nematodes based on the data of 12 protein-coding genes (BI/MP/ML).
The genus Ortleppascaris was removed from the genus Dujardinascaris by Sprent (Reference Sprent1978) and placed in a new genus Ortleppascaris according to morphological characteristics (Sprent, Reference Sprent1978). Recently, Dujardinascaris has been attributed to the Heterocheilidae (Lakshmi & Sudha, Reference Lakshmi and Sudha2000; Masova et al., Reference Masova, Barus, Seifertova, Malala and Jirku2014) or Anisakidae (Moravec & Jirku, Reference Moravec and Jirku2014) when some species of this genus were reported, and Ortleppascaris was attributed to the Ascarididae (Silva et al., Reference Silva, da Silva, Melo, Giese, Furtado and Santos2013) or just Ascarididea (Sprent, Reference Sprent1977, Reference Sprent1978; Zhao et al., Reference Zhao, Wang, Tu, Zhou and Wu2016) when some species were reported. So, it is not very clear to which family the genera Otleppascaris and Dujardinascaris should be attributed. In the trees inferred by the BI, MP and ML methods, the newly sequenced O. sinensis clustered distinctly as a single clade. In addition, this clade consistently clustered with the Anisakidae group with high BI, MP and ML bootstrap values (100, 89 and 54, respectively). Our analyses indicate that O. sinensis cannot be allocated to the families Anisakidae and Ascarididae, and may be attributed to another family – Heterocheilidae – which had been supported by some morphological and molecular data (Lakshmi & Sudha, Reference Lakshmi and Sudha2000; Masova et al., Reference Masova, Barus, Seifertova, Malala and Jirku2014). Although our results have indicated that O. sinensis belongs to the family Heterocheilidae, we should be cautious because no other mtDNA sequences of Heterocheilidae are available to date. So it is necessary to sequence more mtDNAs of Ortleppascaris nematodes in the future, to test and confirm our conclusion. Furthermore, given that, in the present study, the phylogenetic analysis was based only on mtDNA sequences of 12 PCGs, it is still necessary to employ more molecular datasets of the Ascaridida nematodes, or to expand taxon sampling from the lineage, to examine the phylogenetics and genome evolution of the Ascaridida.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0022149X17000542
Acknowledgements
We greatly appreciate the Chinese alligator raisers for providing guidance and technical assistance during our sample collection.
Financial support
Funding support was provided by the Chinese National Natural Science Foundation (nos 31272337 and 31472019), Anhui Provincial Natural Science Foundation of China (no. 1608085MC77) and Key Program in the Youth Elite Support Plan in Universities of Anhui Province (no. gxyqZD2016171).
Conflict of interest
None.







