



Introduction
The light rare earth elements (LREE; La to Sm) and the heavy rare earth elements (HREE; Eu to Lu) are now classified as critical raw materials by the European Commission (European Commission, 2017) due to the growing demand for these strategic elements. This has created a heightened interest in understanding the origin of economic REE deposits (Williams-Jones et al., Reference Williams-Jones, Migdisov and Samson2012; Wall, Reference Wall and Gunn2014).
Currently the largest and highest grade REE deposits are associated with carbonatite rocks (Chakhmouradian and Wall, Reference Chakhmouradian and Wall2012; Chakhmouradian and Zaitsev, Reference Chakhmouradian and Zaitsev2012; Verplanck, Reference Verplanck2017). Carbonatite deposits are commonly LREE-enriched (Woolley and Kempe, Reference Woolley, Kempe and Bell1989; Rankin, Reference Rankin, Linnen and Samson2005; Verplanck et al., Reference Verplanck, Farmer, Kettler, Lowers, Koenig and Blessington2014), but a few carbonatite deposits, such as Huanglongpu (central Shanxi Province, China), the Lofdal complex (north-western Namibia) and Bear Lodge (USA), show relative HREE enrichment (Xu et al., Reference Xu, Campbell, Allen, Huang, Qi, Zhang and Zhang2007; Wall et al., Reference Wall, Niku-Paavola, Storey and Axel2008; Andersen et al., Reference Andersen, Clark, Larson and Neill2016). Many REE carbonatite deposits display evidence of hydrothermal reworking that can influence the final REE distribution and mineralogy (e.g. Smith et al., Reference Smith, Henderson and Campbell2000, Reference Smith, Campbell and Kynický2015; Shu and Liu, Reference Shu and Liu2019; Cangelosi et al., Reference Cangelosi, Broom-Fendley, Banks, Morgan and Yardley2019). Economic concentrations of REE are commonly hosted by minerals precipitated during hydrothermal alteration, rather than the early magmatic REE minerals (Wall, Reference Wall and Gunn2014).
This investigation follows from the recent work of Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018) on the Huanglongpu calcite carbonatites. They investigated the HREE enrichment of the carbonatites by looking at the progression from magmatic REE-bearing minerals to later hydrothermal REE-bearing minerals. They demonstrated that each subsequent REE-bearing mineral stage resulted in preferential leaching of the LREE and enrichment in the HREE. This is associated with a shift to more silica-rich hydrothermal conditions leading to quartz growth, and finally with sulfate mineralisation. They documented magmatic HREE enrichment seen in late-stage magmatic calcite. Subsequently, hydrothermal alteration of magmatic REE phases, notably monazite-(Ce), was accompanied by growth of secondary REE minerals, in particular britholite-(Ce) and Ca–REE fluorocarbonates, which show relative HREE enrichment compared to magmatic phases.
The Huanglongpu carbonatite is mined as a molybdenum deposit, but is considered to be part of a key REE-producing area of China, although its reserves and grades for the REE are unpublished (Kynický et al., Reference Kynický, Smith and Xu2012). The presence of quartz with associated fluid inclusions, together with hydrothermal HREE-rich minerals, make it suitable for investigating the role of secondary hydrothermal fluid in the REE enrichment of carbonatites.
The Huanglongpu carbonatite deposit
Geological setting
The Huanglongpu deposit lies in the north-western part of the Qinling orogenic belt in central China (Xu et al., Reference Xu, Campbell, Allen, Huang, Qi, Zhang and Zhang2007; Fig. 1). This orogenic belt hosts numerous ore deposits and is divided by the Shangdan suture into two parts: North and South Qinling. The northern border of the North Qinling belt is defined by a normal fault associated with the Cenozoic rift basin to the north. The southern Qinling belt is further divided by the Mianlue suture which was extensively reworked by Late Mesozoic thrusting (Xu et al., Reference Xu, Kynický, Chakhmouradian, Qi and Song2010). A comprehensive summary of the North and South Qinling belt lithologies is given in Xu et al. (Reference Xu, Kynický, Chakhmouradian, Qi and Song2010). The Qinling orogenic belt was incorporated into Rodinia during the Grenvillian orogeny, followed by rifting, then subduction-related tectonics in the Palaeozoic and early Mesozoic, leading to Cretaceous to Palaeogene reactivation, and finally Neogene extension. The detailed tectonic history can be found in Ratschbacher et al. (Reference Ratschbacher, Hacker, Calvert, Webb, Grimmer, McWilliams, Ireland, Dong and Hu2003).
Fig. 1. (a) Geological map of the Huanglongpu Mo-district from Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018). (b,c) Examples of Dashigou open pit dykes, located around WGS84 410530 49S 3803439. (b) Set of parallel calcite carbonatite dykes at the Dashigou open pit. (c) Calcite carbonatite dyke with central quartz lenses and fenetised margins.
The Qinling orogenic belt is an important metallogenic belt, hosting the most important Mo ore camp in China and including several world-class Mo deposits (Mao et al., Reference Mao, Xie, Bierlein, Qu, Du, Pirajno, Guo, Li and Yang2008). The mineralisation is mostly hosted by granitic porphyry bodies with a few porphyry-skarn deposits. However, in the Huanglongpu area (Fig. 1) the Mo mineralisation is associated with unusual carbonatite veins. The Huanglongpu area contains an extensional structure hosting ore bodies that extend over a distance of 6 km, mainly controlled by a northwest-striking fault zone (Xu et al., Reference Xu, Kynický, Chakhmouradian, Qi and Song2010; Fig. 1a). There are five ore fields currently being mined (Fig. 1a): Yuantou, Dashigou, Shijiawan I and II and Taoyuan, with a total reserve of 0.18 million tonnes of MoS2. Apart from Shijiawan I, which is hosted by porphyry and has intruded into the Neoarchean Taihua gneiss, these are associated with carbonatite dykes. Most of the carbonatite ore fields are also related spatially to porphyry and porphyry skarn Mo deposits which yield Re–Os ages ranging from 148 Ma to 112 Ma (Mao et al., Reference Mao, Xie, Bierlein, Qu, Du, Pirajno, Guo, Li and Yang2008). However, the carbonatite molybdenite yields Re–Os ages from 209 Ma to 221 Ma and monazite-(Ce) U–Pb and Th–Pb ages from 208.9 ± 4.6 Ma and 213.6 ± 4.0 Ma (Huang et al., Reference Huang, Wu, Du and He1994; Stein et al., Reference Stein, Markey, Morgan, Du and Sun1997; Song et al., Reference Song, Xu, Smith, Kynický, Huang, Wei, Li and Shu2016) indicating that the mineralisation of the carbonatites is not related to overprinting by the porphyry-related systems. The samples analysed in this investigation are from the Dashigou open pit.
The Dashigou open pit
Individual molybdenite-bearing calcite carbonatite dykes at Dashigou are generally between 0.1 and 1 metre in width, and extend for several tens of metres (Fig. 1b). The main set of dykes predominantly dips N to NNW at steep angles, 50–88°N, (Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018; fig. 1b). Regardless of the range of dyke orientations observed by Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018), including some east-dipping conjugate dykes, they are understood to be closely contemporaneous (Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018; fig. 3e). The country rock gneiss has been fenitised next to the carbonatites (Fig. 1c), and now consists mainly of K-feldspar, phlogopite, pyrite and calcite, with minor amounts of monazite-(Ce) and titanite. However, the fenite was apparently unaffected by the later hydrothermal alteration which corroded and altered monazite-(Ce) in the samples we studied.
Mineralogy of the calcite carbonatites
The carbonatite dykes are dominated by calcite and are referred to here as calcite carbonatite. The majority of the dykes contain central quartz lenses (Fig. 1c). A few dykes show evidence for multiple opening events (Table 1). The calcite carbonatite dykes consist of 50 to 80 vol.% calcite, with typical individual crystals ranging from less than 0.1 cm to 2 cm in size, together with sulfates (celestine–baryte), sulfides (molybdenite, pyrite, galena and sphalerite), K-feldspar, phlogopite, albite, REE minerals and apatite. Quartz only occurs as lenses and is only observed in the calcite carbonatite dykes and not the surrounding fenite. The lenses are composed of quartz grains, ranging from less than 100 μm up to 3 cm, containing μm-sized birefringent inclusions discussed later in this manuscript. Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018) described the paragenesis of the calcite carbonatite minerals. Molybdenite can occur as disseminated grains, as fracture infill within the calcite carbonatite, with other sulfides, or in fracture infill within the fenitised gneiss. Celestine–baryte was inferred to be hydrothermal in origin and Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018) documented a crosscutting relationship with the calcite carbonatite dykes. Pyrite and sphalerite are later than cogenetic molybdenite and pyrite.
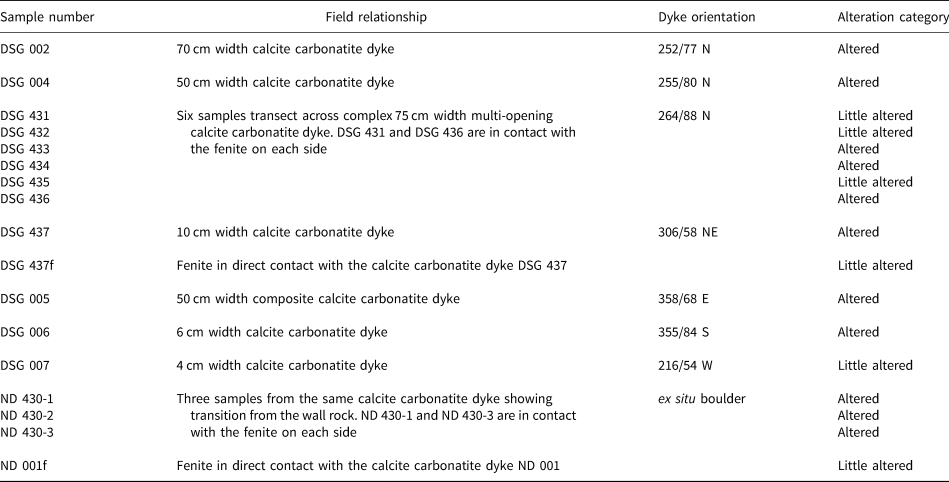
Table 1. Summary of the Dashigou open pit samples used in this work. The alteration categories are based on the predominant REE minerals. ‘Little altered’ refers to samples with predominantly magmatic REE minerals and ‘altered’ refers to those with significant hydrothermal REE minerals (Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018). Only DSG 007 and DSG 434 samples do not host quartz lenses.
The REE mineral assemblages of the calcite carbonatite dykes (Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018) preserve a range of replacement textures making it possible to establish a history of REE mineral growth. The initial, magmatic stage associated with the calcite carbonatites consists of monazite-(Ce), bastnäsite-(Ce), parisite-(Ce) and aeschynite-(Ce) possibly with magmatic burbankite-(Ce); all these minerals are relatively enriched in LREE. Subsequent hydrothermal events first led the monazite-(Ce) to be altered to produce a second generation of apatite, and this was in turn replaced and overgrown by britholite-(Ce) accompanied by the crystallisation of allanite-(Ce). Bastnäsite-(Ce) and parisite-(Ce) were replaced by synchysite-(Ce) and röntgenite-(Ce). Aeschynite-(Ce) was altered to uranopyrochlore and then pyrochlore with uraninite inclusions (Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018). These hydrothermal replacement minerals show HREE enrichment relative to the magmatic precursors.
The magmatic calcite shows some late HREE enrichment indicating that the Dashigou magmatic carbonatite system evolved to HREE-richer conditions (Xu et al., Reference Xu, Campbell, Allen, Huang, Qi, Zhang and Zhang2007; Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018). The composite dykes observed in the field suggest a sequence of calcite carbonatite pulses.
There is significant variation in the extent of hydrothermal alteration between calcite carbonatite samples, with magmatic REE phases well preserved in some, but mostly overprinted in others. Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018) concluded that the hydrothermal alteration was linked to the deposition of some of the quartz including the fluid inclusions which they host.
Methodology
Samples were collected from the Dashigou open pit in June 2016, within a 100 metre radius around location WGS84 49S, 410530E, 3803439N (Fig. 1a). The open pit exposes calcite carbonatite dykes intruded into country rock gneiss (Fig. 1). Four samples were also collected from the Yuantao mine dump a few kilometres north of the Dashigou open pit (WGS84 49S, 409886E, 3804124N). The two fenite samples used in this study were in direct contact with the associated calcite carbonatite samples.
Mineral and fluid-inclusion parageneses were established using optical petrography followed by scanning electron microscopy (SEM) at the University of Leeds using a FEI Quanta 650 field-emission gun scanning electron microscope operated at 20 kV employing back-scattered electron (BSE) and cathodoluminescence (SEM-CL) imaging.
Oxygen isotope measurements were undertaken on selected polished areas of some carbonatite samples mounted in an epoxy block. Prior to being mounted, the selected samples were mapped using BSE. Stable-oxygen-isotope measurements were carried out at the Edinburgh Ion Microprobe Facility (EIMF) using a Cameca 1270 mass spectrometer operating with a primary Cs+ beam, in multi collector mode, operating at conditions of ~5 nA with a net impact energy of 20 KeV (+10 kV primary and –10 kV secondary). The beam size was ~12 μm; standards were: calcite (EIMF) and baryte (Stern S0237). The instrument was calibrated before each session and its drift was corrected by starting and finishing a session by analysing 5 to 10 points of the reference standards and then 5 points every ten analyses. Linear least squares was applied as the correction for drift. Reproducibility of the calcite and baryte standards can be found in Appendix 1 (Supplementary materials – see below).
Raman spectroscopy was carried out in the GeoResources Laboratory, at the University of Lorraine, France. The fluid-inclusion phases were analysed separately to identify solids and Raman-active solution and gas components. Analyses were performed using a LabRAM Raman microspectrometer (Horiba Jobin Yvon) equipped with a Linkam THMS600 microthermometry stage. The excitation beam was a monochromatic green laser at 514 nm (ionised argon 74.53 nm, with a working temperature of –128°C and a Pn of 500 mW). For each solid and gas analysis a spectrum was taken directly next to the fluid or solid inclusion to allow subtraction of the quartz and air N2 peaks from the sample spectrum. The subtractions were made in Horiba LabSpec software, and CrystalSleuth software by RRUFF (Laetsch and Downs, Reference Laetsch and Downs2006) was used to identify the solids. The gas proportions within the fluid inclusions were estimated using the method of Dubessy et al. (Reference Dubessy, Poty and Ramboz1989).
Microthermometry measurements were carried out at the University of Leeds using a Linkam MDS600 Heating Freezing Stage attached to a TMS93 temperature programmer and controller and an LNP cooling system mounted on an Olympus BX50 microscope. Double polished wafers were between 200–300 μm thick. The stage was calibrated using synthetic fluid-inclusion wafers containing pure water fluid inclusions (0°C and 374.1°C) and H2O–CO2 fluid inclusions (–56.6°C and 31.1°C) provided by Ronald Bakker, Leoben University, Austria.
Bulk rock analyses were carried out by Activation Laboratories Ltd. with the sample preparation undertaken at the University of Leeds. Samples were powdered using a steel pestle and mortar and an agate TEMA barrel and sieved to <200 μm. The methods 4Litho and 4F-Total sulfur were used on the calcite carbonatite and country rock samples. The methods and detection limits are detailed in Appendix 1.
Results
Calcite carbonatites
The calcite carbonatite samples have been divided into two broad alteration categories, little altered and altered, based on the extent to which new REE minerals have replaced the original magmatic ones (Table 1). No correlation of alteration with the different dyke orientations was observed. The little-altered samples host predominantly magmatic REE minerals. Monazite-(Ce) is the main primary magmatic REE mineral in the calcite carbonatite samples, and is usually fairly pristine in the little-altered samples (Fig. 2a), although in some instances it can show overgrowth and fracture-fill of late magmatic molybdenite (Fig. 2b). In the altered samples (Table 1) monazite-(Ce) is at least partially corroded and is sometimes fully replaced by new HREE-enriched phases (Fig. 2c), with later HREE-enriched phases in fractures (Fig. 2d). The HREE-enriched phases are predominantly Ca-REE-fluorocarbonates, uraninite, xenotime-(Y) and churchite-(Y) together with unidentified Y and REE-bearing silicate minerals with a variable amount of Ca and Fe, and unidentified Ti, Nb, U, Y and REE-bearing oxide phases with a variable amount of Fe and Ca (Fig. 2c) (Table 2). Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018) described the REE mineral paragenesis, demonstrating that the hydrothermal REE mineralisation becomes more HREE enriched with each subsequent hydrothermal event.
Fig. 2. Back-scattered electron images of magmatic monazite-(Ce) with different degrees of alteration. (a) Magmatic monazite-(Ce) and K-feldspar enclosed by hydrothermal celestine–baryte in a little-altered calcite carbonatite (DSG 431). (b) Magmatic phlogopite and monazite-(Ce) with overgrowth and fracture fill of molybdenite (DSG 431). (c) Magmatic monazite-(Ce) partially replaced by late magmatic molybdenite and an unidentified HREE-rich phase in an altered calcite carbonatite (DSG 434). (d) Magmatic monazite-(Ce) hosted by pyrite with a hydrothermal Y-rich phase in fractures and at the interface of monazite-(Ce) and pyrite grains, note associated hydrothermal celestine–baryte; altered calcite carbonatite (DSG 004). The red arrows in (c) and (d) indicate analysed points where the listed elements were the dominant constituents of unidentified hydrothermal phases. Abbreviation: cls-brt – celestine–baryte; mnz – monazite-(Ce); kfs – K-feldspar; mo – molybdenite; phl – phlogopite; cc – calcite; U – uraninite; Pb – lead metal; py – pyrite.
Table 2. List of minerals encountered in this investigation and their occurrences.
Xu et al. (Reference Xu, Campbell, Allen, Huang, Qi, Zhang and Zhang2007) first reported that the Huanglongpu carbonatites are anomalously rich in the HREE, exhibiting a distinctive flat REE pattern. Xu et al. (Reference Xu, Kynický, Chakhmouradian, Qi and Song2010) investigated calcite carbonatites from the Yuantou, Shijiawan and the Dashigou deposits and reported that total REE concentrations could exceed 3000 ppm. They found that ΣLREE/(HREE+Y) ratios ranged from 0.6 to 2.8. We have obtained a wider range of Σ(LREE/HREE+Y) ratios in this study, from 1.1 to 17.3, correlated with the alteration categories described above, with the little-altered calcite carbonatite samples showing ΣLREE/HREE ratios ranging from 9.9 up to 17.3 and the altered calcite carbonatite samples ratios ranging from 1.1 to 3.8 (Table 3). ΣHREE + Y totals also overlap with those reported by Xu et al. (Reference Xu, Campbell, Allen, Huang, Qi, Zhang and Zhang2007; Reference Xu, Kynický, Chakhmouradian, Qi and Song2010).
* DSG 437f is the fenitised country gneiss associated with sample 437. ‘<' below detection limit of the instrument (detailed in Appendix 1). LREE = La to Sm, HREE = Eu to Lu + Y. Ge, Sb, In, TI and Ta are under 2 ppm; As, Be, Sn, Cs and Hf are under 10 ppm and Ag and Th are under 20 ppm. Note that the total below 100% reflects the lack of data for Mo, Sr and F, which could be an important component of some samples. Compositional data for ND samples are detailed in Appendix 2.
The REE chondrite-normalised patterns for four little-altered calcite carbonatite samples in this study (DSG 431, DSG 431-2, DSG 007 and DSG 435; Tables 1, 3) are shown by blue lines on Fig. 3 and do not show the HREE-enrichment documented by Xu et al. (Reference Xu, Kynický, Chakhmouradian, Qi and Song2010). Instead, they exhibit a LREE-enriched pattern typical of carbonatites worldwide (Woolley and Kempe, Reference Woolley, Kempe and Bell1989) and similar to that of the fenites (Fig. 3, green lines). The fenite REE pattern reflects its REE mineralisation consisting mainly of unaltered monazite-(Ce), which has been observed in other carbonatite associated fenites (e.g. Thair and Olli, Reference Thair and Olli2013; Midende et al., Reference Midende, Boulvais, Tack, Melcher, Gerdes, Dewaele, Demaiffe and Decrée2014; Trofanenko et al., Reference Trofanenko, Williams-Jones, Simandl and Migdisov2016). In contrast, the altered calcite carbonatites in this study (Table 1, shown in red on Fig. 3) are HREE-enriched and plot close to the samples described by Xu et al. (Reference Xu, Kynický, Chakhmouradian, Qi and Song2010) with the most HREE-enriched samples showing a positive Y anomaly (Fig. 3).
Fig. 3. REE chondrite-normalised patterns (McDonough and Sun, Reference McDonough and Sun1995) for calcite carbonatites (little hydrothermal alteration in blue and altered in red) and associated fenites (green lines). Samples with results from Xu et al. (Reference Xu, Kynický, Chakhmouradian, Qi and Song2010) for the Dashigou open pit (alteration not specified, black lines) are shown for comparison. For details of the DSG samples investigated see Table 1.
Quartz lenses within calcite carbonatite dykes
Irrespective of the host dyke's orientation, SEM-CL imaging reveals the presence of three quartz generations making up the quartz lenses that occur within them (Fig. 4). The dominant generation is the earliest, Qz-1; it is moderately luminescent and shows some poorly-defined growth zonation. Generation Qz-2 occurs as non-luminescent overgrowths and porosity infills, merging into fracture fills cutting grains of Qz-1. The last generation, Qz-3, is also a non-luminescent quartz and occurs as thread-like fracture fills cutting both earlier generations. While Qz-1 lacks fluid inclusions and appears patchy and strained in cross polars, both Qz-2 and Qz-3 are cloudy and contain fluid inclusions.
Fig. 4. SEM-CL image showing the paragenesis of the three quartz generations within a calcite carbonatite dyke, note the irregular zoning of the primary quartz (Qz-1) (DSG 005 001).
Fluid-inclusion paragenesis and hydrothermal HREE mineralisation
Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018) showed that secondary quartz grew at the same time as the later hydrothermal REE minerals that are enriched in the HREE. The relationship between HREE minerals and Qz-2 and Qz-3 partially replacing magmatic calcite is shown in Fig. 5. HREE phases occur at the interface between calcite and Qz-2 (Fig. 5a) and in calcite fractures associated with celestine–baryte mineralisation related to Qz-3 (Fig. 5b).
Fig. 5. BSE images of HREE enriched calcite carbonatite sample DSG 433. (a) HREE mineral hosted in hydrothermal Qz-2 at the interface with earlier calcite. (b) Mineralisation of a fracture in calcite by hydrothermal HREE minerals and celestine–baryte, inferred to be associated with the partial replacement of the calcite by quartz. Abbreviations: ‘Fcb-LREE’ – LREE fluorcarbonate; ‘Xtm-Y’ – Xenotime-(Y); ‘Cc’ – calcite; ‘Cls-brt’ – celestine–baryte.
Calcite and quartz stable oxygen isotope data
The samples analysed are from two distinct dykes and data is presented in Table 4. Secondary quartz data is from both Qz-2 and Qz-3 (non-luminescent quartz, Fig. 4) as these could not be distinguished during the analyses. Because of the difficulty in finding exact points for analysis in the ion probe, traverses were made across areas with all quartz generations to ensure that primary and secondary generations would be analysed. It is clear from the results that both Qz-1 and secondary quartz (Qz-2 and Qz-3) have similar compositions with tightly constrained populations of data that differ by no more than the analytical uncertainty (c. 0.25‰). Average compositions are close to 10‰ SMOW (e.g. DSG 002 Qz-1 δ18O ranges from 9.72‰ to 10.43‰ and secondary quartz δ18O ranges from 9.57‰ to 10.18‰; Table 4). Magmatic calcite has similar δ18O contents to quartz, averaging to 9.66‰ for sample DSG 437 and 10.34‰ for sample DSG 002. These figures are similar to calcite analysed by Bai et al. (Reference Bai, Chen and Jiang2019) from the Huanglongpu and the Huangshui'an carbonatites of the Lesser Qinling Orogen (δ18O ranges from 7.10‰ to 9.48‰).
Table 4. Summary of the oxygen isotope data from calcite and quartz, all as δ18O SMOW.
Fluid-inclusion petrography and microthermometry
Three samples of quartz lenses from altered carbonatites were selected for the fluid-inclusion study (Fig. 3; Table 5): DSG 002, DSG 437 and DSG 433 (Table 1). Fluid inclusions range in size from <5 μm up to 70 μm but are commonly between 10 μm to 40 μm. Due to the cloudiness of Qz-2 and Qz-3, we were unable to collect freezing measurements such as TmCO2 and TmCLA. Fluid inclusions are abundant in Qz-2 and mainly occur as clusters or in fluid-inclusion-rich zones in cloudy quartz (Fig. 6a), more rarely in planar arrays and linear trails. The fluid inclusions usually have quite irregular shapes (Fig. 6a,b,d) and are predominantly LCO2 + VCO2 + Lw and LCO2 + VCO2 + Lw + S(s) type fluid inclusions (where LCO2 = liquid CO2, VCO2 = gas CO2, Lw = aqueous liquid and S(s) = from one to five solids). They show variable proportions of CO2 relative to the aqueous liquid, even within the same fluid-inclusion cluster (Fig. 6a). Some inclusions have a homogenised CO2 bubble at room temperature and are referred to as LCO2 + Lw fluid inclusions (Fig. 6a; Table 5; Appendix 2). About 15% of the fluid inclusions in Qz-2 contain one or more solids (Fig. 6a–f; Tables 2,5). These solids are frequently birefringent and individual fluid inclusions can contain up to five different solids, but there is only ever one opaque solid (Fig. 6d). On rare occasions, fluid inclusions are not CO2-bearing and only comprise an aqueous solution and solid(s) (Lw + S) with the solid taking up most of the inclusion volume. The Qz-2 generation may also contain small (<15 μm) birefringent solid inclusions near the fluid inclusions, within the same cluster or fluid-inclusion trail. These solid inclusions are of comparable size to the solids observed within the fluid inclusions (Fig. 6a).
Fig. 6. Microphotographs of fluid inclusions hosted in Qz-2 (a–f) and Qz-3 (g–i). (a) LCO2 + VCO2 ± Lw ± S fluid inclusions hosted in cloudy quartz showing variation of the CO2 and aqueous phase ratio, the fluid inclusion on the top right show the gas bubble taking up most of the fluid inclusions volume while the fluid inclusion in the top middle show a rather small gas bubble, with a homogenised CO2-bearing fluid inclusions (LCO2 ± Lw). The LCO2 + VCO2 ± Lw + S inclusion contains an arcanite solid. Note the anhydrite solid inclusion in the quartz has a similar size to the trapped solid (DSG 002 4). (b) LCO2 + VCO2 ± Lw + 3S fluid inclusion hosting sulfate and calcite solids (DSG 433 2f) with the corresponding cross polar image. (c) LCO2 + VCO2 ± Lw + 2S fluid inclusion hosting two sulfate solids and a LCO2 ± Lw fluid inclusion (DSG 437 8i) with the corresponding cross polar image. (d) LCO2 + VCO2 ± Lw + 4S fluid inclusions clearly showing the CO2 vapour bubble within the CO2 liquid. The fluid inclusion hosts two identified sulfates, one unidentified solid and one opaque (DSG 433 2a). (e) Three LCO2 + VCO2 ± Lw ± S fluid inclusions cluster with one with a trapped anhydrite solid (DSG 002 13a). (f) LCO2 + VCO2 ± Lw + 2S fluid inclusion hosting two sulfate solids, similar to (b) most of the space is taking up by the solids (DSG 433 4d). (g) Thin healed fractures hosting LCO2 + VCO2 ± Lw fluid inclusions cross cutting fluid-inclusion free Qz-1 (DSG 437). (h) LCO2 + VCO2 ± Lw fluid inclusions in a planar array showing variation of the CO2 and aqueous phase ratio (DSG 433 33b). (i) Wide healed fracture hosting Lw and Lw + V in the same planar array (DSG 433 29). Abbreviations: ‘cc’ – calcite; ‘SO4’ – unidentified sulfate phase.
Table 5. Summary of the different fluid-inclusion types and their characteristics measured by microthermometry and Raman spectroscopy.
Microthermometric data for Qz-2 fluid inclusions show a scatter of ThCO2 from +12°C to +30.9°C, yielding a density of the CO2 phase of 0.85 to 0.47 g cm–3 (Valakovich and Altunin, Reference Valakovich and Altunin1968). CO2 always homogenised to the liquid phase.
In Qz-3, fluid inclusions occur in planar arrays decorating healed fractures (Fig. 6g–i) or as linear fluid-inclusion trails in finer healed fractures (<50 μm). Overall, the Qz-3 fluid inclusions are smaller than those in Qz-2 ranging from <5 μm to 25 μm. These fluid inclusions tend to have similar shapes within a single array, either elongate or with a more rounded shape (Fig. 6h–i). A wider variety of fluid-inclusion types are hosted by Qz-3 (Fig. 7) but they are predominantly of LCO2 + VCO2 + Lw type, similar to those in Qz-2, but with more variability in the relative proportion of the CO2 and aqueous phase (Fig. 6h). Some of these fluid inclusions have a homogenised CO2 bubble below room temperature. In aqueous inclusions (Lw + V) there is a reasonably constant liquid/vapour ratio (c. 80:20). Lastly, there are monophase Lw inclusions which are small (<15 μm) and tend to occur in wider healed fractures, typically from 100 μm to 400 μm (Fig. 6i). They are clearly secondary. Lw and Lw + V type fluid inclusions can occur in the same fluid-inclusion trail and in this instance the vapour bubble proportion is usually lower at ~10% (Fig. 6i). Small (<15 μm) birefringent solid inclusions are also hosted by Qz-3, however they tend to be less abundant than in Qz-2.
Fig. 7. Simplified paragenetic diagram illustrating the paragenetic sequence associated to the later quartz generations in the Huanglongpu carbonatites. The REE minerals paragenesis is after Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018). Bar thickness represents minerals and fluid inclusions abundance. Abbreviations: ‘FI’ – fluid inclusion; ‘qz’ – quartz.
For Qz-3, only five measurements of ThCO2 on the LCO2 + VCO2 + Lw inclusions could be performed, ranging from 26.4°C to 29.8°C corresponding to CO2 densities of 0.70 g cm–3 to 0.56 g cm–3 (Valakovich and Altunin, Reference Valakovich and Altunin1968). Twenty Lw + V fluid inclusions show a temperature of total homogenisation (ThTOT) ranging from ~120°C to 222°C. The Lw inclusions fail to nucleate a vapour bubble despite cooling to a temperature of –130°C for 5 min, something that is commonly indicative of low trapping temperature.
The sequence of the different types of fluid inclusions within Qz-3 is often ambiguous as cross-cutting relationships have not been observed. Nonetheless, in Qz-3 the Lw + V inclusions show a max ThTOT of 222°C, and this is likely to be higher than the trapping temperature of the Lw inclusions, suggesting that they are probably not contemporaneous. The non CO2-bearing fluid inclusions are inferred to be the youngest population (Fig. 7).
Raman spectroscopy
About 200 fluid inclusions were analysed from Qz-2 and Qz-3 in the three samples investigated (Table 5; Appendix 2). Compared to Qz-2, Qz-3, fluid inclusions are less suitable for Raman spectroscopy and microthermometric analysis as the majority are smaller than 20 μm. A summary of the fluid inclusion Raman spectroscopy data is provided in Appendix 2. When possible, the same fluid inclusions were analysed for solution, solid and gas. Both solid-bearing and solid-absent inclusions were analysed for solution components and gave comparable results.
Qz-2 fluid inclusions
The aqueous liquid phase of approximately 100 CO2-bearing fluid inclusions was analysed by Raman spectroscopy. Sulfate (SO42–) ions were detected in solution in 90% of the fluid inclusions, from the peak at 981 cm–1. A few inclusions without detectable sulfate in solution have sulfate solids and it is likely that where sulfate was not found this was a result of the small volume of liquid available. HSO4– was not detected during the analysis (1050 cm–1), precluding very acidic conditions (pH < 2).
In approximately 52 CO2-bearing fluid inclusions, the vapour phase composition was determined. It is dominated by CO2 and N2 with CO2 between 68% to 100%. Ninety percent of the fluid inclusions have a CO2 content ≥95%. H2S, CH4 and H2 were only detected as trace constituents (<1%) of the vapour phase. No difference in vapour phase composition was detected between solid-bearing inclusions and the inclusions without solids.
The majority of the solids hosted in the fluid inclusion in Qz-2 are sulfates (Fig. 8). They include arcanite, anhydrite, celestine, aphthitalite and some other unidentified sulfates (Table 2); a few calcite crystals were also found (Figs 6a–f; 9). Note that celestine and glauberite have very similar Raman spectra and they can be hard to differentiate; when this was the case they are noted as celestine/glauberite in Appendix 2. Whewellite, thénardite, anglesite, dolomite, molybdenite, gypsum, muscovite and an unidentified phosphate mineral were also detected in fluid inclusions (Table 2). The opaque solids could not be identified as they moved under the laser beam.
Fig. 8. Frequency histogram showing the different minerals identified by Raman spectroscopy within the fluid inclusions and hosted as solid inclusion in the quartz in Qz-2 and Qz-3. Abbreviation: ‘n.i.’ is ‘non-identified’. Note that the celestine/glauberite spectra are counted in the ‘n.i. sulfate’ bar.
Fig. 9. Laser Raman spectroscopy spectra of some trapped solids hosted in Qz-2 fluid inclusions from the Dashigou calcite carbonatites note that the background signal from the quartz were subtracted. (a) Arcanite spectrum of a LCO2 + VCO2 + Lw + 3S, the solid on the right is calcite and the solid in the middle is an unidentified sulfate solid (DSG 433 2f-1). (b) Anhydrite spectrum of a LCO2 + VCO2 + Lw + S (DSG 002 13a-1). (c) Celestine spectrum of a LCO2 + VCO2 + Lw + 2S, the celestine crystal is accompanied with an unidentified opaque (DSG 437 8b-1). (d) Aphthitalite spectrum in a LCO2 + VCO2 + Lw + 3S, the opaque and the small transparent solid are unidentified (DSG 433 6a).
Solid inclusions also occur hosted directly in the quartz and these are predominantly sulfates (mainly anhydrite with one unidentified sulfate) and calcite.
Qz-3 fluid inclusions
Sulfate was only detected in about half of the 27 LCO2 + VCO2 + Lw fluid inclusions analysed from Qz-3 (Table 5) and, as with Qz-2, fluid inclusions with and without detectable sulfate in solution occur within the same inclusion trails. Like Qz-2, CO2 is the main component of the vapour phase, but in these inclusions is always >99%, with N2 occurring in trace amounts only. The majority of the solid inclusions hosted directly in Qz-3 are calcite with some unidentified phosphate and sulfate particles (Fig. 8).
The aqueous portions of two Lw fluid inclusions were analysed and SO4 was detected in both. About 75% of the 16 Lw fluid inclusions also had detectable sulfate in solution (Table 5).
Compared to Qz-2 fluid inclusions, Qz-3 fluid inclusions lack sulfate solids and sulfate is not always detected in solution. Likewise, there are fewer solid sulfate inclusions in Qz-3.
Fluid-inclusions origin and reconstructed composition
The LCO2 + VCO2 ± Lw ± S(s) fluid inclusions in the Qz-2 and Qz-3 generations have variable CO2/aqueous phase ratios (Fig. 6a,h), from some that are nearly pure CO2 inclusions to others than contain multiple solids, occurring in the same inclusion trails. This suggests that the fluid inclusions result from heterogeneous trapping of different phases that coexisted at the time of fluid-inclusion formation, including an aqueous liquid, supercritical CO2 and sulfate solids. Although the solids could be daughter minerals, formed after trapping during cooling of a homogeneous fluid, the compositional variability (Figs 5; 7) and the fact that they show no signs of dissolution during heating before the inclusions decrepitate between 225°C to 325°C, makes this unlikely. Accidental trapping is the preferred interpretation, particularly as some of the same solid phases also occur in the quartz as solid inclusions of similar size (Fig. 6a). We infer that these solids were present in the fluid while Qz-2 was being precipitated. Hence, the composition of these inclusions does not represent any single fluid. Regardless of this limitation, we have attempted to estimate the compositions of a number of the individual inclusions.
Due to the chemically complex sulfate-rich fluid and sulfate solids within the fluid inclusions, interpretation of the microthermometric measurements is problematic due to the lack of knowledge of the precise chemical system. The presence of sulfate crystals in these fluid inclusions precludes conventional estimation of their dissolved load as NaCl equivalent salinity. Instead, we used visual proportion estimates to calculate the bulk composition of individual solid-bearing fluid inclusions (Shepherd et al., Reference Shepherd, Rankin and Alderton1985; Fig. 6a; Appendix 2). Due to the clear evidence of heterogeneous trapping, these estimates represent the composition of the material trapped in the fluid inclusions, not a true fluid composition.
Fluid inclusion DSG 002 4 (Fig. 6a) is a LCO2 + VCO2 + Lw + S fluid inclusion hosting a single crystal of arcanite consisting of 15% of the inclusion by volume, with 15% CO2-rich vapour phase and 70% arcanite-saturated aqueous phase (the volumes were estimated using visual proportion estimates as presented in Appendix 2). These proportions give a bulk fluid-inclusion composition of 33 wt.% K2SO4 equivalent or ~3 molal K2SO4. Some other solid-bearing fluid inclusions have multiple sulfate crystals taking up >15% of the fluid-inclusion volume and must therefore have an even higher sulfate content (Fig. 6). The sulfate-bearing LCO2 + VCO2 + Lw fluid inclusions hosted in Qz-2 have similar petrographic characteristics to the LCO2 + VCO2 ± Lw ± S(s) fluid inclusions. Hence, their salinities have also been estimated as equivalent K2SO4 using the visual estimate proportion method assuming a K2SO4 saturated solution. The majority of these fluid inclusions have the CO2 bubble occupying from 10% to 60% of the fluid inclusion. This corresponds to a salinity between 6–10 wt. % K2SO4 equivalent, or <0.7 molal K2SO4.
Interpretation and discussion
Origin of the quartz lenses
The systematic localisation of the quartz lenses within the dyke cores (Fig. 1) indicates they are genetically linked to the calcite carbonatite and formed in the same stress field, which is consistent with the lack of quartz lenses in the surrounding rocks. The quartz lenses are not intergrown with the calcite indicating they are not conventional magmatic phases.
Both Qz-2 and Qz-3 are believed to be of hydrothermal origin due to the volume for volume replacement texture observed, their characteristic non-luminescent CL emission (Fig. 4) and the presence of fluid inclusions. The Qz-3 Lw + V fluid inclusions ThTOT temperatures point to low hydrothermal temperatures (120–222°C).
The origin of the original quartz (Qz-1 in this study) from the Huanglongpu calcite carbonatites, which accounts for just over 50% of the quartz lenses in our samples overall, has been discussed by Song et al. (Reference Song, Xu, Qi, Zhou, Wang and Kynický2015). They suggested a magmatic origin of the quartz from Si enriched carbonatitic liquids which might have been produced by intensely fractional crystallisation of non-silicate minerals. Bai et al. (Reference Bai, Chen and Jiang2019) expanded on the work of Song et al. (Reference Song, Xu, Qi, Zhou, Wang and Kynický2015) and concluded that quartz precipitated mainly before calcite, which is consistent with the calcite veins locally crosscutting the quartz lenses observed in this study, but must also be reconciled with the fact that quartz lenses only occur within calcite carbonatite dykes. Bai et al. (Reference Bai, Chen and Jiang2019) also favoured a magmatic origin, but they ascribed the coarse, nearly monomineralic quartz grains to a supercritical C–H–O–Si magmatic fluid intermediate between a hydrous melt and an aqueous solution. Our stable oxygen isotope data (Table 4) indicates very small fractionation between quartz and calcite, which is indicative of temperatures in excess of 500°C (Zheng, Reference Zheng1999) and is consistent with a magmatic environment (Table 4). We conclude that the quartz veins formed from a fluid of magmatic origin, but were subsequently reworked by hydrothermal activity.
Conditions of hydrothermal activity
There is no direct measurement of the temperature of Qz-2 and Qz-3 precipitation but the quartz luminescence and the fluid inclusions provide some indications. The brighter luminescence of Qz-1 compared to the later quartz generations indicates a higher temperature for growth. The later quartz generations exhibit low CL emissions, typical of hydrothermal quartz (Rusk and Reed, Reference Rusk and Reed2002; Rusk et al., Reference Rusk, Reed, Dilles and Kent2006). Both Qz-2 and Qz-3 show very similar luminescence (Fig. 4) and on the basis of the literature on non-luminescent hydrothermal quartz precipitation, they have probably precipitated at temperatures below ~450°C.
The CO2-bearing fluid inclusions hosted in Qz-2 and Qz-3 decrepitated upon heating before homogenisation. It is likely that decrepitation only occurs above the temperature of trapping and, if so, the maximum trapping temperature is below the range 225 to 325°C. The homogenisation temperature ThTOT of the Lw + V fluid inclusions hosted in Qz-3 give a temperature range from 120 to 220°C which may be close to the true trapping temperature as the system was experiencing significant fluid flow. This is consistent with the low luminescence from Qz-2 and Qz-3 (Fig. 4) and with the association of hydrothermal bastnäsite-(Ce) with them. Shu and Liu (Reference Shu and Liu2019) estimated that, in the Dalucao carbonatites, hydrothermal bastnäsite-(Ce) formed between 147°C and 323°C while the pervasive hydrothermal bastnäsite-(Ce) stage of the Maoniuping carbonatite deposit also crystallised at low temperature, between 160°C to 240°C (Zheng and Liu, Reference Zheng and Liu2019). Both these studies invoke a fluid with high (SO4)–2 content, alongside Cl– and F–. It should be noted that the similarity in oxygen isotope composition between different quartz generations (Table 4) suggests that the hydrothermal fluid from which they grew had an oxygen composition dominated by interaction with magmatic silicates under similar conditions to those at which the hydrothermal quartz was precipitated.
Possible sources of hydrothermal fluids and their dissolved components
This investigation has demonstrated that the fluid inclusions in the hydrothermal quartz (Qz-2 and Qz-3) are very variable. Where solids are present they vary in both abundance and mineralogy between adjacent inclusions. The ratio of CO2 vapour phase to aqueous fluid is also variable. We also note that the sulfate-rich fluid could not have also transported all the associated cations, as baryte and celestine are nearly insoluble (Blount, Reference Blount1977; Monnin, Reference Monnin1999). We conclude from these lines of evidence that the population of solid-bearing inclusions results from heterogeneous trapping, but the issue of how such a complex, multi-phase fluid came to be present must still be explained.
We note however that similar CO2-bearing fluid inclusions with sulfate solids and opaque crystals hosted in quartz and calcite from a quartz-bearing carbonatite deposit in the north Qinling region were attributed by Song et al. (Reference Song, Xu, Smith, Kynický, Huang, Wei, Li and Shu2016) to trapping of a carbonatite fluid exsolved at subsolidus conditions. Sulfate-rich fluids have also been found in the Maoniuping REE deposit, from the Himalayan orogenic belts, China within a similar carbonatite setting (Xie et al., Reference Xie, Hou, Yin, Dominy, Xu, Tian and Xu2009, Reference Xie, Li, Hou, Cooke, Danyushevsky, Dominy and Shuping Yin2015). They described extremely sulfate-rich melt-fluid inclusions in fluorite and suggested a sulfate-rich supercritical orthomagmatic fluid originated from the unmixing of the carbonatite melt. Alkali-sulfate salt melt inclusions have been identified in carbonatites by Panina and Motorina (Reference Panina and Motorina2008).
There are two difficulties in applying these models to the examples described here. Firstly the heterogeneous character of the inclusions makes it very unlikely that their contents correspond to any single fluid. Secondly, at Dashigou, no melt inclusions or other sulfate-rich phases are present in the early, high temperature Qz-1. Instead, we infer that the variety of the fluid inclusions provides evidence for at least three different types of fluids associated with hydrothermal Qz-2 and Qz-3: a sulfate-rich aqueous fluid, a more dilute aqueous fluid and a carbonic fluid. Although the sulfate content of these hydrothermal fluids might have been derived directly from a carbonatite parent, this cannot be demonstrated, and it is possible that the sulfate is derived from oxidation of reduced sulfur under surface conditions. Acid sulfate geothermal waters are common in many modern volcanic geothermal systems formed at similar temperatures to those inferred for the hydrothermal activity here (Lewis et al., Reference Lewis, Palmer, Sturchio and Kemp1997; Smith et al., Reference Smith, Jenkin, Naden, Boyce, Petterson, Toba, Darling, Taylor and Millar2010). Boiron et al. (Reference Boiron, Moissette, Cathelineau, Banks, Monin and Dubessy1999) described sulfate-rich fluid inclusions associated with retrograde quartz veins from Ouro Fino (Brazil) and pointed out that the composition of the fluid was similar to some present day geothermal systems where feldspars have been destroyed and so cannot buffer the Na/K ratio. However, S isotope data from sulfides and sulfates are consistent with a magmatic sulfur source with δ34S of ~1‰ (Huang et al., Reference Huang, Wang, Nie and Jiang1984; Song et al., Reference Song, Xu, Smith, Kynický, Huang, Wei, Li and Shu2016).
The more dilute fluid present in many Qz-3 fluid inclusions is also consistent with the introduction of a fluid of geothermal origin, as it is common in modern geothermal fields for fluids with different ligand concentrations to occur within the same system (Henley and Hedenquist, Reference Henley, Hedenquist, Henley, Hedenquist and Roberts1986; Lewis et al., Reference Lewis, Palmer, Sturchio and Kemp1997).
The carbonic fluid observed in the majority of the fluid inclusions hosted in Qz-2 and Qz-3 could have resulted from the dissolution of calcite within the carbonatite (Fig. 10, model 1). Nonetheless, CO2-rich fluids have also been commonly described as orthomagmatic fluids resulting from carbonatite magma unmixing (Rankin, Reference Rankin, Linnen and Samson2005; Bühn et al., Reference Bühn, Rankin, Schneider and Dulski2002).
Fig. 10. Potential models for the origin of the fluid inclusions cogenetic with the HREE mineralisation in the calcite carbonatite dykes at the Dashigou open pit.
It is unlikely that metal and sulfate ions now present in Qz-2 inclusions were introduced in a single fluid. It is clear from the varied mineralogy of the inclusions that the proportions of major cations in sulfate inclusions are very variable, and in any case the bulk fluid composition cannot have existed as a single phase under the conditions inferred for the hydrothermal activity, if at all. The Maoniuping REE carbonatite deposit host sulfate-rich multisolid daughter-crystal fluid inclusions (Xie et al., Reference Xie, Li, Hou, Cooke, Danyushevsky, Dominy and Shuping Yin2015; e.g. ADC and ADV fluid inclusions), which behaved differently as they did meet the expectations for a single-phase origin. However, unlike these fluid inclusions, none of the CO2-bearing fluid inclusions at Dashigou homogenised upon heating, which is evidence for heterogeneous trapping.
Model for the hydrothermal alteration at Huanglongpu
Two possible models for the hydrothermal alteration and HREE enrichment of the calcite carbonatites at Dashigou, based on inclusion mineralogy and fluid-inclusion results are presented in Fig. 10. They are intended to demonstrate how a sulfate-rich fluid is able to interact with a source of cations to produce a multi-phase fluid.
Model 1 is based on the reaction of a single acid sulfate fluid with the calcite carbonatite and its hosts. This generates CO2 and precipitate sulfates by the neutralisation of the acid fluid by wallrock reactions (e.g. H2SO4 + CaCO3 = CaSO4 + CO2 + H2O). The sulfate minerals present in the fluid inclusions and in quartz are mainly salts of K (arcanite), Ca (anhydrite), Sr (celestine) and Na (aphthitalite) (Fig. 9), and these cations are readily available within wallrock calcite carbonatite dykes or the surrounding fenite (e.g. H2SO4 + 2KAlSi3O8 = K2SO4 + Al silicates). In this model, local production of CO2 explains why it is associated closely with the sulfate fluid.
Model 2 involves the mixing of two externally-derived fluids, one sulfate-rich and one CO2-rich, with cations carried by the carbonic fluid, and does not explicitly require chemical interaction with the carbonatite or fenite. This model can also result in the precipitation of sulfate salts, but requires cations to be introduced in a carbonic fluid even though they, and the CO2, are already available in the rocks.
Irrespective of the origin of the initial alteration and precipitation of sulfates, the growth of Qz-3 appears to be associated with the introduction of a more dilute fluid to the system and this is also shown in Fig. 10.
While Qz-2 occurs as thread-like infills linked by a cobweb texture, Qz-3 always occurs infilling healed fractures (Fig. 4). We propose a late fracturing event led to the introduction of the more dilute water, leading to Qz-3 precipitation and the less saline fluid inclusions. Loss of CO2 at this stage is likely as aqueous Lw + V and Lw fluid inclusions become common (Fig. 10).
Hydrothermal REE enrichment
The HREE enrichment of the Dashigou carbonatite leading to the uncommon flat REE pattern (Fig. 3) is believed to be due to a combination of factors. Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018) proposed a HREE-enrichment model in which magmatic, HREE-enriched calcite with a relatively flat REE pattern (their fig. 7a) provided a baseline source for the secondary REE mineralisation. The magmatic calcite has lower REE contents than the magmatic REE phases, but it accounts for over 50% of the carbonatite deposit and is more reactive than the REE minerals, making calcite a viable HREE-enriched source. The primary magmatic LREE-rich mineralisation consisted of monazite-(Ce) (e.g. sample HLP15, ΣLREE/(HREE+Y)= 587470, table 4) and fluorocarbonate (bastnäsite-(Ce) and parisite-(Ce)) mineralisation. These early REE minerals show steep, LREE enriched, chondrite-normalised patterns as demonstrated by their high ΣLREE/(HREE + Y) ratio (Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018; fig. 8A–B). The sulfate-rich fluid leached the REE from the magmatic carbonatite without fractionation and then precipitated secondary HREE-enriched phases with flat REE patterns (e.g. britholite-(Ce), Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018; sample HLP10, ΣLREE/(HREE + Y)= 11, fig. 8C-E; table 6). The hydrothermal REE minerals are still Ce-dominant but have much higher HREE content than found typically in carbonatite related systems.
Hydrothermal Qz-2 and Qz-3 grew alongside secondary REE minerals as the carbonatites became enriched in HREE (Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018), suggesting that the fluid inclusions contain information about the fluid responsible for the REE transport and deposition. The associated fluid inclusions have high sulfate contents and Qz-2 and Qz-3 emplacement correlates overall with widespread growth of sulfate minerals and deposition of secondary quartz in the calcite carbonatite dykes.
Rare earth element transport is also likely to be an important factor for HREE-enrichment. Migdisov and Williams-Jones (Reference Migdisov and Williams-Jones2008, Reference Migdisov and Williams-Jones2014) and Migdisov et al. (Reference Migdisov, Williams-Jones, Brugger and Caporuscio2016) identified two categories, ligands leading to REE transportation (Cl– and SO42–) and ligands leading to REE deposition (F–, OH–, CO32–, HCO3– and PO42–). At Dashigou, sulfate is inferred to be the main complexing agent for REE transport from the fluid-inclusion data. The flat REE profile of the hydrothermally altered calcite carbonatites (Fig. 3) is consistent with experimental studies on the REE–SO42– complexes at hydrothermal temperatures from 100°C to 400°C (Migdisov and Williams-Jones, Reference Migdisov and Williams-Jones2008, Reference Migdisov and Williams-Jones2014; Migdisov et al., Reference Migdisov, Williams-Jones, Brugger and Caporuscio2016). These studies agree that sulfate is a non-selective ligand as it does not fractionate the LREE from the HREE. The stability of REE–SO42– complexes is not strongly dependent on atomic number and they have similar formation constants across the lanthanide series. In contrast, HREE–Cl2+ complexes show an overall lower stability than REE-sulfate complexes and as this decreases along the lanthanide series, a chloride-dominated fluid will fractionate the LREE from the HREE. In contrast a sulfate fluid is able to transport the LREE and the HREE equally. One case study on a natural carbonatite system has demonstrated previously the importance of REE sulfate transport at low temperature below 240°C (Zheng and Liu, Reference Zheng and Liu2019). Sulfate transport has also been observed in sedimentary basin systems at low hydrothermal temperature between 100°C and 120°C (Richter et al., Reference Richter, Diamond, Atanasova, Banks and Gutzmer2019).
The chloride content of the Dashigou fluids were not measured but no trace of Cl-bearing salts were observed suggesting only a minor amount of this ligand is present in the fluids. The precipitation of sulfate minerals within the calcite carbonatite dyke as the sulfate fluid encountered Ca, Na and K is inferred to have destabilised the REE–SO42– complexes and triggered the deposition of the hydrothermal REE minerals. While sulfate transport does not account for the HREE enrichment specifically, it plays an important role in linking REE mineralisation to the growth of sulfate minerals.
The final HREE enrichment factor is thought to be the stabilisation of secondary minerals whose crystal structures tend to favour HREE, in particular HREE-enriched fluorocarbonates, uraninite, xenotime-(Y), churchite-(Y) and other minerals which were not always possible to identify; Smith et al. (Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018) also reported britholite-(Ce) and Ca–REE fluorocarbonate mineralisation. These fractionate the HREE out of the fluid they are crystallising from. An additional mechanism that could also have concentrated the HREE is the co-precipitation of hydrothermal gangue LREE enriched phase such as sulfate minerals.
In summary, a HREE-enriched source is probably important for the final HREE enrichment. The late-stage magmatic HREE enrichment of the calcite, the main gangue mineral of the carbonatite, provides a source baseline allowing a potential secondary HREE enrichment (Xu et al., Reference Xu, Campbell, Allen, Huang, Qi, Zhang and Zhang2007; Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018; Bai et al., Reference Bai, Chen and Jiang2019). However, it is also possible that HREE-enrichment is enhanced at the hydrothermal stage. This might have happened indirectly, by selective removal of LREE in a Cl fluid (Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018), although no evidence for such a fluid has been seen, or directly through transport and precipitation of REE. Sulfate-REE complexation facilitates non-selective transport of the REE, and precipitation of REE minerals might then occur in response to precipitation of sulfate minerals. Stabilisation of secondary HREE-enriched minerals in the hydrothermal environment could have selectively concentrated HREE.
Conclusions
The calcite carbonatites at Dashigou can be divided into two groups on the basis of hydrothermal alteration of magmatic minerals, including monazite-(Ce); little-altered calcite carbonatites exhibit a normal carbonatite LREE-enriched REE pattern, but more altered dykes have enhanced levels of HREE and show an unusual flat REE pattern (Fig. 3). These observations suggest that the rocks became enriched in HREE in the course of the hydrothermal alteration which was accompanied by the growth of secondary minerals which favoured incorporation of HREE (Smith et al., Reference Smith, Kynický, Xu, Song, Spratt, Jeffries, Brtnicky, Kopriva and Cangelosi2018). Alteration took place in at least two phases whose conditions are not well-defined but are between 450°C and 120°C. The hydrothermal fluid responsible for the alteration was remarkably sulfate-rich, and the bulk composition of heterogeneously-trapped fluid inclusions can exceed 30 wt.% K2SO4 equivalent. The REE were transported as sulfate complexes and probably precipitated due to the precipitation of sulfate gangue minerals. The simplest model for the process involves interaction of an externally-derived acid sulfate hydrothermal fluid with carbonatite and fenite (Fig. 10). Calcite and silicates neutralised the acidity of the sulfate-rich fluid, generating CO2 and acquiring cations (K, Ca and Na) that precipitated sulfates. Late fracturing was accompanied by renewed quartz precipitation from a more dilute fluid lacking CO2.
Overall, the HREE enrichment of the Dashigou carbonatite is believed to be due to a combination of factors. Late-magmatic HREE enrichment of the calcite, the main mineral of the carbonatite, provided a source baseline. Secondary HREE-enrichment was made possible by a fluid rich in sulfate, a non-selective REE ligand able to transport and concentrate the REE equally, but in complexes that are destabilised by the precipitation of sulfate minerals. Final HREE-enrichment was achieved by the growth of secondary REE minerals which favour HREE, notably HREE-enriched fluorocarbonates, uraninite, xenotime-(Y) and churchite-(Y).
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2019.78.
Acknowledgements
We thank Richard Walshaw and Duncan Hedges for help with the SEM imaging and the EPMA analyses at the University of Leeds. We are grateful to John Craven for the help with the SIMS analyses. Cheng Xu and Song Wei (University of Peking) organised access to the Dashigou open pit and guided us in the field. Finally, thanks to Marie-Camille Caumont from the University of Lorraine, who supervised the Raman spectroscopy analyses. This work was supported by the NERC SoS:RARE project (NE/M01147X/1) and NERC and the British Geological Survey (IMF639/1017).

















 Open access
Open access