



Comorbidity between mental disorders is the rule rather than the exception. Autism and psychosis are two specific conditions that co-occur more frequently than is currently appreciated by clinical services, both as disorders (8–12%) and as traits (25–31%). Traditionally, diagnostic separation of autism and psychosis has been encouraged by the distinct age at onset: early childhood in autism and late adolescence or young adulthood in psychosis. The Research Domain Criteria (RDoC) project addresses this concern, proposing that different symptom expressions might represent age-adjusted variation in shared dispositions. Although psychosis and autism have overlapping traits, such as social–communicative and emotional deficits, and unusual thinking and interests,Reference Hommer and Swedo1 diagnostic hierarchies in current clinical practice view autism and psychosis as mutually exclusive. If a person receives an autism diagnosis as a child, they are less likely subsequently to receive a psychosis diagnosis, even if new symptoms emerge. In fact, DSM-5 and ICD-11 diagnose schizophrenia in the presence of autism only if prominent delusions or hallucinations are present.
Impaired inhibitory processing in autism and psychosis
Human and animal studies of autism and psychosis consistently report common pathophysiology involving impaired inhibitory processing in the brain. For example, although samples are often small, in both autism and psychosis, post-mortem studies report a reduction in neural markers associated with inhibitory processing. One proposed mechanism for this reduction in inhibitory processing involves reduced expression of enzymes GAD65 and GAD67, which are responsible for synthesising gamma-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the human brain. Alternatively, impaired inhibitory processing may be attributed to reduced GABA receptor subunits and/or deficits in a specific subclass of inhibitory interneurons, namely parvalbumin-positive (PV+) interneurons.
Genetic studies and mouse models corroborate this thesis and offer insights into the shared and distinct symptomatology of autism and psychosis. Susceptibility to autism has been linked to genes encoding proteins of the neuroligin–neurexin complex, including CNTNAP2, which are important for inhibitory synaptic development and transmission. Susceptibility to psychosis has been linked to a number of genes, such as NRG1, implicated in the development of cortical PV+ interneurons. Mice with knockout of the CNTNAP2 gene display – among other alterations – a reduced number of PV+ interneurons, which are thought to be linked to core behavioural deficits (e.g. deficits in communication and social behaviour, repetitive behaviours) as well as hyperactivity and epileptic seizures. Similarly, deletion of the NRG1 receptor ErbB4 has been associated with a 30% decrease in the number of PV+ interneurons. Whereas some genetic variations may be more strongly associated with a specific disorder (e.g. CNTNAP2 with autism), genetic studies support the view that GABAergic dysfunction is common to both autism and psychosis, potentially offering shared treatment strategies for specific symptoms and deficits across both disorders. However, even symptomatology that appears superficially similar (such as deficits in social cognition) may be explained by distinct neurobiological mechanisms and be driven by different motivational, emotional and cognitive processes.
This association between psychiatric conditions and abnormal interneuron function is further supported by in vivo studies. Although neural inhibition cannot be measured directly in vivo using non-invasive methods, indirect measures can be acquired to test specific predictions. For example, in both autism and psychosis, changes in gamma oscillations, associated with PV+ interneuron activity, have been measured using electroencephalography (EEG) and magnetoencephalography (MEG). Impaired inhibitory processing has also been reported in autism and psychosis by indexing GABAergic deficits using paired-pulse transcranial magnetic stimulation (TMS), a type of non-invasive brain stimulation that can be used to dissociate inhibition mediated by GABAA and GABAB receptors.
To index inhibitory processing in vivo, an alternative approach involves using proton magnetic resonance spectroscopy (1H-MRS) to quantify the concentration of GABA within a voxel of interest. Although interpreting the functional significance of this technique is not straightforward, 1H-MRS gives a measure of the total concentration of GABA within a localised brain region and is particularly sensitive to ‘unbound’ GABA, which arguably correlates with neurotransmitter and neuromodulator pools of GABA. When applied to patient populations, 1H-MRS reveals either a reduction in GABA levels in autism, or no change, relative to controls. Although comparable observations have been made in psychosis, meta-analyses report no difference in GABA levels relative to controls. Notably, these meta-analyses/reviews include data acquired from a number of different brain regions (e.g. medial prefrontal cortex or other frontal areas, occipital, parietal and temporal cortices, striatum, cerebellum), limiting our ability to assess the relevance of anatomy to these 1H-MRS measures of GABA. Moreover, some studies report higher GABA levels, which may reflect differences in exposure to psychopathology, duration of illness, pharmacological treatment or simply differences in the source of the GABA quantified using 1H-MRS signal. Despite these challenges, 1H-MRS has the potential to provide in vivo insight into inhibitory dysregulation in both autism and psychosis, if studies are conducted with sufficient statistical power and data quality, along with appropriate control for potentially confounding variables.
Although a chemical impairment in GABA does not necessitate a functional impairment in inhibition, taken together, post-mortem studies, animal models, in vivo studies and clinical investigations do support the view that inhibitory processing is impaired in both autism and psychosis. Yet, exceptions to this general phenomenon must be critically appraised. One important factor to consider are the changes in inhibitory processing across development. Notably, GABA switches from an excitatory to an inhibitory neurotransmitter early in development, with continued maturation of the GABAergic system late into adolescence. For example, given that interneuron diversity is determined during development through an interplay between genetic and environmental factors, comparable GABAergic dysfunction during development and adulthood may affect the structure and function of neural circuits in profoundly different ways. Similarly, developmental age may be a crucial factor to consider when measuring inhibitory processing and seeking to explain differences and inconsistencies across studies. Finally, autism and psychosis both co-occur with epilepsy, a third disorder characterised by major disruption to inhibitory processing.
What are the implications of modified inhibitory function?
In the healthy brain, excitation and inhibition (E/I) are balanced at both a local and global level, a consequence of homeostatic processes that ensure neuronal excitability is maintained within a narrow dynamic range.Reference Barron, Vogels, Behrens and Ramaswami2 The E/I balance thus accounts for the temporal precision of neural computation, where signal propagation is rapidly quenched by inhibition. Although the E/I balance is regularly disturbed during new learning, experimental evidence and theoretical modellingReference Vogels, Sprekeler, Zenke, Clopath and Gerstner3 indicates a critical role for inhibitory synaptic potentiation in restoring and maintaining this balance, potentially via PV+ interneurons. At a behavioural and cognitive level, similar homeostatic inhibitory mechanisms may be engaged in habituation and suppression of unattended stimuli.Reference Barron, Vogels, Behrens and Ramaswami2
When subtle disturbances to the E/I balance are not corrected by homeostatic mechanisms, the consequences are likely far-reaching: small changes in signal gating can be amplified by unstable neural networks, resulting in profound changes to cognition. At the extreme, near persistent E/I imbalance may cause epilepsy – a disorder that has prevalence rates of 22% in autism and 5.6% in psychosis. More subtle perturbations in the E/I balance are thought to account for cognitive impairments observed in autism and psychosis (i.e. impaired masking of irrelevant perceptions, memories and behaviours). Such phenotypes can be mimicked in theoretical models and in the healthy adult brain if GABA levels are temporarily reduced using transcranial direct current stimulation (tDCS), which induces spontaneous memory expression and memory interference.Reference Koolschijn, Emir, Pantelides, Nili, Behrens and Barron4 However, given that the E/I balance dynamically fluctuates, the effect of impaired inhibitory processing likely depends on neurodevelopmental stage and contextual factors.
Furthermore, the precise neural pathway affected by reduced inhibition may lead to changes in perception and memory via overweighting either of sensory input or of prior expectation. For example, deficits in autism may be explained by weak priors and overweighting of bottom-up sensory information. This may result in hypersensitivity, taking the form of sensory overload and in turn lead to an impaired capacity to adequately update subsequent predictions, resulting in behavioural rigidity. Positive psychotic symptoms, on the other hand, may be attributed to overly strong top-down priors, with delusions and hallucinations described as a prediction error generated via a mismatch between predictions generated from prior knowledge and from received sensory input. Using a Bayesian framework that rests on the use of hierarchical generative models, the relative weight, or ‘precision’, ascribed to sensory evidence and prior expectation may explain distinct symptoms in autism and psychosis (e.g. delusions and hallucinations versus sensory overload and repetitive behaviours), but also account for shared symptoms reported across both disorders (e.g. difficulties with social–emotional and face processing; social withdrawal) (Fig. 1). At a physiological level, the relative weighting of sensory evidence and prior expectation may be set by the precise balance between neural excitation and inhibition at different levels in the cortical hierarchy. In particular, inhibitory interneurons that provide feedforward inhibition (such as PV+ interneurons) are thought to exert gain control over incoming signals, whereas vasoactive intestinal polypeptide-expressing (VIP+) interneurons, which mediate disinhibition of pyramidal cells by targeting other inhibitory interneurons, may determine the weight of top-down priors. Impaired inhibitory processing may thus explain symptoms attributed to both strong and weak priors. Accordingly, the precise symptomatology observed in autism and psychosis could map onto deficits in a particular subclass of inhibitory interneuron, each of which is characterised by a distinct developmental trajectory.
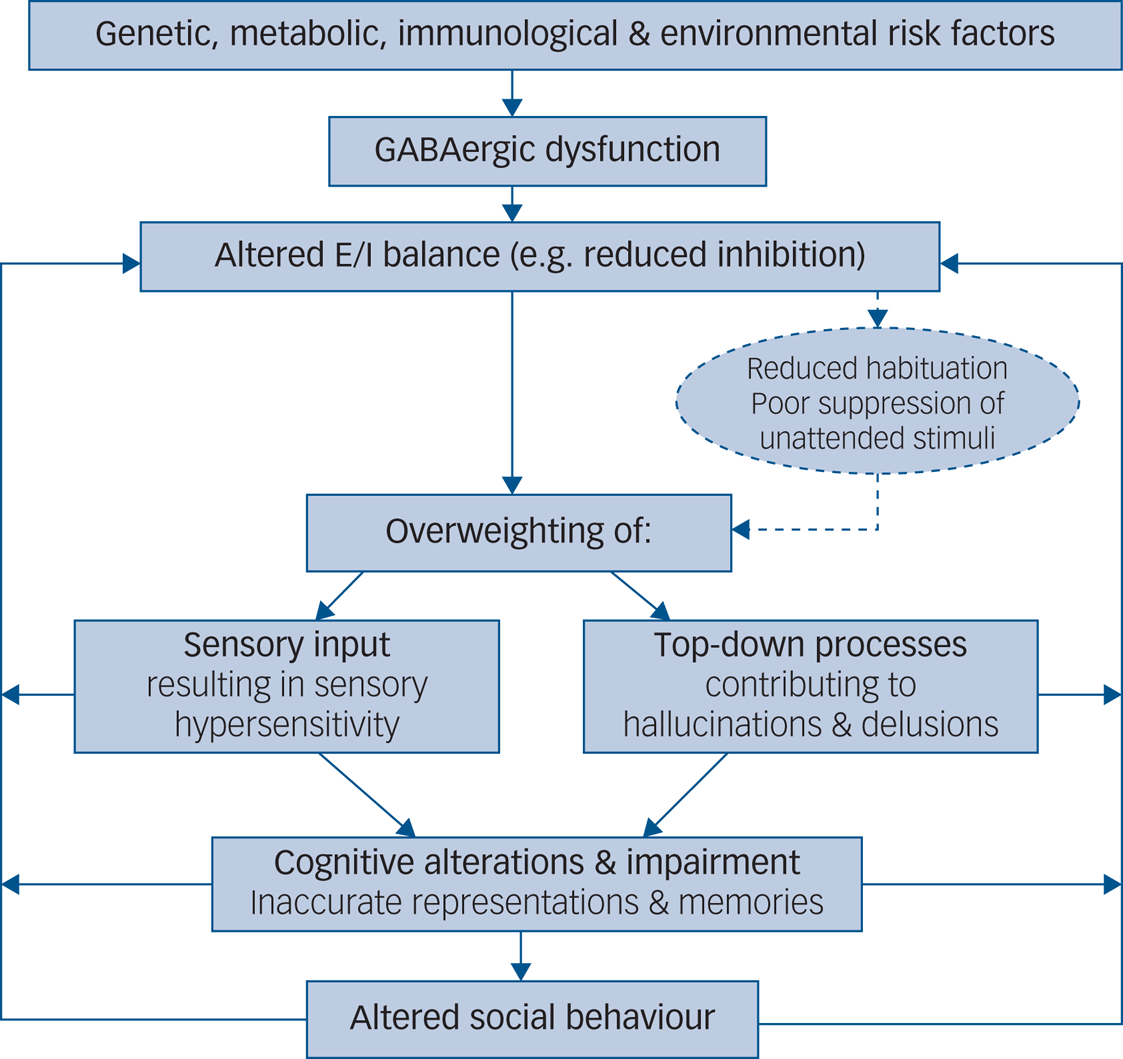
Fig. 1 Aetiological model of symptomatology in autism and psychosis elicited by GABAergic dysfunction. E/I, excitation/inhibition.
Pharmacological implications and other challenges
For both autism and psychosis, it is crucial that we develop new treatments that target early pathophysiological mechanisms and elements of the phenotype that we are currently unable to manage. On the basis of findings from clinical research studies, the GABAergic system has been targeted with some potential for improving clinical symptoms,Reference Foss-Feig, Adkinson, Ji, Yang, Srihari and McPartland5 especially social cognition. For example, GABAA and GABAB receptor agonists (clonazepam, baclofen) have yielded promising behavioural effects in mouse models in autism, reversing social deficits. Similarly, peripubertal diazepam administration in rats reduces hyperactivity and anxiety in models of schizophrenia, via reduced spontaneous firing rates in the amygdala and prevention of a hyperdopaminergic state. Although animal-modelling studies have helped to establish the neural mechanisms underlying the pathogenesis of both disorders, the full complexity of psychiatric disorders can rarely, if ever, be modelled, limiting the predictive power for clinical trials performed in humans.
By contrast, studies in humans suffer from other challenges, such as difficulties controlling for confounding factors and limited statistical power. Nevertheless, some pharmacological agents show promising results from clinical observations. For example, bumetanide (a diuretic that reinforces GABAergic inhibition) can improve emotional face processing in adolescents with autism. Furthermore, GABAA receptor agonists may have particular value in treating catatonia – a state of immobility and near-unconsciousness – which is another common feature in both autism and psychosis. However, paradoxical reactions (e.g. increasing anxiety and aggression) have been observed in some individuals with autism treated with diazepam.
In addition to pharmacological manipulations, a number of other alternative treatments have been proposed that involve modulating cortical excitability. For example, insulin-like growth factor 1 (IGF-1) restores neuronal plasticity by reducing cortical GABA levels, and was found to reduce social impairment and repetitive behaviours in individuals presenting with autism symptoms. Lastly, certain neurosteroids may be used to enhance GABAA receptor function and may be effective in subgroups of children with autistic traits.
Conclusions
Co-occurrence of autism and psychosis traits is a significant clinical problem, specifically in the early stages of illness, when there is significant diagnostic ambiguity. Both disorders appear to be characterised by GABAergic alterations, which might be associated with cognitive deficits such as memory impairment or increased memory interference. Impaired inhibitory processing may further affect other, more diagnosis-specific, symptoms such as sensory hypersensibility and positive psychotic symptoms. A number of therapeutic avenues have been tested that show varying degrees of promising results, but what is missing from the literature is an exploration of patients presenting with both clinically relevant autism and psychosis to explore shared and distinct disease-related and therapeutic mechanisms. Thus far, it has not been possible to integrate results from different methods into a coherent picture.
Outlook
In humans, recent advances in in vivo neuroimaging include the development of functional 1H-MRS, 1H-MRS imaging and more accurate measures of GABA with ultra-high-field imaging. When combined with other imaging modalities and techniques, such as functional magnetic resonance imaging, TMS and tDCS, neuronal inhibition may be assessed and manipulated, providing a means to relate GABA levels to neural mechanisms that underlie cognitive traits. These techniques thus provide a foundation for establishing more nuanced measures of the pathophysiology underlying phenotypes attributed to E/I imbalance, and may be applied as diagnostic tools across neurodevelopment. If combined with clinical studies that assess patients with homogeneous symptoms as opposed to a common diagnosis, these tools may reveal unique insight into the shared pathophysiology underlying autism and psychosis. In parallel, advances in mouse genetics and optical imaging can characterise the microcircuit mechanism, including the contribution of different interneuron subtypes.
Together these measures have the potential to establish stratification markers across patients diagnosed with autism and psychosis, to tailor treatment in a manner that redefines contemporary diagnosis. Investigating inhibitory processing may therefore help establish whether autism and psychosis should be understood as separate disorders that sometimes co-occur, or a unitary condition where shared risk factors and pathogenetic mechanisms are modulated by neurodevelopment.
Author contributions
K.H. drafted the manuscript and all authors contributed substantially to the conception and revision of the work. All authors approved the final version of the manuscript.
Declaration of interest
M.R.B. reports personal fees from the Medical Defence Union, from Oxford University Press, and from Cambridge University Press, outside the submitted work.
ICMJE forms are in the supplementary material, available online at https://doi.org/10.1192/bjp.2020.163.




eLetters
No eLetters have been published for this article.