



Introduction
Acute appendicitis is a common acute surgical emergency, with an estimated 17.7million cases worldwide per year (Wickramasinghe et al., Reference Wickramasinghe, Xavier and Samarasekera2021) and a lifetime risk of appendicitis of approximately 1 in 8 (Lee et al., Reference Lee, Park and Choi2010). Multiple studies (Bhangu et al., Reference Bhangu, Søreide, Di Saverio, Assarsson and Drake2015; Shahmoradi et al., Reference Shahmoradi, Zarei, Beiranvand and Hosseinnia2021) have attempted to investigate the aetiology of acute appendicitis and yet there is a lack of consensus on the aetiology. Direct luminal obstruction has been the conventional theory for the pathogenesis of acute appendicitis, commonly associated with faecoliths, lymphoid hyperplasia, parasites and very rarely caecal or appendiceal malignancy. Environmental factors such as seasonal alterations in temperature also affect the incidence and course of acute appendicitis (Wolkomir et al., Reference Wolkomir, Kornak, Elsakr and McGovern1987).
Appendix as a potential microbial reservoir, allowing repopulation of the gastrointestinal tract has been theorised but relevant data are scarce (Bhangu et al., Reference Bhangu, Søreide, Di Saverio, Assarsson and Drake2015). The importance of the gut microbiome is increasingly being recognised in gut-related diseases, especially its role in inflammation (Duvallet et al., Reference Duvallet, Gibbons, Gurry, Irizarry and Alm2017; Glassner et al., Reference Glassner, Abraham and Quigley2020; Jackson et al., Reference Jackson, Mongodin, Davenport, Fraser, Sandler and Zeichner2014; Oh et al., Reference Oh, Pimentel, Leite, Celly, Villanueva-Millan, Lacsina, Chuang, Parodi, Morales, Weitsman, Singer-Englar, Barlow, Zhai, Pichestshote, Rezaie, Mathur and Pimentel2020). Several studies using bacterial cultures of paediatric populations have highlighted the association of various microbiota in the pathogenesis of acute appendicitis (Elhag et al. Reference Elhag, Alwan, Al-Adnani and Sherif1986; Hattori et al., Reference Hattori, Yuasa, Ikegami, Nishiyama, Takeuchi, Miyake, Kuno, Miyata, Fujino and Minami2019; Swidsinski et al., Reference Swidsinski, Dorffel, Loening-Baucke, Theissig, Ruckert, Ismail, Rau, Gaschler, Weizenegger, Kuhn, Schilling and Dorffel2011). The advancement of high throughput sequencing of the human microbiome has led to a growth in knowledge of the microbiome and the role of dysbiosis in disease. Despite this, there has been limited research undertaken on adult appendicitis. The aim of this study is to characterise the microbiome of adult acute appendicitis using 16sRNA gene sequencing in a prospective cohort.
Methods
Participants
Patients who presented with acute appendicitis and subsequently underwent laparoscopic appendectomy, between March and September 2020, were recruited prospectively at Christchurch Hospital, Canterbury, New Zealand. Demographics and relevant clinical data were collected from electronic hospital health system. Participants were 18 years and older, with acute appendicitis confirmed on histology for inclusion in the study. Patients were excluded if they had antibiotic for more than 24 hours prior to surgery or in the previous 6 weeks. Appendicectomies performed in conjunction with concurrent colorectal or gynaecological disease were also excluded.
Rectal swabs were taken using DNA/RNA Shield Collection tubes with swabs (Zymo Research, Irvine, CA). Rectal swab is minimally invasive and is a suitable sampling method as proven from previously published pilot study (Turner et al., Reference Turner, O’Grady, Hudson, Morgan, Frizelle and Purcell2022). Patients had a rectal swab taken pre-operatively. Healthy volunteers had rectal swabs taken by a single clinician.
Appendicitis is categorised based on histological findings; complicated appendicitis is defined as appendicitis with perforation, gangrene and/or peri-appendicular abscess formation whereas these features are absent in simple/uncomplicated appendicitis. Complicated appendicitis is known to be associated with worse outcomes, prolonged hospitalisation and increased cost and therefore it would be useful to identify the microbiome-based markers, as a predictor of complicated disease.
This study had approval from the University of Otago Human Ethics Committee (H20/045). All participants provided written informed consent upon enrollment.
DNA extraction
All swabs were stored in DNA/RNA lysis tubes (Zymo Research, Irvine, CA) to maintain optimum stability of the DNA. DNA was extracted from samples using ZymoBIOMICS micro-DNA Kits (Zymo Research, Irvine, CA), according to the manufacturer’s instructions. In brief, samples homogenised using ZR BashingBead followed by centrifugation at >10,000 rpm for a minute. Supernatant was added to DNA Binding buffer and transferred Zymo-Spin IC-Z Column in a collection tube and centrifuged at maximum for 1 minute. The column was washed several times and DNA eluted in 20 μl DNase/RNase-free water. Finally, DNA concentration and 260/280 nm absorbance were measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). DNA was stored at −20C prior to 16S rRNA sequencing.
16S rRNA sequencing
Sequencing 16S rRNA region was carried out as per previous publication (Turner et al., Reference Turner, O’Grady, Hudson, Morgan, Frizelle and Purcell2022). Specifically, Nextera XT DNA Index kit (Illumina, San Diego, CA) was used for library preparation of the V3–V4 hypervariable regions of the 16S rRNA gene, using a single-step PCR with dual index primers (16SF_V3: 5′-TATG GTAATTGGCC TACGGGA GGC AGCAG-3′ and 16SR_V4: 5′AGTCAGTCAGCCGGA CTACH VGGGTWTCTAAT-3′). Illumina sequencing adaptors and barcodes were then added using limited cycle PCR. The libraries were pooled by equal molarity before loading on the Illumina MiSeq platform. PhiX was used at 20 per cent of the prepared libraries. Paired-end reads of length 250 bp were generated.
Sequences of appendicitis samples are available from the NCBI Sequencing Read Archive as BioProject PRJNA843336 while sequences of healthy samples are available as Bioproject PRJNA648177.
Microbiome analysis
Quality control
DADA2 (v1.18.0) was used to filter, trim, join sequencing reads, and remove chimaeras to obtain amplicon sequence variants (Callahan et al., Reference Callahan, McMurdie, Rosen, Han, Johnson and Holmes2016). The SILVA database (v132) was utilised to assign taxonomy to the amplicon sequence variants (Quast et al., Reference Quast, Pruesse, Yilmaz, Gerken, Schweer, Yarza, Peplies and Glockner2013). The taxonomy, sample metadata, and sequences were combined into a phyloseq object for subsequent analysis (McMurdie and Holmes, Reference McMurdie and Holmes2013).
Phylum-level differences
Taxa identified were agglomerated to the Phylum level and percent abundances were obtained per samples. Wilcoxon tests were used to compare Phylum differences between groups. A p-value of <0.05 is considered significant.
Diversity analysis
We rarefied samples to the smallest library size (26,882). We used phyloseq’s estimate richness function for Observed (richness) and Shannon (richness and evenness) measures, and compared differences of the alpha diversity measures by Wilcoxon tests. A p-value of <0.05 is considered significant.
We used principal coordinate analysis (PCoA) to test for overall differences in the microbiome between groups, using UniFrac distances between samples.
Differential abundance
DESeq2 was used to identify differentially abundant genera between healthy and appendicitis groups. Only genera that had 10 per cent abundance in 10 per cent of samples from each group were used. We considered a fold-change between comparisons significant if it has adjusted p-value of <0.05.
The bioinformatics analysis may be accessed at https://gitlab.com/alsulit08/adult_acute_appendicitis.
Results
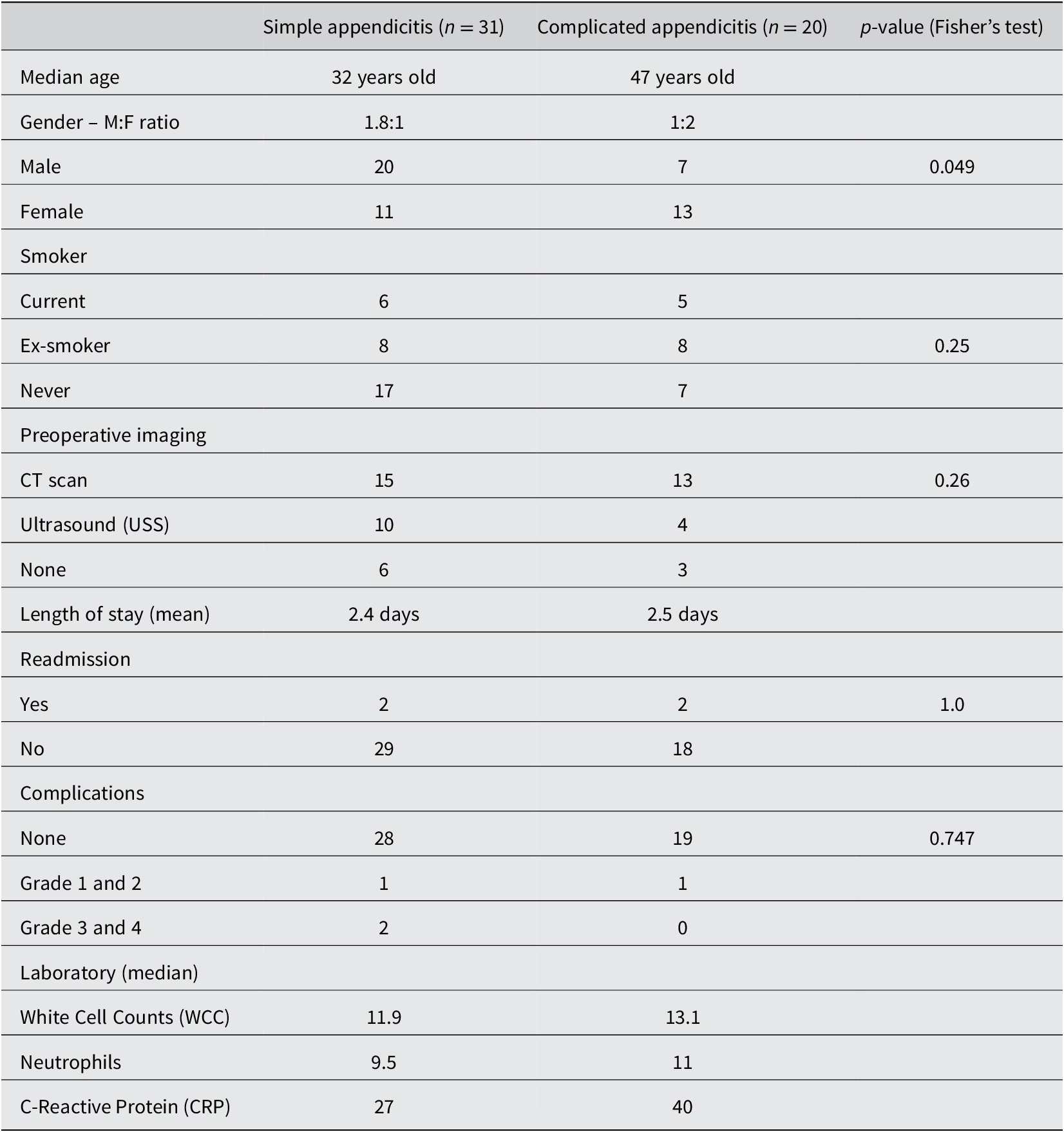
A total of 60 patients with acute appendicitis and 20 healthy controls were recruited for the study. There were only 58 rectal samples in appendicitis group had sufficient materials for analysis. Of the 78 total samples, we have a total of 7,465,619 reads for analysis after DADA2 quality control, with a range of 26882–195877 reads and median 83,122.5. There were 6,569 amplicon sequence variants (ASVs) identified in the 78 samples. The median age was 35.5 years old in appendicitis group and 32.5 years old in the control group. The age range is between 18 and 74 years old. In the appendicitis group, participants were predominantly male with NZ European ethnicity. There were 31 patients with simple appendicitis and 20 patients (41 per cent) with complicated appendicitis (perforated and/or gangrenous). Patients were older and had slightly higher inflammatory markers in the complicated appendicitis subgroup (Table 1).
Table 1. Demographics of the appendicitis group.
Comparison of the microbiome of acute appendicitis and healthy group
Bioinformatic analysis showed a statistically significant difference in the alpha diversity between the groups (p < 0.0001), with samples from healthy controls having higher alpha diversity. Unweighted UniFrac analysis showed clustering of samples by appendicitis or health controls (Figure 1A). Wilcoxon tests on phyla between healthy and appendicitis sample showed a significant difference of Firmicutes, Actinobacteria and Proteobacteria phyla percent abundance (Figure 1B). Further classification at the genus level show loss of beneficial gut commensals, including Blautia, Lachnospiraceae, Faecalibacterium and Ruminococcus in swabs from appendicitis patients, compared to healthy controls (Figure 1C).
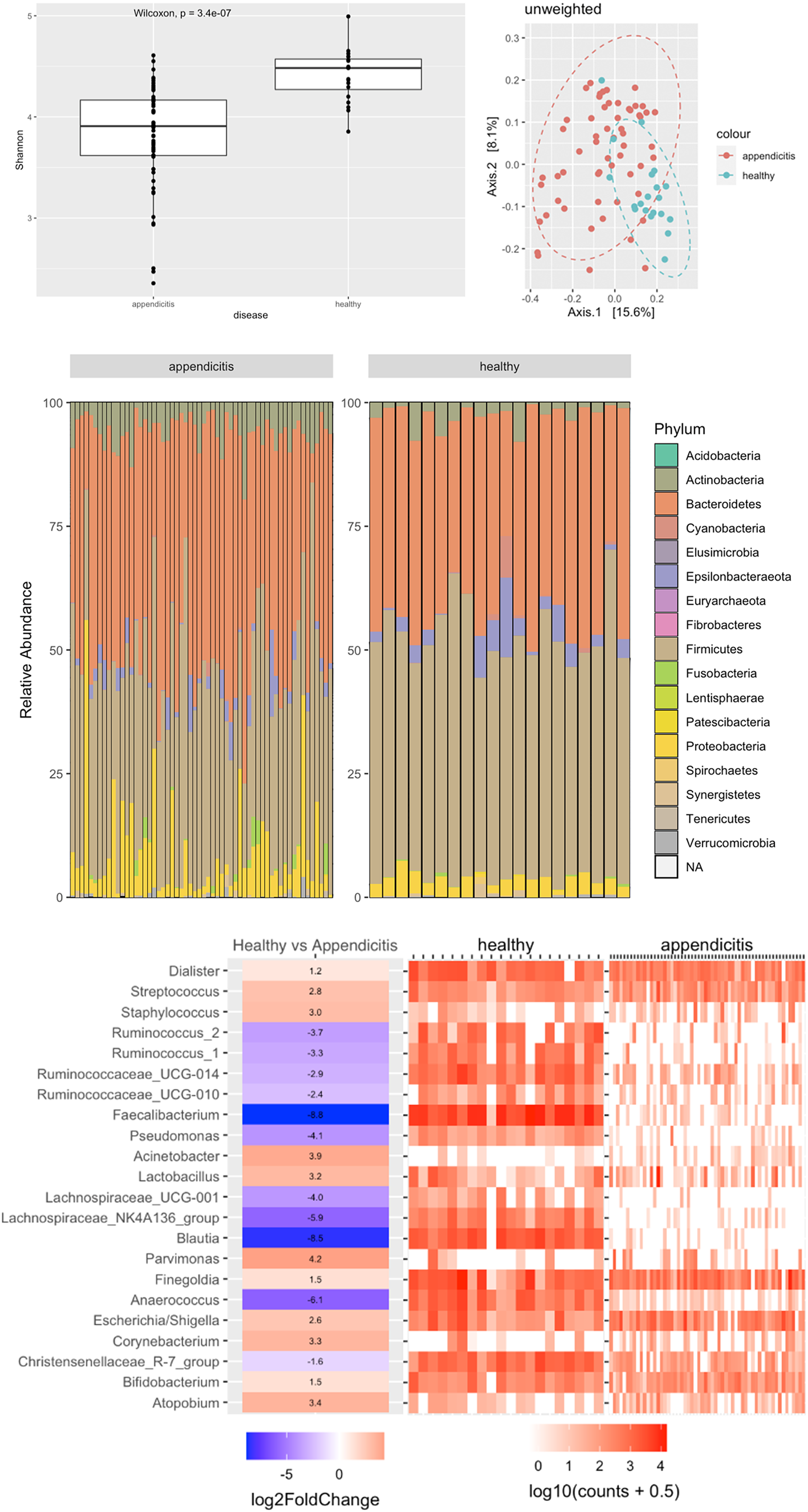
Figure 1 (A) Alpha diversity and PCoA of rectal swabs between appendicitis and healthy controls. (B) Phyla abundance between the appendicitis and healthy groups. (C) Comparison between appendicitis and healthy samples at genus level. A positive log fold change means the genus more abundant in appendicitis and a negative log fold means the genus is more abundant in healthy samples.
Comparison of the microbiome between simple and complicated appendicitis
Shannon diversity analysis showed no significant differences in alpha diversities between simple and complicated appendicitis in rectal samples (p = 0.98) (Figure 2). Phylum-level abundance were similar for both simple and complicated acute appendicitis (Figure 3). Further classification at the genus level showed no difference between the two groups.
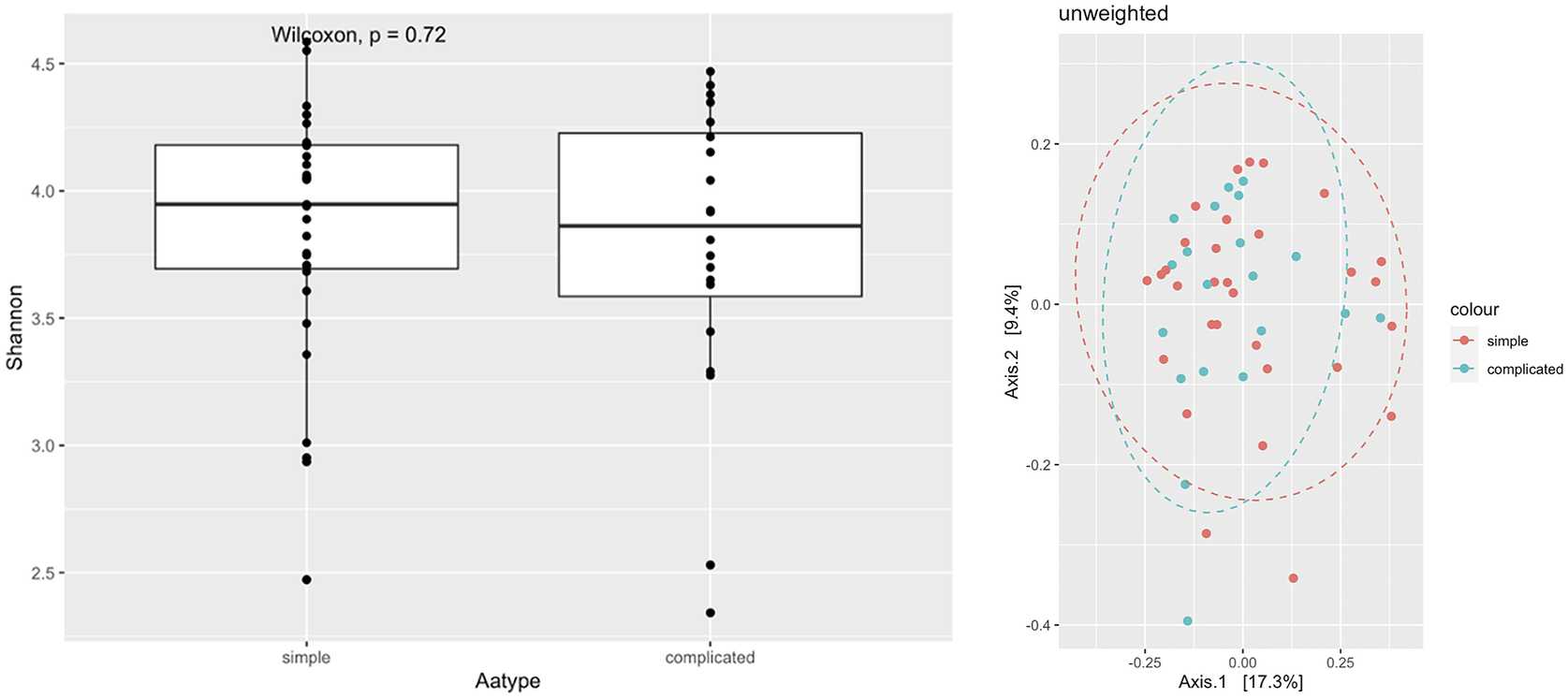
Figure 2 Alpha diversity and PCoA of rectal swabs comparing simple and complicated appendicitis.

Figure 3 Phyla distribution between simple and complicated appendicitis.
Discussion
In this study, there was a difference between the microbial composition as assessed by rectal swabs of those with acute appendicitis and healthy controls. Metagenomics sequencing analysis showed a reduction in diversity and loss of commensal bacteria in people with appendicitis. The loss of commensals including phyla important to gut homeostasis, such as Lachnospiraceae, Blautia and Faecalibacterium, which may play a role in the cascade leading to the development of acute appendicitis. There were no significant microbiome differences found at both phylum and genus level, between patients with simple and complicated appendicitis.
Despite the frequency of acute appendicitis, we know very little for certain about its aetiology. Microbiological causes have been suggested in various studies. Guinane et al. (Reference Guinane, Tadrous, Fouhy, Ryan, Dempsey, Murphy, Andrews, Cotter, Stanton and Ross2013) first provided insight into the microbial composition of the appendix in a study of seven patients, aged 5–25 years old, using 16S-rRNA sequencing. Analysis of faecal and tissue samples revealed that appendices contained a high bacterial biomass and were postulated to be a reservoir for commensal bacterial. Across the seven samples, 15 different phyla were detected, dominated by five major phyla – Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria and Fusobacteria. In our study of an adult cohort, we found similar dominant phyla with the exception of Fusobacteria, due to different sampling methodologies.
Rectal swabs were used as these can be taken without delay when the patient is admitted, unlike faecal samples, where there could be a delay of up to 24 hours for sample collection. This is important as patients are routinely administered antibiotics on admission and antibiotics have been shown in multiple studies (Dudek-Wicher et al., Reference Dudek-Wicher, Junka and Bartoszewicz2018; Mu and Zhu, Reference Mu and Zhu2019) to affect the gut microbiome. Previously published findings from our pilot study (Turner et al., Reference Turner, O’Grady, Hudson, Morgan, Frizelle and Purcell2022) and others (Budding et al., Reference Budding, Grasman, Eck, Bogaards, Vandenbroucke-Grauls, van Bodegraven and Savelkoul2014; Reyman et al., Reference Reyman, Van Houten, Arp, Sanders and Bogaert2019; Short et al., Reference Short, Hudson, Besasie, Reveles, Shah, Nicholson, Johnson-Pais, Weldon, Lai, Leach, Fongang and Liss2021) have shown that rectal swabs are a suitable method of analysing changes in the gut microbiome between patient groups.
Several other paediatric studies of appendicitis suggest that Fusobacterium is associated with acute appendicitis (Salo et al., Reference Salo, Marungruang, Roth, Sundberg, Stenstrom, Arnbjornsson, Fak and Ohlsson2017; Schulin et al., Reference Schulin, Schlichting, Blod, Opitz, Suttkus, Stingu, Barry, Lacher, Buhligen and Mayer2017; Swidsinski et al., Reference Swidsinski, Loening-Baucke, Biche-ool, Guo, Dörffel, Tertychnyy, Stonogin and Sun2012; Zhong et al., Reference Zhong, Brower-Sinning, Firek and Morowitz2014). Salo et al. (Reference Salo, Marungruang, Roth, Sundberg, Stenstrom, Arnbjornsson, Fak and Ohlsson2017) noticed an increased abundance of Fusobacterium and a decreased in Bacteroides in complicated appendicitis compared to uncomplicated appendicitis. A further study of intraluminal samples by Schulin et al. (Reference Schulin, Schlichting, Blod, Opitz, Suttkus, Stingu, Barry, Lacher, Buhligen and Mayer2017) reported that Fusobacterium necrophorum was mainly found in catarrhal appendicitis, Pseudomonas endodontalis in phlegmonous appendicitis and F. nucleatum in gangrenous appendicitis. In contrast, in our study, Fusobacterium was detected, but was not a predominant phylum in either healthy or appendicitis groups. These findings may be due to differences between paediatric populations in these other publications and the adult population under investigations here; an adult population may have more resilient microbiome and dysbiosis may not be as pronounced.
A growth in human microbiome research has led to better understanding of disease associated microbiome change. A study on the aetiology of appendicitis in 277 children showed that Escherichia coli was the predominant bacteria followed by Streptococci (Richardsen et al., Reference Richardsen, Schob, Ulmer, Steinau, Neumann, Klink and Lambertz2016). In addition, Zhong et al. (Reference Zhong, Brower-Sinning, Firek and Morowitz2014) performed the 16S ribosomal gene sequence analysis to profile the microbiota of the luminal fluid of the appendix in children with appendicitis and normal appendices, and found an increased relative abundance of the pathogen containing genera, Fusobacterium and Prevotella with a corresponding depletion of Bacteroides (11.4 vs. 52.9 per cent). In our study, Actinobacteria and Proteobacteria phyla are increased in appendicitis patients with a concomitant increase in the pathogen containing Parvimonas, Acinetobacter, E. coli, Corneybacterium and Streptococci genera (Figure 1B,C). The apparent differences in microbiome changes between paediatric and adult population might be due to different methodologies in the various studies or other factors (ie. dietary or lifestyle factors) which may influence the dysbiosis in acute appendicitis.
Analysis of differential abundance at the genus level in our study also revealed a loss of commensal bacteria in appendicitis samples compared to healthy samples. These included Blautia, Faecalibacterium and Lachnospiracaea, genera that contain short-chain fatty acids (SCFA) producing bacteria. SCFAs are important metabolites that are an energy source for epithelial cells and also contribute to gut barrier function (Loh and Blaut, Reference Loh and Blaut2012; Parada Venegas et al., Reference Parada Venegas, De la Fuente, Landskron, Gonzalez, Quera, Dijkstra, Harmsen, Faber and Hermoso2019) and play an anti-inflammatory role in the gut (Blaak et al., Reference Blaak, Canfora, Theis, Frost, Groen, Mithieux, Nauta, Scott, Stahl, van Harsselaar, van Tol, Vaughan and Verbeke2020; Machiels et al., Reference Machiels, Joossens, Sabino, De Preter, Arijs, Eeckhaut, Ballet, Claes, Van Immerseel, Verbeke, Ferrante, Verhaegen, Rutgeerts and Vermeire2014; Segain et al., Reference Segain, de la Bletiere, Bourreille, Leray, Gervois, Rosales, Ferrier, Bonnet, Blottiere and Galmiche2000). Our observed reduction of microbial diversity has been described in other inflammatory conditions such as inflammatory bowel disease and acute appendicitis (Loh and Blaut, Reference Loh and Blaut2012). Ott (Reference Ott2004) performed a 16S rDNA study of mucosa associated colonic microflora and found a reduction of microbial diversity in both Crohn’s disease and Ulcerative colitis. This demonstrates that gut inflammation has a strong association with changes in gut microbiome. This preliminary study looks at the overall changes in pattern of bacterial dysbiosis in relation to gut inflammation, neither at individual bacteria level nor specific aetiology for acute appendicitis.
This is one of the few published studies on the microbiome of acute appendicitis in adult population. This preliminary study looked at broad changes in taxa in the gut microbiomes between patients with acute appendicitis and healthy controls, and between patients with complicated versus uncomplicated appendicitis, using a well establish method of analysis (16S rRNA sequencing). We have used rectal swabs instead of faecal sampling, a methodology we have previously verified (Turner et al., Reference Turner, O’Grady, Hudson, Morgan, Frizelle and Purcell2022). The methodology of this study does not however allow us to determine whether gut microbiome dysbiosis was a cause of appendicitis or just secondary to the pathology. Despite the relatively large cohort size in this study, future large-scale studies are necessary to validate the findings presented here and suggest perform functional analysis to establish bacterial signatures associated with acute appendicitis.
Conclusion
In conclusion, this study has added knowledge to the current literature on microbiome studies of acute appendicitis in adult population. We have found a reduction in diversity and loss of commensals in the microbiome of those with acute appendicitis. The loss of commensals bacteria may play a role in the cascade leading to the development of acute appendicitis.
Acknowledgement
The authors would like to thank University of Otago administrative staffs.
Notes on contributors
The authors thank our University of Otago Statistician and Laboratory staffs.
Disclosure statement
The authors have no known conflicts of interest to report.
Author contributions
Conceptualisation: F.F.; Data curation: M.S.L.; Formal analysis: A.S., R.P.; Investigations: A.S., R.P.; Methodology: M.S.L., A.S., R.P.; Project administration: F.F.; Resources: M.S.L.; Software: A.S.; Supervision: R.P., F.F.; Visualisation: M.S.L., A.S.; Writing – original draft: M.S.L.; Writing – review and editing: F.F., R.P.
Research Transparency and Reproducibility
Protocols, materials and relevant data are available for readers and can be access by contacting corresponding author.





 Open access
Open access