


Introduction
One of the best-understood and most studied learning paradigms is classical cued fear conditioning where a neutral stimulus is repeatedly paired with an aversive unconditioned stimulus (US) such as an electric shock. Over time the neutral stimulus alone elicits a learnt or conditioned fear response that resembles innate or unconditioned responses. Repeated presentation of the unreinforced conditioned stimulus (CS) leads to a decrease of the acquired fear response or extinction. A key brain region for the acquisition of conditioned fear is the amygdala (LeDoux, Reference LeDoux2000) whereas extinction has been related to the inhibition of amygdalar activation by the prefrontal cortex (Herry et al. Reference Herry, Ferraguti, Singewald, Letzkus, Ehrlich and Lüthi2010). In post-traumatic stress disorder (PTSD) and other anxiety disorders the enhanced acquisition and delayed extinction of fear responses are viewed as a major aetiological mechanism (Bremner et al. Reference Bremner, Vermetten, Schmahl, Vaccarino, Vythilingam, Afzal, Grillon and Charney2005; Delgado et al. Reference Delgado, Olsson and Phelps2006; Rauch et al. Reference Rauch, Shin and Phelps2006; Blechert et al. Reference Blechert, Michael, Vriends, Margraf and Wilhelm2007). Recent evidence further suggests that insufficient prefrontal activation during extinction might be important for the maintenance of fear symptoms and stimulation of these brain areas could have therapeutic effects (Amano et al. Reference Amano, Unal and Paré2010). Anxiety disorders such as PTSD are also characterized by alterations in the function of the hypothalamic–pituitary–adrenal (HPA) axis to a hypoactive mode with reduced basal and stress-related cortisol levels as well as increased suppression after the dexamethasone-suppression test (Yehuda, Reference Yehuda2006; Wessa & Rohleder, Reference Wessa and Rohleder2007).
The heritability of the components of fear conditioning has been estimated at 35–45% (Hettema et al. Reference Hettema, Annas, Neale, Kendler and Fredrikson2003) and several genes involved in the regulation of plasticity and emotional reactivity have been found associated with the conditionability of fear responses (Lonsdorf et al. Reference Lonsdorf, Weike, Nikamo, Schalling, Hamm and Ohman2009). Besides altered fear conditioning, anxiety disorders such as PTSD are also characterized by a dysfunction of the HPA axis, which is regulated by corticotropin-releasing hormone (CRH) and glucocorticoids. They have been implicated in fear conditioning in animal models as well as in PTSD in humans (de Kloet et al. Reference de Kloet, Vermetten, Geuze, Lentjes, Heijnen, Stalla and Westenberg2008; Rodrigues et al. Reference Rodrigues, LeDoux and Sapolsky2009). For example, CRH injection into the central nucleus of the amygdala and CRH injection into the medial prefrontal cortex both result in increased anxiety-related behaviour (Timpl et al. Reference Timpl, Spanagel, Sillaber, Kresse, Reul, Stalla, Blanquet, Steckler, Holsboer and Wurst1998), while mice lacking the CRH receptor (CRHR1) gene display reduced anxiety-related behaviour (Müller et al. Reference Müller, Zimmermann, Sillaber, Hagemeyer, Deussing, Timpl, Kormann, Droste, Kühn, Reul, Holsboer and Wurst2003). In PTSD, elevation of CRH was found in the cerebrospinal fluid, indicating a possible role in altered brain activity (Baker et al. Reference Baker, West, Nicholson, Ekhator, Kasckow, Hill, Bruce, Orth and Geracioti1999; Jaferi & Bhatnagar, Reference Jaferi and Bhatnagar2007; de Kloet et al. Reference de Kloet, Vermetten, Geuze, Lentjes, Heijnen, Stalla and Westenberg2008) in fear learning. These observations strengthen the assumption of a tight connection between the amygdaloid CRH system and HPA functioning in fear conditioning as well as in stress-related disorders, such as PTSD, in humans.
The glucocorticoid receptor (GR) is a main regulator of the HPA axis and has been shown to influence endocrine and behavioural measures of fear in various animal studies (Bremner et al. Reference Bremner, Licinio, Darnell, Krystal, Owens, Southwick, Nemeroff and Charney1997; de Kloet et al. Reference de Kloet, Vreugdenhil, Oitzl and Joëls1998). Pharmacological GR (NR3C1) manipulations, for example, of amygdaloid GRs were shown to modulate processes of fear conditioning (Yang et al. Reference Yang, Chao and Lu2006) and to be related to the establishment of fear memories (e.g. Oitzl et al. Reference Oitzl, Fluttert, Sutanto and de Kloet1998), and to facilitated extinction of conditioned fear (Yang et al. Reference Yang, Chao and Lu2006). GR receptors in the prefrontal cortex might be related to a failure to extinguish established fear responses (Tronel & Alberini, Reference Tronel and Alberini2007) and to an enhancement of emotional memory consolidation, suggesting that these effects reflect an interaction of the medial prefrontal cortex and the basolateral amygdala (Roozendaal et al. Reference Roozendaal, McReynolds, Van der Zee, Lee, McGaugh and McIntyre2009).
However, it is not known if the reported changes in brain activity during fear conditioning and the altered HPA axis activity seen in PTSD patients reflect consequences of the traumatic event or might be interpreted as predisposing vulnerability factors increasing the susceptibility for the disorder. The latter has been proposed in a study that examined firefighters before the experience of traumatic events. Reduced extinction of an aversively conditioned corrugator electromyographic response predicted more than 30% of the variance of PTSD symptoms following trauma (Guthrie & Bryant, Reference Guthrie and Bryant2006). A twin study conducted with combat veterans and their non-combat-exposed sibling revealed that structural changes in the brain are pre-existing familial vulnerability factors (Gilbertson et al. Reference Gilbertson, Shenton, Ciszewski, Kasai, Lasko, Orr and Pitman2002; Pitman et al. Reference Pitman, Gilbertson, Gurvits, May, Lasko, Metzger, Shenton, Yehuda and Orr2006). As a formal genetic study attributes moderate heritability to all components of the fear-conditioning process (Hettema et al. Reference Hettema, Annas, Neale, Kendler and Fredrikson2003), it is of interest to identify the genetic variations responsible at the molecular level. The identified genes could be regarded as vulnerability factors making subjects more susceptible for stress-related disorders.
We used an imaging genetics approach in two independent samples (sample 1: n=60, sample 2: n=52) of healthy individuals to investigate the influence of genetic variation of HPA axis-related genes [CRH receptor 1 (CRHR1), GR (NR3C1)] on the acquisition and extinction of cued fear responses. The products of both genes are among the most important molecules of the HPA axis and were repeatedly reported to be involved in processes of fear conditioning in animal studies (CRHR1, e.g. Radulovic et al. Reference Radulovic, Rühmann, Liepold and Spiess1999, Kikusui et al. Reference Kikusui, Takeuchi and Mori2000, Otagiri et al. Reference Otagiri, Wakabayashi and Shibasaki2000; NR3C1, e.g. Cordero et al. Reference Cordero, Kruyt, Merino and Sandi2002, Yang et al. Reference Yang, Chao and Lu2006, Kohda et al. Reference Kohda, Harada, Kato, Hoshino, Motohashi, Yamaji, Morinobu, Matsuoka and Kato2007, Kolber et al. Reference Kolber, Roberts, Howell, Wozniak, Sands and Muglia2008), which was the reason to include these two in the present study. In addition, the first genetic findings for PTSD exist for the Bcl1 variant (rs41423247) of the GR gene. The GG genotype of this single nucleotide polymorphism (SNP) was associated with low basal cortisol levels in PTSD (Bachmann et al. Reference Bachmann, Sedgley, Jackson, Gibson, Young and Torpy2005) and with more long-term traumatic memories and higher PTSD symptom scores (Hauer et al. Reference Hauer, Weis, Papassotiropoulos, Schmoeckel, Beiras-Fernandez, Lieke, Kaufmann, Kirchhoff, Vogeser, Roozendaal, Briegel, de Quervain and Schelling2011). We employed functional magnetic resonance imaging (fMRI) and used a linear regression approach (according to Ressler et al. Reference Ressler, Mercer, Bradley, Jovanovic, Mahan, Kerley, Norrholm, Kilaru, Smith, Myers, Ramirez, Engel, Hammack, Toufexis, Braas, Binder and May2011), which permitted the comparison of groups with respect to degree of genetic variation with and without minor alleles. We examined amygdala activation as an indicator for acquisition and prefrontal activation as an indicator for extinction and also included connectivity analyses. To control for successful conditioning, we assessed skin conductance responses (SCRs) and self-report measures.
Method
Participants
A total of sixty persons [38 male, 22 female, mean age 21.25 (s.d.=3.02, range 19–37) years, all right-handed] recruited in schools for ambulance rescue workers as part of a longitudinal study were examined in study 1 (sample 1) and 52 persons [32 male, 20 female, mean age 22.27 (s.d.=3.67, range 18–37) years, all right-handed] from the same population were recruited for study 2 (sample 2). Participants with past traumatic events as assessed by the German version of the Posttraumatic Diagnostic Scale (Griesel et al. Reference Griesel, Wessa and Flor2006) were excluded. All participants were medication free and had no physical or mental disorders. The study was approved by the Ethics Committee of the Medical Faculty Mannheim, Heidelberg University, and written informed consent was obtained from all participants, who were paid for participation.
Psychological assessment
Standardized clinical assessment with the Structured Clinical Interview for DSM-IV Axis I (First et al. Reference First, Spitzer, Gibbon and Williams1997b ) and Axis II (First et al. Reference First, Gibbon, Spitzer, Williams and Benjamin1997a ) was performed to exclude persons with mental disorders. Psychological assessment was identical for all subjects and included the Center for Epidemiological Studies Depression Scale (Radloff, Reference Radloff1977), the trait section of the State-Trait Anxiety Inventory (Spielberger et al. Reference Spielberger, Gorsuch and Lushene1970), the Perceived Stress Scale (Cohen et al. Reference Cohen, Kamarck and Mermelstein1983) and the Childhood Trauma Questionnaire (Bernstein & Fink, Reference Bernstein and Fink1998) (see Table 1). Since we did not find any differences in scores on the psychological measures depending on the genotype, we did not focus on these data in the Results section.
Table 1. Clinical characteristics of the participants
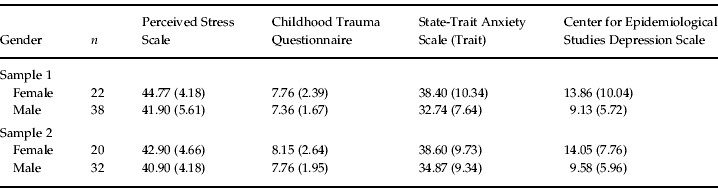
Data are given as mean (standard deviation).
SCR and self-report data
The SCRs were recorded from two electrodes placed on the thenar and hypothenar eminence of the participants' right hand using a sampling rate of 16 Hz and a VarioPort recording system (BECKER MEDITEC, Germany). Data analysis was performed using EDA-PARA software (F. Schäfer, Germany) and followed the guidelines of Fowles et al. (Reference Fowles, Christie, Edelberg, Grings, Lykken and Venables1981) . Trials were visually inspected for artifacts and SCR amplitudes were quantified as the maximum response in the time window of 1–4 s (first interval response) and 5–9 s (second interval response; Prokasy & Ebel, Reference Prokasy and Ebel1967) after stimulus onset and were measured in microSiemens (μS). SCR amplitudes below 0.05 μS were classified as zero responses. SCR data were normalized using a logarithmic [log (1+SCR)] transformation.
After each conditioning phase, participants verbally rated the emotional valence and arousal of the CSs (1=very calm to 9=very arousing, 1=very pleasant to 9=very unpleasant) as well as the CS–US contingency (1=no CS–US contingency to 9=perfect CS–US contingency). All auditory or visual instructions for the experimental procedure were standardized. Communication was realized via headphones with attached microphones.
SCRs and self-report data were analysed separately using Predictive Analytic Software (PASW) for Windows, version 18.0.1 (SPSS Inc., USA). Both SCRs and self-reports showed successful conditioning and extinction in samples 1 and 2. Since differences in the genotype groups could not be observed for either measure, we present only the fMRI analyses in the Results section.
DNA extraction, selection of SNPs and genotyping
Venous blood samples were obtained from all participants. Genomic DNA was isolated with the QIAamp DNA extraction kit (www.qiagen.com/). For genetic characterization of the NR3C1 and CRHR1 genes, we selected SNPs with potential functionality from the literature as well as tagging SNPs from the HapMap database and literature. For the NR3C1 gene, we chose the potentially functional variants N363S (rs6195) (e.g. Jewel & Cidlowski, Reference Jewell and Cidlowski2007), BclI (rs41423247) (Stevens et al. Reference Stevens, Ray, Zeggini, John, Richards, Griffiths and Donn2004) and Tth111I (rs10052957) (Rosmond et al. Reference Rosmond, Chagnon, Chagnon, Pérusse, Bouchard and Björntorp2000). Tagging SNPs for the NR3C1 gene were selected by a blockwise strategy from HapMap data, using haplotypes above 5% frequency in HaploView (e.g. Barrett et al. Reference Barrett, Fry, Maller and Daly2005). NR3C1 transcript NM_000176, which covers 123.8 kbp on chr5, contained only one large haplotype block, which is tagged by four haplotype tagging SNPs, i.e. rs33389, rs4986593, rs10482672 and rs190488 (HapMap Rel 16c, NCBI B34 assembly, dbSNP b124). Tagging SNPs for the CRHR1 gene, rs1876831 and rs242938, were selected from the literature, based on detailed linkage disequilibrium information of CRHR1 SNPs from several publications (e.g. Treutlein et al. Reference Treutlein, Kissling, Frank, Wiemann, Dong, Depner, Saam, Lascorz, Soyka, Preuss, Rujescu, Skowronek, Rietschel, Spanagel, Heinz, Laucht, Mann and Schumann2006; Wassermann et al. Reference Wasserman, Wasserman, Rozanov and Sokolowski2009; Grabe et al. Reference Grabe, Schwahn, Appel, Mahler, Schulz, Spitzer, Fenske, Barnow, Lucht, Freyberger, John, Teumer, Wallaschofski, Nauck and Völzke2010; Nelson et al. Reference Nelson, Agrawal, Pergadia, Wang, Whitfield, Saccone, Kern, Grant, Schrage, Rice, Montgomery, Heath, Goate, Martin and Madden2010). The tagging markers were shown to be sufficient to provide information on the brain activation during fear conditioning and captured 66% of the markers of the NR3C1 and 45% of SNPs of the CRHR1 genes according to HapMap release 24 (threshold r 2 ⩾ 0.8, allele frequency ⩾5%). Genotyping was performed using an Applied Biosystems Prism 7900HT RealTime PCR system. Table S1 (see Supplementary material) lists the IDs of Applied Biosystems Assays-on-DemandTM and Primer/Probe sequences of Assays-by-Design. Allele frequencies and genotype counts for the polymorphisms that contributed significantly to the association signals in the two samples are shown in Table 2.
Table 2. Allele frequencies and genotype counts for the polymorphisms that contributed significantly to the association signals in the two samples

HWE, Hardy–Weinberg equilibrium.
a Test for deviation from HWE (Wigginton et al. Reference Wigginton, Cutler and Abecasis2005).
Design of the fear-conditioning experiment
Perception and pain threshold as well as pain tolerance (three repetitions in an ascending series) were assessed for the calculation of the intensity of the US, which was painful electrical stimulation (Digitimer, UK) at the thumb of the right hand set to a level of 80% of pain tolerance. Two different geometric shapes (a square and a rhombus) in different colours (blue or yellow) were presented as cue stimuli with colours and shapes counterbalanced across subjects. During habituation 10 CS+ (neutral stimulus that would later be followed by the aversive US), 10 CS− (neutral stimulus that would later be followed by the absence of the US) and four US were presented in random order. The acquisition was divided into two phases, each consisting of nine CS+ paired with the US, nine CS+ not paired with the US and 18 CS− (never paired with the US) in random order. The extinction phase consisted of 18 CS+ and 18 CS− trials. The CS+ was reinforced in 50% of the trials with a shock duration of 2.7 s before the end of the CS+ projection (see Fig. 1). After each phase the participants rated valence and arousal on a self-assessment manikin. CS–US contingency was rated on a scale from 0 (US will not follow the CS) to 100 (US will definitely follow the CS).
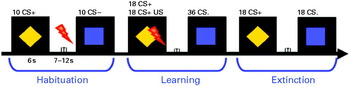
Fig. 1. Fear-conditioning paradigm. CS+, Neutral conditioned stimulus (yellow rhombus) later followed by an aversive unconditioned stimulus (US), an electric shock (red flash); ITI, inter-trial interval; CS−, neutral stimulus (blue square) later followed by the absence of the US.
Statistical analysis
Effects of genetic markers on amygdala activation during fear conditioning and medial prefrontal activation during fear extinction were assessed using a linear regression approach (Ressler et al. Reference Ressler, Mercer, Bradley, Jovanovic, Mahan, Kerley, Norrholm, Kilaru, Smith, Myers, Ramirez, Engel, Hammack, Toufexis, Braas, Binder and May2011). SNPs were coded using an additive model, i.e. using the number of minor alleles of the respective markers as predictors. Age and gender were also tested but revealed no significant effects and were thus not included in the further analyses. For the first sample a model was built including all of the above-mentioned SNPs as predictors. For the second sample the same model was tested, but this time omitting the markers that had not obtained a nominally significant p value in the first sample. In these models the minor alleles of the nominally significant markers uniformly showed up as associated alleles. In order to build a summary score of all SNPs, genotype was coded by the total number of minor alleles across all markers. Scores were built across both genes and for the NR3C1 gene separately. This permitted the comparison of groups with respect to the degree of genetic variation with and without minor alleles for brain activation (see the legend of Fig. 2 for details). All significance levels were set to p<0.05.

Fig. 2. (a) Increased activity in the left amygdala elicited by CS+ versus CS- in the first half of the acquisition as a function of NR3C1 genotype, coded 0 for no minor allele (
![]() ), 1 for one or two minor alleles (□), 2 for more than two minor alleles (
), 1 for one or two minor alleles (□), 2 for more than two minor alleles (
![]() ) (for sample 1, group 0: n=26, group 1: n=26, group 2: n=8; for sample 2, group 0: n=16, group 1: n=30, group 2: n=6). CS+, neutral conditioned stimulus later followed by the aversive unconditioned stimulus (US); CS-, neutral stimulus later followed by the absence of the US. Values are means, with 95% confidence intervals (CIs) represented by vertical bars. * p<0.05. (b) Genotype-dependent differential activation of the prefrontal cortex during extinction involving CRHR1 and NR3C1 genotypes (coded 0 for no minor allele (
) (for sample 1, group 0: n=26, group 1: n=26, group 2: n=8; for sample 2, group 0: n=16, group 1: n=30, group 2: n=6). CS+, neutral conditioned stimulus later followed by the aversive unconditioned stimulus (US); CS-, neutral stimulus later followed by the absence of the US. Values are means, with 95% confidence intervals (CIs) represented by vertical bars. * p<0.05. (b) Genotype-dependent differential activation of the prefrontal cortex during extinction involving CRHR1 and NR3C1 genotypes (coded 0 for no minor allele (
![]() ), 1 for one minor allele (□), 2 for more than one minor allele (
), 1 for one minor allele (□), 2 for more than one minor allele (
![]() ) (for sample 1, group 0: n=12, group 1: n=24, group 2: n=23; for sample 2, group 0: n=13, group 1: n=20, group 2: n=19). BA, Brodmann area. Values are means, with 95% CIs represented by vertical bars. * p<0.05. (c) T maps revealing increases in functional coupling for the contrasts between genotype group 2 versus groups 0 and 1 during the early acquisition phase (left panel) and genotype-dependent functional coupling during early acquisition between the left amygdala and prefrontal cortex (right panel). Group 0, no minor allele (
) (for sample 1, group 0: n=12, group 1: n=24, group 2: n=23; for sample 2, group 0: n=13, group 1: n=20, group 2: n=19). BA, Brodmann area. Values are means, with 95% CIs represented by vertical bars. * p<0.05. (c) T maps revealing increases in functional coupling for the contrasts between genotype group 2 versus groups 0 and 1 during the early acquisition phase (left panel) and genotype-dependent functional coupling during early acquisition between the left amygdala and prefrontal cortex (right panel). Group 0, no minor allele (
![]() ); group 1, one minor allele (□); group 2, more than one minor allele (
); group 1, one minor allele (□); group 2, more than one minor allele (
![]() ); AU, arbitrary units at the target-region peak voxels. Values are means, with 95% CIs represented by vertical bars. (d) t Maps revealing increases in functional coupling for the contrasts between the genotype groups during the extinction phase (left panel) and coupling strength for the extinction phase between the left prefrontal cortex and left amygdala (right panel). Group 0, no minor allele (
); AU, arbitrary units at the target-region peak voxels. Values are means, with 95% CIs represented by vertical bars. (d) t Maps revealing increases in functional coupling for the contrasts between the genotype groups during the extinction phase (left panel) and coupling strength for the extinction phase between the left prefrontal cortex and left amygdala (right panel). Group 0, no minor allele (
![]() ); group 1, one minor allele (□); group 2, more than one minor allele (
); group 1, one minor allele (□); group 2, more than one minor allele (
![]() ). Values are means, with 95% CIs represented by vertical bars.
). Values are means, with 95% CIs represented by vertical bars.
fMRI
Neuroimaging was performed during classical aversive delay cued conditioning in a 1.5 T Magnetom Vision scanner (Siemens Medical Solutions, Germany). Contiguous transversal T2*-weighted echo-planar images (EPI) with blood oxygenation level-dependent (BOLD) contrast were used (echo time 45 ms, flip angle 90°) that covered the whole brain (35 slices, slice thickness 3 mm, 1 mm gap, field of view=220×220 mm2, 64×64 matrix). The effective repetition time was 3.77 s per volume. A total of 560 volumes were recorded. Statistical parametric mapping (SPM) software was used for image processing and analysis (SPM2, http://www.fil.ion.ucl.ac.uk/spm/). The images were slice-time corrected for phase shift during volume acquisition, realigned to the first image for motion artefacts, spatially normalized to a standard EPI template, spatially smoothed with an isotropic Gaussian kernel with a full width at half-maximum of 10 mm, and temporal high-pass filtered (cut-off 128 s). Specific effects were tested by applying linear contrasts to the parameter estimated for each event. Contrast images of interest were calculated for each subject, and the resulting contrast images were entered into a second-level random-effects analysis to produce group results (one-sample t test). For each phase (early acquisition, late acquisition, and extinction) contrasts between CS+ and CS−, i.e. CS+>CS−, were calculated. We used a threshold of p<0.001 for the entire brain (uncorrected, extent threshold k=5 voxels). Additionally, according to our a priori hypothesis that alterations of amygdala activation during acquisition and prefrontal cortex activation during extinction should be associated with HPA axis-related genes (e.g. Yang et al. Reference Yang, Chao and Lu2006; Tronel & Alberini, Reference Tronel and Alberini2007), we adopted a region-of-interest (ROI) approach with small volume correction [p<0.05, family-wise error (FWE) corrected], also corrected for the number of ROIs. Regions were defined using the MARINA software package (http://www.bion.de/). To detect the association between genotype and fMRI activation in the amygdala and in the prefrontal cortex on a voxel-by-voxel basis, the contrast images of all subjects (percentage signal change of CS+ versus CS−) were included in a regression analysis with SPM.
Functional coupling analysis
Connectivity analyses were performed to determine genotype-dependent changes in functional coupling between the amygdala, the hippocampus and the prefrontal cortex that are related to the acquisition and extinction of the learned response. Functional coupling between seed regions (spheres with radius=6 mm, coordinates based on the current sample for acquisition phase: x, y, z: −24, −6, −18; for extinction phase: x, y, z: −36, 60, 0) and target regions was determined for the early acquisition phase and the extinction phase separately using standard SPM methods. For the early acquisition phase the left amygdala served as the seed region and the prefrontal cortex as the target region. For the extinction phase, the left prefrontal cortex [Brodmann area (BA) 10] (seed region) and left amygdala (target region) were used for the functional coupling analysis.
From both seed regions fMRI time series were extracted and used as regressors in a subsequent single-subject analysis, where the movement-related covariates were additionally included. These contrasts were used to carry out a random-effects analysis to determine functional coupling between groups assigned to different genotypes using two-sample t tests.
We report T values small-volume corrected using the following ROIs: prefrontal cortex (coordinates x=0, y=52, z=−3; sphere with radius=9 mm, see Heinz et al. Reference Heinz, Wrase, Kahnt, Beck, Bromand, Grüsser, Kienast, Smolka, Flor and Mann2007) and left amygdala (created using the MARINA software package). Peak activations were correlated with subjective and endocrinological variables using Pearson correlations.
Results
Acquisition- and extinction-related significant BOLD signal differences between CS+ and CS− were found in a number of brain regions (see Supplementary Table S2 and Fig. 2).
A linear regression (Ressler et al. Reference Ressler, Mercer, Bradley, Jovanovic, Mahan, Kerley, Norrholm, Kilaru, Smith, Myers, Ramirez, Engel, Hammack, Toufexis, Braas, Binder and May2011) including all tested variants yielded highly significant associations of the minor alleles of several SNPs with fear conditioning. These SNPs were then included in a score-based analysis (see Fig. 2 and Table 3). We observed in the left, but not the right, amygdala a positive correlation between the peak BOLD signal change in the amygdala elicited by CS+ (danger signal) versus CS− (safety signal) during early acquisition and the number of minor alleles of NR3C1 rs33389, NR3C1 Bcl1 (rs41423247) and NR3C1 rs4986593 [r=0.48, F(3, 55)=6.01, p=0.001, see also Fig. 2]. For extinction, lack of differential activation in the prefrontal cortex was associated with an increasing number of minor alleles of CRHR1 rs242938, CRHR1 rs1876831, NR3C1 rs6195 and NR3C1 Tth111I (rs10052957), suggesting impaired extinction with increased genetic minor allele variants [r=0.60, F(4, 54)=6.24, p<0.001].
Table 3. Peak voxel values of functional coupling analysis in sample 1 showing increased coupling for the genotype group coded 2 as compared with genotype groups 0 and 1Footnote a
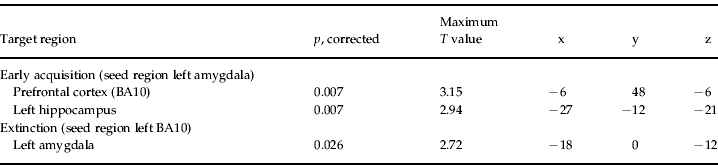
BA, Brodmann area; MNI, Montreal Neurological Institute.
a Coordinates are stated in MNI space.
During early acquisition, the subjects with several minor alleles showed significantly more functional coupling of the BOLD signal between the left amygdala and prefrontal cortex compared with those with only one or no minor allele on the NR3C1 gene. For extinction, functional coupling strength between the left prefrontal cortex and left amygdala was most pronounced for the subjects with at least two minor alleles on the NR3C1 and CRHR1 genes. No significant association with single genetic markers was seen.
We sought to replicate the association of these polymorphisms with amygdala and prefrontal cortex activation during acquisition and extinction in a second independent sample of 52 individuals from the same population (for sample characteristics, see Tables 1 and 2). Of the three NR3C1 polymorphisms, two were again significant predictors of differential amygdala activation in the early acquisition phase [NR3C1 rs33389, NR3C1 rs4986593: r=0.45, F(2, 42)=5.45, p=0.008] when all variables were entered, but NR3C1 Bcl1 (rs41423247) was also significant by itself when a stepwise solution was employed [r=0.41, F(1, 44)=8.82, p=0.005]. The same four SNPs [CRHR1 rs242938, CRHR1 rs1876831, NR3C1 N363S (rs6195), NR3C1 Tth111I (rs10052957)] were again significant for differential activation (i.e. delayed extinction) in the prefrontal cortex (BA10) during extinction [r=0.47, F(4, 43)=2.79, p=0.04].
These findings could also be found when combining both samples. Genetic variants were unrelated to skin conductance or self-report data of fear conditioning.
Discussion
The present study is the first to show a significant contribution of HPA axis-related genes to brain activation during fear conditioning and extinction in humans. Thus, more minor alleles in the NR3C1 gene were related to enhanced amygdala activation during conditioning and more minor alleles in the NR3C1 and CRHR1 genes were associated with reduced prefrontal activation during extinction, suggesting an accumulation of the effects. In addition, enhanced amygdala–prefrontal connectivity during acquisition, suggesting better fear memory consolidation, and higher amygdala–prefrontal interaction during extinction, suggesting sustained fear, were genotype related.
The development of PTSD after trauma exposure might depend on altered processes of fear learning. An inability to habituate to aversive stimuli and a reduction in inhibition of fear memories have often been reported (Rauch et al. Reference Rauch, Shin and Phelps2006; Yehuda & LeDoux, Reference Yehuda and LeDoux2007; Jovanovic et al. Reference Jovanovic, Norrholm, Blanding, Davis, Duncan, Bradley and Ressler2010; Jovanovic & Ressler, Reference Jovanovic and Ressler2010; Shin & Liberzon, Reference Shin and Liberzon2010). The amygdala was previously shown to be involved in the elaboration of conditioned fear responses (Davis, Reference Davis1992), while the prefrontal cortex was shown to regulate the activity of the amygdala in a top-down process and to inhibit the extinction of conditioned fear responses (Milad & Quirk, Reference Milad and Quirk2002; Peters et al. Reference Peters, Kalivas and Quirk2009). The present data are in accordance with animal data that reported a close association of CRH1 receptors in the amygdala and fear conditioning (Yang et al. Reference Yang, Chao and Lu2006) and a close association of GRs and fear acquisition (Kolber et al. Reference Kolber, Roberts, Howell, Wozniak, Sands and Muglia2008). The finding that significant amygdala activation was only present during early acquisition also supports previous reports on time-dependent activation of the amygdala during conditioning (e.g. Büchel et al. Reference Büchel, Morris, Dolan and Friston1998). Thus, the amygdala seems to play a larger role in the initiation than the maintenance of the response. In addition, we did not find similar genotype-related differences for subjective indicators of conditioning or skin conductance as we found for brain activation. This is not unusual and related to the fact that brain activation may be more closely related to genetic factors than subjective ratings or peripheral responses and may thus be more suitable as an intermediate phenotype (e.g. Meyer-Lindenberg, Reference Meyer-Lindenberg2010). Our data strengthen the hypothesis that HPA axis functioning and stress play a role in the development of anxiety disorders (McFarlane et al. Reference McFarlane, Barton, Yehuda and Wittert2011) and are in accordance with the findings described above that genes involved in glucocorticoid signalling are differentially expressed in PTSD (Yehuda et al. Reference Yehuda, Cai, Golier, Sarapas, Galea, Ising, Rein, Schmeidler, Müller-Myhsok, Holsboer and Buxbaum2009). The observed pattern corresponds to the deficits in patients with PTSD who retain responses to trauma cues, fail to extinguish cue-relevant associations and show altered genotypes of HPA axis-related genes (Rauch et al. Reference Rauch, Shin and Phelps2006; Yehuda et al. Reference Yehuda, Flory, Pratchett, Buxbaum, Ising and Holsboer2010).
Although the minor allele cannot be regarded as relevant to the analysed conditioning and clinical characteristics a priori, the distribution of alleles in the model applied to the first sample showed that the minor alleles accumulated for the observed neural activation patterns during conditioning. The reason for our observation that the associated alleles are the minor alleles (allele frequency of ⩽40.4%) is unclear. From an evolutionary point of view, both minor and major alleles can confer susceptibility to a trait, and based on selection for a trait or drift, the allele frequency of a marker is subject to change over time (Jobling et al. Reference Jobling, Hurles and Tyler-Smith2003; Keller & Miller, Reference Keller and Miller2006). However, for two of the analysed markers [NR3C1 Bcl1 (van Rossum et al. Reference van Rossum, Koper, van den Beld, Uitterlinden, Arp, Ester, Janssen, Brinkmann, de Jong, Grobbee, Pols and Lamberts2003), NR3C1 N363S (Huizenga et al. Reference Huizenga, Koper, De Lange, Pols, Stolk, Burger, Grobbee, Brinkmann, De Jong and Lamberts1998)] the minor alleles have been reported to increase glucocorticoid sensitivity (which was suggested to occur in PTSD, e.g. Rohleder et al. Reference Rohleder, Joksimovic, Wolf and Kirschbaum2004; Yehuda et al. Reference Yehuda, Golier, Yang and Tischler2004), but this seems not to apply to all tissues (Koper et al. Reference Koper, Stolk, de Lange, Huizenga, Molijn, Pols, Grobbee, Karl, de Jong, Brinkmann and Lamberts1997; Kumsta et al. Reference Kumsta, Entringer, Koper, van Rossum, Hellhammer and Wüst2008). This strengthens our observation of the association of the minor alleles with trait-like factors of increased fear memory consolidation and failed extinction reflected by amygdala–prefrontal activation patterns.
Supplementary material
For supplementary material accompanying this paper, visit http://dx.doi.org/10.1017/S0033291712000359.
Acknowledgements
Support for this study was provided by grant no. SFB636/C1 from the Deutsche Forschungsgemei-nschaft to H.F.
Declaration of Interest
None.








 Open access
Open access