1. Introduction
In contrast to central Antarctica, only few glacio chemical records are available from Antarctic ice shelves. Due to their simple topography, the relatively high snow-accumulation rate and the immediate access to the ocean, these snow fields are expected to be particularly useful in deriving high-resolution records of coastal Antarctic snow chemistry. Addressing the question of the biogeochemical cycles of nitrogen and sulphur components, representative deposition fluxes of these species into the Southern Ocean, which are difficult to access, may be quantified by determining the deposition fluxes on to the adjacent ice shelves.
The Filchner-Ronne Ice Shelf, extending about 500 km from the coast to the margin of the Antarctic inland ice, is of particular importance in this context, because it lies adjacent to the biologically highly productive Weddell Sea. With the aim of evaluating the dynamics and mass balance of the Filchner-Ronne Ice Shelf~ numerous glaciological studies in the framework of the international Filchner-Ronne Ice Shelf Programme have been undertaken there.
During several German glaciological surveys in the years 1985–92, an extensive series of snow-pit samples and shallow firn cores has been recovered from the central Filchner-Ronne Ice Shelf (Reference Graf, Moser, Oerter, Reinwarth and StichlerGraf and others, 1988, Reference Graf, Miller and Oerter1991; Reference Oerter, Drücker, Kipfstuhl, Nixdorf, Graf and OerterOerter and others, 1992b). Here, we present an overview of the results of the chemical analyses of 14 firn cores, each covering a time period in the order of 20 years, and five snow pits for 15 different sampling sites. This detailed data base appears to be appropriate for determining the spatial significance of any trends in the chemical composition of the surface firn. This information is directly necessary for the interpretation in terms of temporal changes of the chemical depth profiles of two intermediate-depth ice cores recovered on the Filchner- Ronne Ice Shelf.
Therefore, emphasis will be on the spatial distribution and particularly on the continental effects of the mean concentrations and deposition fluxes of sea salt, nitrate and the biogenic sulphur compounds. In addition, the seasonality of these chemical trace species, which are used in combination with the stable isotope stratigraphy to derive precise and temporal representative snow-accumulation rates, will be briefly outlined. Detailed results of the isotopic analyses have been given by Reference GrafGraf and others (1994) and specific aspects of the isotopic and chemical information on the two Berkner Island ice domes have been outlined by Reference WagenbachWagenbach and others (1994).
2. Sampling
The chemical analyses reported here were undertaken on snow-pit samples and shallow firn cores collected during several German glaciological surveys on the Filchner-Ronne lee Shelf during the summer field campaigns 1985–86, 1989–90 and 1991–92. An overview of the various drilling sites on both ice shelves is shown on the map in Figure 1. All firn cores were transported back in a frozen state to the Alfred- Wegener-Institut für Polar-und Meeresforschung in Bremerhaven, where the core samples were split into two parts: one was prepared for high depth resolution analysis of stable isotopes of water by the GSF in München-Neuherberg, and the second one was used for chemical analysis at the Institut für Umweltphysik in Heidelberg. The firn cores from the drilling locations marked in Figure 1 by solid circles were prepared for high depth resolution (3–5 cm) analysis to resolve the seasonality in the chemical stratigraphy. Cores from locations marked by open circles were analysed at lower depth resolution of 20–30 cm only.
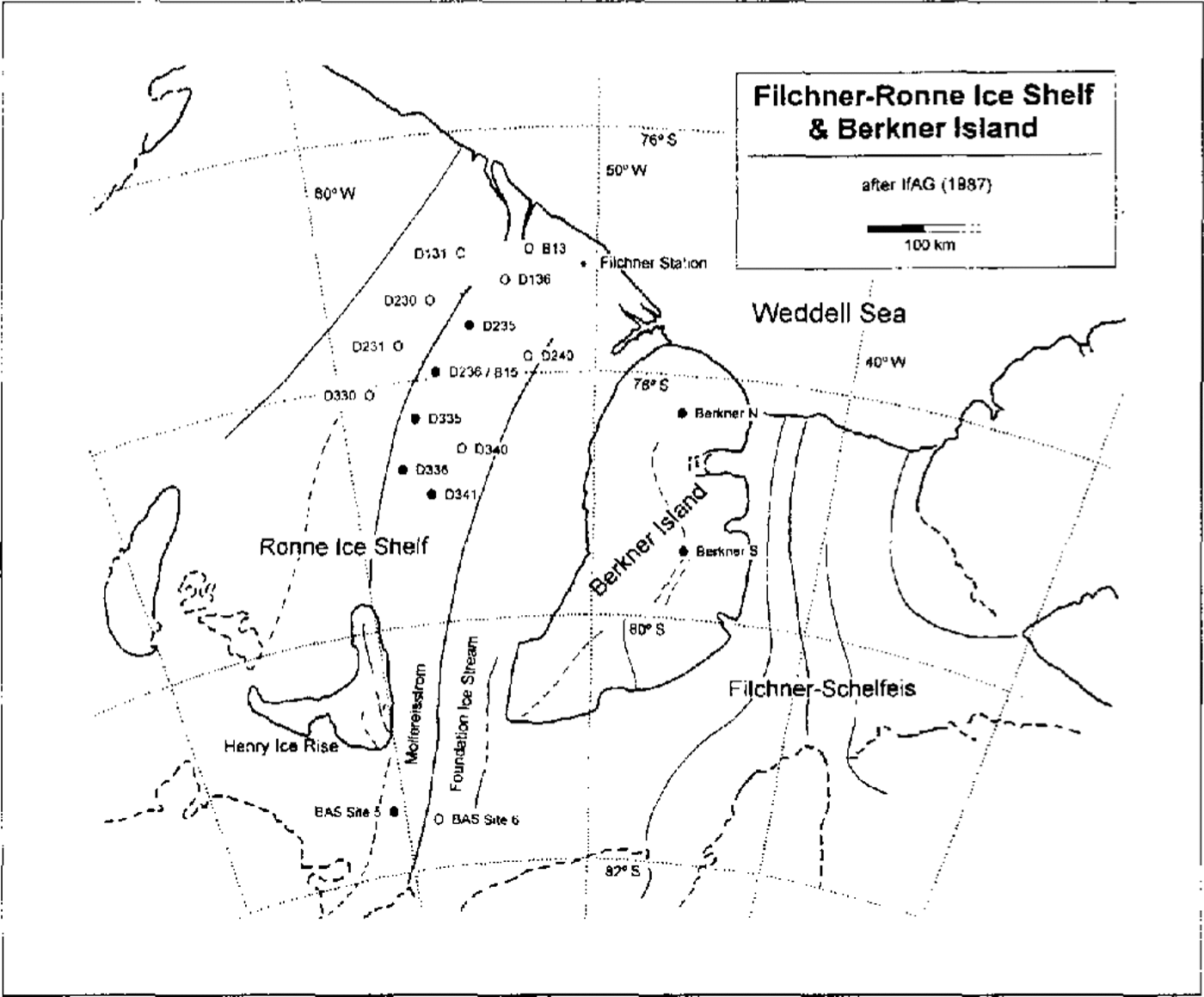
Fig. 1. Map of the Filclner-Ronne Ice Shelf after IfAG (1987). Solid circles are assigned to firn cores analysed at high depth resolution (3–5 cm), open circles refer to firn cores analysed in lower depth resolution (20–30 cm).
3. Analytical procedures
Outer parts were removed carefully from the snow and firn core samples by contamination-free handling procedures in a cold laboratory. The resulting samples were melted in pre-cleaned polyethylene bags.
All ion chromatographic measurements were made using a Dionex 4500i chromatograph equipped with Dionex columns and chemical suppression. For most samples, a gradient technique on a Dionex AS4A column with NaOH as eluent was used to analyse all anions in one run (methanesulphonate, chloride, bromide, nitrate and sulphate). Sodium was analysed only on part of all firn cores either by ion chromatography, using a Dionex CS 10 column, or by flame atomic emission spectrometry. Excluding bromide, the detection limit for the species with lowest concentrations, methanesulphonate, is about 0.5 ppb for a sample volume of 1000μI which is fully appropriate for most of the snow samples. Lower concentrated samples were partly analysed using a pre-concentration column and 5 ml sample volume. The analytical precision for all ion measurements reported here is typically 5%.
Prior to the fim-core sub-sampling for detailed chemical analyses, continuous d.c. conductivity profiles were recorded in the cold laboratory following the method introduced by Reference HammerHammer (1980) (ECM method). A plane surface is prepared on the firn core with a microtome knife. Along this surface, two carbon-brush electrodes are moved with constant velocity applying a voltage of 1500V. In Antarctic ice cores, the resulting ECM signal has been interpreted as an indicator of strong acid concentration (Reference Legrand, Petit and KorotkevichLegrand and others, 1987; Reference Moore, Wolff, Clausen and HammerMoore and others, 1992).
4. Chemical characterization of the filchner-ronne ice shelf
Table 1 summarizes the mean concentrations of methanesulphonic acid (hereafter denoted as MSA), non-sea-salt sulphate (nss sulphate), chloride and nitrate for the sampling sites on the central Filchner-Ronne Ice Shelf. For consistency in the evaluation of the chemical depth profiles being present in either high or low depth resolution, all values are calculated as mass weighted, arithmetic means.
Generally, chloride, nitrate and nss-sulphate concentrations fall into ranges which are consistent with chemical data obtained hom other Antarctic ice shelves, namely the Ross Ice Shelf (Reference Herron and LangwayHerron and Langway, 1979; Reference HerronHerron, 1982), Riiser-Larsenisen (Reference GjessingGjessing, 1984; Reference Neubauer and HeumannNeubauer and Heumann, 1988) and Ekstrømisen (Reference Graf, Miller and OerterMoser, 1991). Compared to the chemical composition of the Antarctic inland ice (Reference Clausen, Langway, Oeschger and LangwayClausen and Langway, 1989), the level of sea salt on the ice shelves is about one order of magnitude higher and that of nss sulphate is only slightly higher, whereas nitrate concentrations do not differ significantly in both regimes.
Typical MSA concentrations for the Filchner-Ronne Ice Shelf are in the range 10–18 ppb, which is slightly higher than the 6 12 ppb observed at Ekstrømisen (Reference MinikinMinikin, 1989). Other ice-shelf data for MSA are to our knowledge not yet available. Concentrations on the Antarctic plateau appear to be slightly lower: for instance, a range of 7–13 ppb was given by Reference Legrand, Feniet-Saigne, Saltzman and andLegrand and others (1992) for a South Pole firn core and lower levels were measured in the Vostok and Dome C ice cores, Reference Legrand, Feniet-Saigne, Saltzman, Germain, Barkov and PetrovLegrand and others, 1991).
Table 1. Mean concentrations of aerosol species and snow-accumulation rates for firn-core and snow-pit samples collected on the Filchner Ronne Ice Shelf. The dates refer to the chemicalfy anafysed depth intervals

Sea-salt species
The chloride to sodium ratio was determined at high depth resolution only for the sampling sites D236 and D335. This ratio is slightly higher in summer layers than the sea-water ratio of 1.80 (Reference Wilson, Riley and SkirrowWilson, 1975), suggesting additional deposition of HCl during this season (Legrand and Delmas, 1988). The mean chloride to sodium mass ratio for both positions is 2.0. To treat all sites consistently, we chose chloride as the sea-salt reference element.
Bromide concentrations were determined incidentally when a gradient technique was used for anion analyses. Against the assumption of finding the winter sea-salt peaks associated with enhanced bromide concentrations according to the sea-water proportion of bromide, in the near-surface layers we generally found a weak seasonal pattern with higher concentrations in summer. These summer values are in the order of 1–3 ppb and essentially in excess of the contribution expected from sea salt. This observation may be partly explained by the deposition of gaseous HBr in an analogous way to HCl. However, bromide seems to be severely affected by a post depositional loss process because the concentrations fall almost always below our detection limit of about 0.5 ppb below 2 or 3 m depth. Therefore, no further use of the bromide data will be made here.
nss-sulphate calculation
Using sodium (or chloride) as a reference element for sea salt, the nss-sulphate concentrations are commonly calculated from the total sulphate and the sea-salt concentration according to Equation (1):
with

However, if the sea-water mass ratio of sulphate to sodium of 0.252 (Reference Wilson, Riley and SkirrowWilson, 1975), here denoted as k, is used in Equation (1), significantly negative nss-sulphate concentrations occur in almost all winter firn layers at each sampling site of the Filchner-Ronne Ice Shelf. This is also true at site D336, which is located about 300 km away from the coast. This phenomenon is also observed in other coastal Antarctic regions, as for example in firn cores from Riiser-Larsenisen (Reference GjessingGjessing, 1989) and the Antarctic Peninsula (Reference Mulvaney, Pasteur, Peel, Saltzman and WhungMulvaney and others, 1992) or in aerosols sampled at Neumayer Station (Reference Wagenbach, Görlach, Moser and MünniehWagenbach and others, 1988). Apparently, sulphate must be depleted, at least in winter, in airborne sea-salt particles, leading to an important underestimation of nss sulphate using Equation (1). The process responsible for the sulphate depletion is still not clear but we expect this to happen because of a chemical fractionation during the production of sea-salt -particles under conditions of low air temperatures.
To compensate for the assumed sulphate fractionation, an empirical approach to the true sulphate to sodium ratio in the local sea-salt aerosol has been established by selecting all winter samples, for which the true nss-sulphate level can be expected to be relatively low and constant (unpublished work of A. Minikin and D. Wagenbach). Variation of k in Equation (1) to give a minimal correlation between (formally) calculated nss sulphate and sodium among these samples then leads to a corrected k *,

which is considerably lower than k but surprisingly constant for all drilling locations analysed here in high depth resolution (Table 2). The apparent sulphate depletion, at least in the winter sea-salt particles, is found to be about a factor of 5. Considering the different sampling processes involved, this value agrees fairly well with the depletion factor of about 4 deduced from the aerosol record at Neumayer Station.
Subsequently, the term nss sulphate refers exclusively to the corrected nss-sulphate concentrations as calculated by the sodium to sulphate ratios, k *, given in Table 2 (for sites not mentioned in Table 2 an arithmetic mean of 0.049 is used for k *). Although there is no clear evidence for the sulphate-fractionation effect in the sea-salt particles to occur also in summer, we have omitted a seasonal dependence of k *. Therefore, the summer nss sulphate concentrations may be systematically overestimated. Because of generally lower sea-salt and maximum nss-sulphate concentrations in the summer firn layers, we estimate this effect to be of the order of 10% or even lower.
In any case, if sea-salt concentrations reach or even exceed I ppm, which may still be the case at distances of about 100 km from the coast, nss-sulphate values measured in bulk or especially in winter samples will be considerably underestimated if the sulphate-fractionation effect is not accounted for.
Table 2. Estimated sulphate to sodium mass ratios in sea-salt aerosol (k *) at Neumayer Station and at various locations on the Filchner-Ronne Ice Shelf

5. Seasonality of chemical species
Serving as a typical example for the firn cores analysed in high depth resolution, the chemical, ECM and δD profiles of site D235 are presented in Figure 2. The ECM and the chemical records were slightly low-pass filtered to emphasize the main features. As is the case at sites D236, D335, D336 and D341 (not shown here) and also at the Berkner Island ice domes, the ECM, nss-sulphate, nitrate and δD profiles reveal a clear annual layering of the firn. Among the chemical records, the nss-sulphate particularly shows most univocal seasonal variations with summer maxima lying very well in phase with the isotopic summer maxima. Therefore, the precise dating of all the firn cores was established by a combination of the isotopic and the nss-sulphate stratigraphy (for details see Reference GrafGraf and others, 1994).
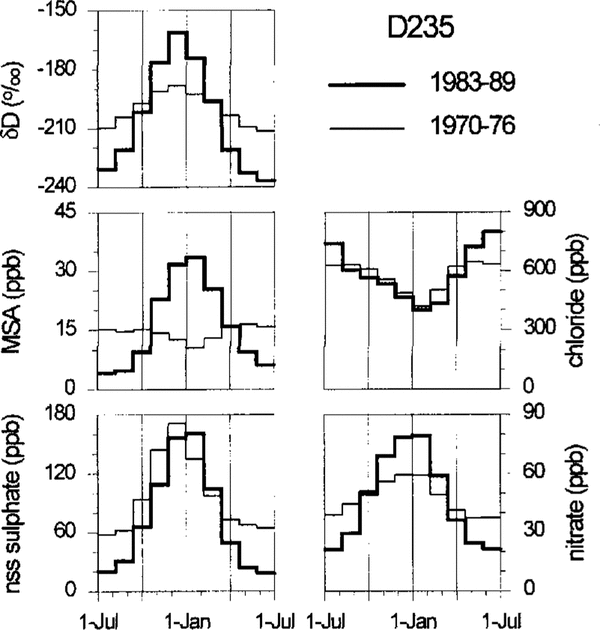
Figure 3 shows for site D235, which again is presented as atypical example for all the sites investigated, the calculated mean annual cycles of chloride, nitrate, MSA and nss sulphate. To obtain these mean seasonal patterns, the periods January-June and July-December were defined in each individual annual layer according to the year-by-year dating mentioned above and then divided into evenly spaced time intervals for subsequent averaging over all selected years. To reveal possible ageing and/or depth effects in the seasonal pattern of these species, the averaging of individual years was carried out for two depth intervals: the upper containing the first 7 years of accumulation as counted from the surface (1983–89), and the lower containing a 7 year period close to the bottom of the firn core (1970–76).

Fig. 2. Profiles of ECM, chemical species and δD for the firn core drilled at site D235. The depth scale is given in water equivalent (w.e.). All records are transformed to equidistant depth intervals (1.5 cm w.e.). The ECM and chemical records are slightly low-pass filtered with a five point Gaussian filter. Summer horizons according to the year-to-year dating are marked with vertical bars.
Chloride
Generally, all chloride depth profiles are characterized by episodic sea-salt events which show up predominantly in the winter firn layers. This seasonal pattern is illustrated in Figure 3 by the mean annual cycle at site D235, showing the chloride minimum in January and the maximum around June-July with no significant difference between both depth intervals. The sea-salt concentration in the firn, therefore, does not directly follow the annual cycle of sea-ice cover which is generally at its minimum around February (Reference ZwallyZwally, 1984). As a consequence, sea-salt particles deposited on the Filchner- Ronne Ice Shelf within the winter accumulation must have been transported over relatively long distances (if not derived from local polynas). The sea-salt signal, therefore, appears to be connected more closely to the large-scale storm activity over the Southern Ocean rather than to the actual sea-ice coverage.

Fig. 3. Mean annual cycles of D, chloride, nitrate, MSA and nss sulfilzate for the firn core drilled at site D235. The thick line is assigned to a near-surface depth interval (corresponding to the years 1989–83) and the thin line to a lower depth interval (1976–70). Note that the apparent higher nss-sulphate base line in the lower core section (which is not observed at the other drill sites) may arise from an averaging effect or a systematic error in the correction for the winter-time nss sulphate.
Nitrate
In all firn cores, nitrate shows a quite distinct seasonal pattern with a maximum in summer (December- January) and minimum in winter (Fig. 3). A comparison of the nitrate urn records with the Neumayer Station aerosol record, however, reveals an important difference for the timing of the absolute nitrate maximum: nitrate sampled by the high-volume aerosol ulters peaks regularly during late spring-early summer with secondary maxima in late winter and early spring (Wagenbach and others, 1988). These secondary maxima are, however, observed only in some years in the firn records. Part of the discrepancies between the seasonal aerosol and the firn nitrate cycles may simply be explained by the much lower time resolution achieved in the firn records. In explaining this phenomenon more rigorously, post-depositional changes of nitrate in surface snow and the mobilization of HNO3 from the aerosol filters during summer by biogenic sulphuric acid should also be considered.
Unlike sea salt or nss sulphate, the amplitude of the annual nitrate cycle is considerably dampened with increasing depth by roughly a factor of 2 in all fim cores. This effect is only partly attributed to a down-core reduction of the depth resolution in relation to the annual layer thickness. Also, possible small phase shifts between the chemical and the isotopic profiles due to sub-sampling errors, which would for example also influence the nss sulphate peaks, cannot explain this smoothing effect. The broadening of the nitrate peaks may be driven by gas phase HNO3 diffusion within the porous firn layer during snow metamorphosis. However, by comparing the nitrate levels in the upper and lower depth intervals of all firn cores, no significant net loss of nitrate could be observed.
nss sulphate and msa
The nss sulphate and, restricted to the near-surface layers, also the MSA profiles show a very pronounced annual cycle with peak-to-base line ratios of up to 10. Within the limitations given by the depth resolution of approximately ten samples per year, the summer peak coincides with the maximum of the stable-isotope content. It is here defined to mark the beginning of each year, which is in agreement with our findings at Neumayer Station where the aerosol record from 1983 to 1992 shows nss-sulphate air concentrations consistently peaking in the first half of January (Reference Wagenbach, Görlach, Moser and MünniehWagenbach and others, 1988).
The direct aerosol measurements at Neumayer Station show a basically congruent annual cycle of MSA and nss sulphate (unpublished data of A. Minikin and D. Wagenbach). This is indeed always the case for the topmost urn layers (for site D235, see Figures 2 and 3) but for depths greater than 3–4 m the phase relation to the nss-sulphate record is almost reversed with MSA peaks frequently occurring in winter firn layers. However, the overall MSA to nss-sulphate ratios do not differ significantly in the selected upper and lower depth intervals MSA winter peaks were previously observed by Reference Mulvaney, Pasteur, Peel, Saltzman and WhungMulvaney and others (1992) in the Dolleman Island ice core and they attributed this phenomenon to a post-depositional migration process which is still unclear.
Ecm
A methodically important result is the suitability of ECM -profiling at the Filchner-Ronne Ice Shelf for preliminary dating purposes even in the low-density firn. It is obvious that the ECM signal is correlated to the summer maxima of the strong acid contributors nss sulphate (H2SO4), nitrate (HNO3) and, partly, MSA and HCl. Furthermore, the sea-salt peaks, frequently occurring in winter layers, are always accompanied by a near-zero ECM current. The combination of both effects leads to the relatively clear annual variation of the ECM signal. Only the ECM profiles of the southernmost drilling sites, BAS sites 5 and 6, reveal no clear seasonal pattern but this is in accordance with the blurred isotopic stratigraphy found there.
6. Spatial variations of the snow chemistry
In order to investigate the spatial trends of the mean concentrations, the sampling sites were combined into three sets, each associated with a different flowline of the ice shelf (see map in Fig. 1): first, the B13–B 15 flowline extending upstream from the B13 site to Möllereisstrom including the sites D 136, D235, D236 (B15), D335, D336 and BAS site 5; secondly, the so-called western flowline (D131, D230, D231 and D330), and thirdly, the eastern flowline (D240, D340, D341 and, again, BAS site 5). From the data given in Figure 4, no significant trend of the mean concentrations of the chemical components in a northwest-southeast direction or parallel to the coast can be deduced. Slightly lower concentration levels generally observed on the western flowline are possibly due to the 10% higher accumulation rates there.
However, going southward, parallel to the flowlines, the chemical firn composition changes systematically with increasing distance from the ice edge. As shown in Figure 4, these changes are consistent within all three data sets. The chloride concentrations drop from about 900 ppb 50 km inland from the ice edge down to 200 ppb at the southernmost sites close to the grounding line. This corresponds to a decrease of about 30% (100km, if the data for the B13–B 15 flowline are parameterized by an exponential fit. MSA also exhibits a quite distinct decrease of about 25%/100 km, whereas the nss-sulphate concentration decreases more gradually by about 10%/100km. Nitrate, on the other hand, reveals an almost opposite pattern: the concentrations appear to show a weak tendency of increasing values towards more continental positions on the ice shelf.
A special situation for the sea-salt (chloride) gradient is encountered in the direct vicinity of the ice edge, where much higher sea-salt concentrations than indicated by the general trend in Figure 4 were observed (Reference Görlach, Wagenbach, Kipfstuhl, Stuckenberg and KohnenGörlach and others, 1985). Also, at Neumayer Station on Ekstf0misen, at a distance of 9 km from the coast, firn core analysis revealed a mean chloride concentration of 40 ppm. Such high concentrations are caused by dry deposition of locally produced, large sea-salt particles, as can be inferred from the much lower mean chloride concentration of 1.5 ppm in fresh snow collected regularly at the same site (Reference TrefzerTrefzer, 1992).

Fig. 4. Spatial trends of chloride, nitrate, MSA and nss sulphate concentrations on the central Filchner-Ronne Ice Shelf. The data points are divided into three sets representing different flowlines on the ice-shelf surface (for details see text). Snow-pit data are not included. The exponential fits were calculated for the data assigned to the B13–B15 flowline.
There are two important questions connected with the continental effects described above:
-
Which information on the main source and on the transport history of the chemical species can be deduced from the systematic changes observed?
-
What are the consequences to be considered in evaluating net temporal changes from chemical depth profiles of intermediate-depth ice cores drilled on the central Filchner-Ronne Ice Shelf?
Sources and transport of chemical species
The continental effects directly reflect the marine source region of the sea-salt and the biogenic sulphur aerosol components in contrast to nitrate, where at least an advection through the upper troposphere is to be expected (Reference Legrand and KirchnerLegrand and Kirchner, 1990). From direct aerosol measurements of nss sulphate and MSA at coastal Antarctic stations, it has already been established that nss sulphate is controlled by the biogenic emission of dimethyl sulphide from the Southern Ocean surrounding Antarctica (Reference Wagenbach, Görlach, Moser and MünniehWagenbach and others, 1988; Reference Prospero, Savoic, Saltzman and LarsenProspero and others, 199l; Reference Savoie, Prospero, Larsen and SaltzmanSavoie and others, 1992). In particular, the existence of the continental effects of the biogenic sulphur species deposited on the Filchner-Ronne Ice Shelf confirms that the main source region of these species is the adjacent Weddell Sea area. This is plausible, because at least the front and central parts of the Filchner-Ronne lee Shelf are directly influenced by marine rather than continental air masses as experienced from the weather conditions in the field and indicated by modelling of the katabatic wind regime over the Antarctic continent (Reference Parish and BromwichParish and Bromwich, 1991).
Assuming a first-order removal process being responsible for the observed near exponential decrease of the chloride, MSA and nss-sulphate concentrations leads to relative residence times of these aerosol species against wet deposition on the ice shelf in the order of 1, 1.3 and 2, respectively (as calculated from exponential fits to all data). Note that, on an ice shelf, the continental effects are not biased by any orographical effects. Surprisingly, the relative residence time of the large-particle sea-salt aerosol is thereby comparable to that of MSA, which is a secondary aerosol component and hence associated with sub-micron particles (Reference Pszenny, Castelle, Galloway and DucePszenny and others, 1989). This indicates, that the sea-salt aerosol deposited in the central Filchner-Ronne Ice Shelf is relatively aged and not primarily of local origin.
The difference in the relative removal rates of MSA and nss sulphate may be a consequence of their different reaction pathways: according to current knowledge, both components constitute the end products in the DMS oxidation scheme but, in contrast to MSA, biogenic nss sulphate is believed to be formed via the gaseous precursor SO2, whose atmospheric lifetime is comparable to that of aerosol particles (Reference Andreae and Buat-MénardAndreae, 1986; Berresheim, 1987). The SO2 may, therefore, be subject to considerable transport over the ice shelf before it is oxidized to sulphate. Compared to MSA, this may lead to the weaker continental effect of nss sulphate observed at the Filchner-Ronne Ice Shelf.
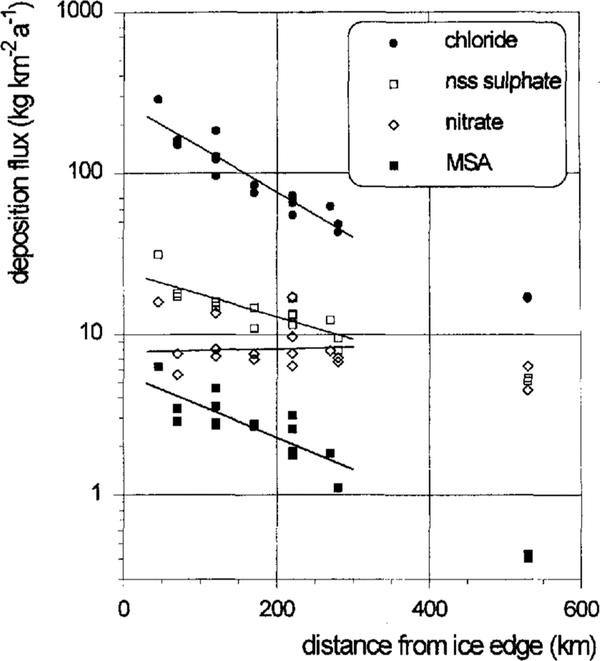
The geochemically relevant deposition fluxes of the aerosol species investigated and shown in Figure 5 were calculated from the mean concentrations and the accumulation rates given in Table 1. Due to the systematic decrease of the accumulation rates along the flowline, the spatial gradients for the sea-salt, nss-sulphate and MSA deposition fluxes are enhanced when compared to the corresponding concentrations. Again, the mean deposition fluxes can be well described by the exponential fits shown in Figure 5. However, no significant trend is observed for the nitrate-deposition fluxes.
The chloride-deposition fluxes in the southernmost parts of the ice shelf do not fit into the exponential decreases observed in the central region. This may suggest a stronger influence of continental air masses being low in sea salt at this site.

Fig. 5. Spatial trends of chloride, nitrate, MSA and nss sulphate deposition fluxes on the central Filchner-Ronne Ice Shelf.
Implications for ice cores b13 and b15
In the two intermediate-depth ice cores drilled on the central Filehner-Ronne Ice Shelf, B13 (215m deep) and B15 (320m), the surface trends in the chemical composition will be reflected in the corresponding depth profiles. Each core penetrated the entire layer of meteoric ice (approximately 150m in both cores) and partly the marine ice (Reference OerterOerter and others, 1992a, Reference Oerter, Drücker, Kipfstuhl, Nixdorf, Graf and Oerterb). Both cores provide a complete record of the precipitation deposited on to the ice shelf over a time-scale estimated to be in the order of 2000 years.
Due to the fast-flowing ice shelf and according to a simple flow model after Reference ThomasThomas (1973), a particle path, for example, from the snow surface at BAS site 5 (the southernmost shallow firn core on the B13–B15 flowline) reaches the drill site of B15 at a depth of 100m and B13 at 105 m, approximately. From the surface down to these depths, the concentrations will systematically decrease according to the surface-concentration gradients. For the B13 core, for example, this would give a decrease of chloride from 900 ppb at the surface by 80% or for nss sulphate from 100ppb by 40%, approximately. However, despite averaging over a time period of at least 20 years, there is a considerable scatter in the surface data from which the continental effects are defined. As deduced from the standard deviations of the residuals to the exponential fits, this relative scatter is estimated to be about 20% for chloride and nss sulphate, and about 30% for MSA. Apart from systematic errors inherent to the particle-path modelling, this ambiguity in the determination of the continental effects alone will generally limit the net temporal changes which can be deduced uni-vocally along ice cores 1113 or B15. This is particularly true for possible systematic long-term temporal trends which will most probably be obscured by the systematic surface trends unless they clearly exceed the observed 20–30% uncertainties given above for sea salt, nss sulphate and MSA.
On the other hand, particularly the strong sea-salt gradient may be used to deduce the origin of ice found at certain depths in the ice cores (Reference Herron and LangwayHerron and Langway, 1979). Similarly, Reference GrafGraf and others (1994) inferred from the stable-isotope record that the bottom part of the meteoric ice in the B13 and B15 cores most probably contains inland ice. Because typical chloride concentrations on the Antarctic plateau are always well below 100 ppb (e.g. Reference Kirchner and DelmasKirchner and Delmas, 1988; Reference Mosley-Thompson, Dai, Thompson, Grootes, Arbogast and PaskievitchMosley-Thompson and others, 1991), this ice should be marked by a significantly lower chloride concentration than the about 200 ppb measured near the grounding line.
Acknowledgement
This study was financially supported by the Deutsche Forschungsgemeinschaft (DFG).