


Under physiological conditions, vascular endothelium contributes to vascular homeostasis by monitoring haemostasis, vascular tone, endothelial permeability and media smooth muscle cell migration(Reference De Caterina1). Leucocyte binding to the endothelium is a common feature of several inflammatory and immunological disorders. Polymorphonuclear leucocytes bind to the endothelium in acute inflammation; meanwhile, monocytes adhesion occurs predominantly in atherosclerosis.
Atherosclerosis is an inflammatory process started by monocyte adhesion and migration into the vascular wall. Vascular endothelium is central in its initiation and propagation(Reference Ross2, Reference Gimbrone3). Leucocyte recruitment into the vascular wall is a multistep process that subsequently includes leucocyte rolling, firm adhesion and transmigration. Different leucocyte subsets and diverse endothelial cell (EC) adhesion molecules are involved. Selectins (L-selectin; P-selectin; E-selectin) are responsible for initial stages of cell rolling. Integrins participate in firm adhesion, and Ig super family mediates transmigration step. Ig super family includes vascular cell adhesion molecule (VCAM-1), intercellular adhesion molecules (ICAM-1; ICAM-2; ICAM-3) and platelet EC adhesion molecule-1(Reference Rao, Yang and García-Cardena4, Reference Mizia-Stec5).
Atherogenesis is also associated with an increase of EC turnover, and it may also result in an accelerated EC apoptosis(Reference Choy, Granville and Hunt6). Apoptosis of EC may be caused by oxidative stress, local inflammatory mediators or by cytolytic attack of activated killer T cells, cytokines and oxidised LDL(Reference Burlacu, Jinga and Gafencu7).
Sex steroids hormones exert diverse effects on vascular system, inducing rapid vasodilatation, anti-thrombotic effect and attenuation of atherosclerosis disease(Reference Caulin-Glaser, García-Cardena and Sarrel8). Oestradiol reduces endothelial activation and leucocyte adhesion molecules expression, and it decreases leucocyte binding to vascular wall(Reference Simoncini, Maffei and Basta9).
Phyto-oestrogens may improve vascular function having favourable effects on surrogate cardiovascular risk markers(Reference Colacurci, Chiàntera and Fornaro10). The phyto-oestrogen family includes lignans and flavonoids such as genistein (Gen) and daidzein, which are mainly present in soya beans and soya-derived foods(Reference Reinli and Block11). Gen shares structural features with the potent oestrogen 17-β-oestradiol, particularly the phenolic ring, which confers ability to bind to oestrogen receptor (ER) and sex hormones binding protein(Reference Barnes12, Reference Kuiper, Carlsson and Grandien13). Gen can exert both oestrogenic and anti-oestrogenic activity. It has been reported that the isoflavone could exert a dual mechanism of action dependent on the dose employed. Below 20 μm, Gen acts via an ER-dependent pathway involving extracellular signal-regulated kinase 1/2 activation, and at higher concentration (>25 μm) its effects are attributed to its tyrosine kinase inhibitor activity(Reference Akiyama, Ishida and Nakagawa14). In postmenopausal women, results of epidemiological studies have suggested that high dietary intake of isoflavones may contribute to a low incidence of high prevalent chronic diseases such as osteoporosis, hormone-related cancers and CVD(Reference Liu, Homan and Dillon15). Although potential effects of isoflavones on human vascular health have widely been investigated in the last decade, the mechanism of action of Gen in vascular wall remains unclear. Dietary supplementation with Gen would have beneficial effects on vascular tone and prevention of atherosclerosis(Reference Dixon and Ferrerira16).
We have previously shown that Gen exhibits anti-platelet activity through its direct action on vascular tissue(Reference Polini, Rauschemberger and Mendiberri17). In rat aortic strips isolated from fertile female Wistar rats, Gen (0·1–100 nm) non-genomically stimulates NO synthase (NOS) and cyclo-oxygenase activities via the ER-dependent pathway. The enhancement of NO and prostacyclin release induced by the isoflavone inhibit platelet activation and aggregation. These effects were also detected in rats deprived of ovarian function(Reference Polini, Rauschemberger and Mendiberri17).
In the present work, we study the direct action of Gen at EC level. We evaluate whether submicromolar concentration of the phyto-oestrogen would modulate endothelial proliferation, monocyte adhesion and cellular apoptosis either in the presence or absence of proinflammatory agents.
Materials and methods
Materials
[3H]Thymidine was purchased from New England Nuclear (Chicago, Des Plaines, IL, USA). Griess reaction solutions were purchased from Britania Laboratories (Buenos Aires, Argentina). Trypsin/EDTA (10 × ), l-glutamine (100 × ), amphotericin B (0·25 mg/ml), penicillin/streptomycin (100 × ) and fetal bovine serum were obtained from PAA Laboratories (Pasching, Austria). RT-PCR RNA kit and Superscript III CellsDirect cDNA synthesis system were purchased from Invitrogen (Carlsbad, CA, USA). Gen, Dulbecco's modified Eagle's medium (DMEM), lipopolysaccharides (LPS from Escherichia coli 0127) and all other reagents were purchased from Sigma Chemical Company (St Louis, MO, USA).
Animals
Sexually mature female Wistar rats (3–5 weeks old) were employed. All animal work was performed at the Unit of Animal Care belonging to the Biology, Biochemistry and Pharmacy's Department of the University. The Animal Care Use Committee approved the protocol used.
Endothelial cells cultures
EC were obtained from aortic rings explants isolated from 3–5-weeks-old Wistar female rats, as previously reported(Reference Cutini, Sellés and Massheimer18, Reference Yeh, Hwang and Liu19). Briefly, the full length thoracic aorta was aseptically removed, and then cut into ring segments (2 mm). Ring explants were seeded in a 60 mm matrix-coated Petri dishes (NUNC) containing DMEM supplemented with 10 % fetal calf serum (FCS), 60 μg/ml penicillin, 100 μg/ml streptomycin, 2·5 μg/ml amphotericin-B and 2 mml-glutamine, and they were incubated at 37°C in 5 % CO2 atmosphere. After 5 d of culture, ring explants were removed, and the remaining cells were allowed to reach confluence. The identity of the EC(Reference Bachetti and Morbidelli20) was determined as follows: (a) by phase-contrast microscope observation of the characteristic morphology of cobblestone-shaped growth in confluent monolayers; (b) by positive immunocytochemistry reactivity to factor VIII and to anti-vimentin, clone V9 using DakoCytomation EnVision system; (c) by the bioability to synthesise NO. Cells from passages 2–7 were used for all experiments.
Measurement of nitric oxide production
EC were seeded on twenty-four multi-well (NUNC) plates at a density of 4 × 104 cells/cm2 and allowed to grow to 90 % confluence, and then exposed to Gen for 5–30 min. Respectively control (vehicle alone) was also processed. Nitrites were measured in the incubation media as a stable and non-volatile breakdown product of the NO released, employing Griess reaction(Reference Rauschemberger, Sellés and Massheimer21). Cells were dissolved in NaOH, and protein content was measured(Reference Lowry, Rosebrough and Farr22). The results were expressed as nmol of nitrites per mg protein.
[3H]Thymidine incorporation assay
EC were seeded on twenty-four multi-well plates (NUNC) at a density of 9 × 104 cells/well in DMEM supplemented with 10 % FCS and were allowed to grow to 60–70 % confluence. Cells were synchronised by placing in serum-free DMEM for 24 h, and further exposed to 10 nm Gen or vehicle control (ethanol < 0·1 %) for 24 h in fresh DMEM containing 1 % FCS. The cells were pulsed with 37 Bq/ml of [3H]thymidine during last 2 h of treatment. Cells were rinsed twice with PBS in order to remove the unincorporated [3H]thymidine. Ice-cold TCA (10 %) was added, and the acid-insoluble material was dissolved with 1 m NaOH. Radioactivity was measured by liquid scintillation, and [3H]thymidine incorporation was normalised to protein content(Reference Cutini, Sellés and Massheimer18, Reference Kyaw, Yoshizumi and Tsuchiya23).
RT-PCR assay
EC were treated with Gen, bacterial LPS (1 μg/ml) or Gen plus bacterial LPS (the isoflavone was added 5 h before bacterial LPS addition). Respective control with vehicle alone (ethanol 0·1 %) was also processed. Total cellular RNA extraction and reverse transcription were performed using Superscript III CellsDirect cDNA synthesis system according to the manufacturer's instructions. Complementary DNA was then amplified by PCR using a programmed thermocycler (Biometra Uno II; Biometra, Göttingen, Germany). PCR cycles were as follows: E-selectin (95°C, 3 min, 94°C, 30 s, 53°C, 45 s, 72°C, 60 s, 72°C, 7 min, 32 cycles); P-selectin (94°C, 3 min, 94°C, 30 s, 62°C, 45 s, 72°C, 45 s, 72°C, 7 min, 38 cycles); VCAM-1 (95°C, 3 min, 95°C, 60 s, 55°C, 60 s, 72°C, 60 s, 72°C, 7 min, 32 cycles). Primers sequences were as follows(Reference Peng, Shelley Chireyath and Yanjv24, Reference Callera, Montezano and Touyz25): E-selectin, forward: 5′ CAA CGT GCA CGT TTG ACT GT 3′, reverse: 5′ AGG TCA AGG CTT GAA CAC TG 3′; P-selectin, forward: 5′ TAA TCC CCC GCA GTG TAA AG 3′, reverse: 5′ AGG TTG GCA ATG GTT CAC TC 3′; VCAM-1, forward: 5′ TAA GTT ACA CAG CAG TCA AAT GGA 3′. The expression of housekeeping gene glyceraldehyde-3-phosphate dehydrogenase was checked for each set of RT-PCR experiments (forward primer: 5′ TCC CTC AAG ATT GTC AGC AA 3′, reverse primer: 5′AGA TCC ACA ACG GAT ACA TT 3′; amplification steps: 95°C, 3 min, 94°C, 30 s, 53°C, 30 s, 72°C, 45 s, 72°C, 7 min, 35 cycles). Negative controls were also processed. PCR amplification products were detected by electrophoresis in agarose gels stained with ethidium bromide. Results were obtained from at least four independent experiments.
DNA fragmentation assay
EC cultured in DMEM containing 1 % FCS were exposed to Gen or vehicle alone. H2O2 (200 μm) was employed as apoptosis inducer. In Gen plus H2O2 treatment, Gen was added at the beginning and H2O2 addition was made in the last 6 h of the treatment. Cells were washed twice with PBS and lysated with 10 mm EDTA, 400 mm NaCl, 1 mg/ml proteinase K, 35 mm SDS and 10 mm Tris–HCl pH 8·2 at 37°C overnight. DNA was extracted using phenol–chloroform–isoamyl alcohol (25:24:1). DNA samples were separated by electrophoresis on 1 % (w/v) agarose gel containing 1 mg/ml of ethidium bromide and photographed on an ultraviolet transilluminator(Reference Ohtsuka, Miyashita and Shirai26). Integrity density was measured employing the image processing and analysis software of the NIH, ImageJ, 1.43 c version(Reference Rasband27).
Cells adhesion assay
Peripheral blood mononuclear cell and monocyte isolation
Separation of peripheral blood mononuclear cell was performed by density gradient(Reference Bøyum28). Heparinised whole blood diluted with PBS (1:1) was carefully layered onto Ficoll-Paque Plus and centrifuged at 400 g. Mixed mononuclear cell interface was collected, and viability was checked with trypan blue. Peripheral blood mononuclear cell were suspended in DMEM-10 % FCS and placed on NUNC Petri dishes (2·0 × 107 cells/ml) for 1 h at 37°C to allow peripheral blood monocyte (PBM) adhesion. Monocytes morphology was determined by May–Grünwald–Giemsa staining. The culture medium containing non-adherent cells was removed. Adhered cells were incubated for 15 min in PBS–EDTA 10 mm, washed and cultured in DMEM-10 % FCS, 60 μg/ml penicillin, 100 μg/ml streptomycin for 72 h. Medium was replaced at 48 h. Adherent cells were detached using a rubber policeman and suspended in DMEM. Absolute number of PBM was counted using an automatised counter. Viability was newly confirmed with trypan blue assay.
Peripheral blood monocyte adhesion assay
EC were starved for 24 h with serum-free medium, and then exposed to Gen, LPS or Gen plus bacterial LPS in DMEM (1 % FCS) as indicated in each experiment. An exact number of PBM were seeded on pretreated EC and incubated for 2 h at 37°C in a humidified 5 % CO2 atmosphere(Reference Pawlowski, Abraham and Pontier29). Supernatants of each well containing non-adhered PBM were collected and counted. The number of adhered PBM to EC was calculated by the difference between total mononuclear seeded and non-adherent PBM. EC and adhered PBM were dyed using Giemsa stain. Images ( × 200) were obtained using an OLYMPUS C7070WZ optical microscope system. Results are expressed as means and standard deviations of number of cells counted.
Endothelial cell migration assay
EC were seeded at a density of 5 × 105 cells/cm2 in 60 mm NUNC dishes with DMEM containing 10 % FCS, and were grown to 90 % confluence. Cells were starved for 24 h with serum-free medium. In order to evaluate EC migration, two scratch wound assays were carried out within the same well in each condition: (a) a strip was scratched using a 200 μl pipette tip(Reference Gojova and Barakat30); (b) a wound was made by pressing a razor blade down on the dish to cut the cell layer. The blade was then gently moved to one side to remove part of the monolayer. Immediately after that, the adhered cells were washed twice with PBS and cultured in fresh DMEM containing 1 % FCS plus Gen or vehicle control. After 48 h of culture, cells were fixed in glutaraldehyde 0·1 % and stained with haematoxylin–eosin. Migration was quantified by counting the number of cell nuclei that migrated at scratched area in at least seven different microscopic fields representative of each culture plate. EC migration was recorded using an OLYMPUS C7070WZ optical microscope system. Results are expressed as means and standard deviations of number of migrated cells/field(Reference Bürk31, Reference Pedram, Razandi and Aitkenhead32).
Statistical analysis
Each experimental condition has been performed in three independent experiments performed by quadruplicate. All data are presented as means and standard deviations. Comparisons between two means were made using Student's t test, and multiple comparisons were made with ANOVA using SSPS Statistical software version 10.0 for Windows. Differences of P < 0·05 were considered significant.
Results
In order to evaluate the effect of Gen on EC proliferation, we employed the [3H]thymidine incorporation technique. Figure 1 shows that Gen significantly increased cell proliferation after 24 h of treatment. This mitogenic effect was seen in a wide range of submicromolar concentration, 0·001–10 nm, (30·80 (sd 2·46) v. 62·3 (sd 6·67); 54·5 (sd 6·20); 49·6 (sd 4·47 × 103) cpm/mg protein, control v. Gen 10− 12, 10− 10 and 10− 8 m, respectively, P < 0·001). At higher concentration (1 μm), the phyto-oestrogen significantly inhibited EC proliferation (30·8 (sd 2·46) v. 9·8 (sd 2·21) × 103 cpm/mg protein, control v. Gen 10− 6 m, P < 0·001). No significant differences in DNA synthesis were observed after 48 h of treatment (not shown). To test whether ER would mediate the proliferative action of Gen, EC were preincubated with the high affinity ER antagonist ICI 182780 for 30 min before phyto-oestrogen treatment. As can be observed in Table 1, the presence of ER antagonist completely suppressed the proliferative effect elicited by Gen.
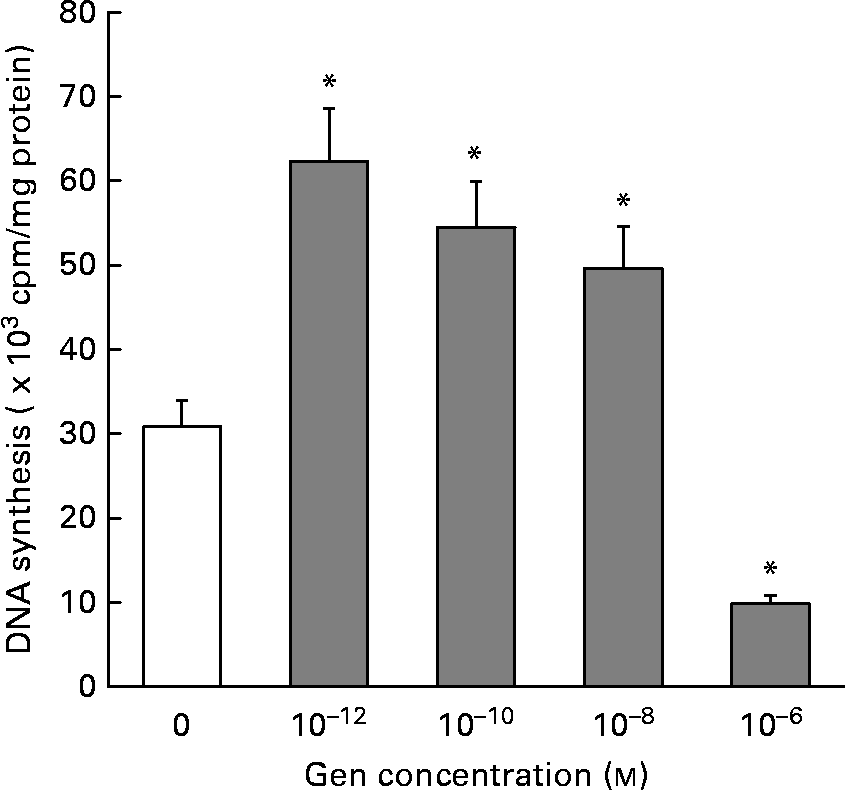
Fig. 1 Effect of genistein (Gen) on endothelial cells (EC) proliferation. Starved EC cultures were incubated with different concentrations of Gen for 24 h. [3H]Thymidine incorporation was measured as described in the Materials and methods section. Results represent the means and standard deviations of three independent experiments (n 4). * Mean values were significantly different (P < 0·001) with respect to control value.
Table 1 Effect of l-nitro-arginine methyl ester (l-NAME) and ICI 182780 on mitogenic action of genistein (Gen)†
(Mean values and standard deviations of three independent experiments (n 4))
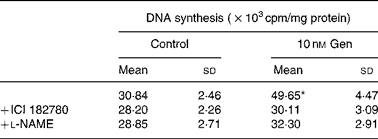
* Mean value was significantly different from that for the vehicle treatment (P < 0·001).
† EC cultures were preincubated with l-NAME (10 μm) or ICI 182780 (1 μm) for 1 h, and immediately after that exposed to 10 nm Gen for 24 h. [3H]thymidine incorporation was measured as described in the Materials and methods section.
It is widely known that NO is an important second messenger of the endothelium. We have previously reported that, in rat aortic strips, Gen stimulates NO production through a non-genomic mechanism of action. Since endothelial and smooth muscle cells coexist in aortic rings, we evaluated if the phyto-oestrogen enhances NO synthesis at endothelial level. NO was measured after rapid stimulation of EC with the isoflavone (Fig. 2). The phyto-oestrogen treatment significantly increased NO production between 5 and 30 min (50, 61, 80, 139 and 48 % above each control value) with similar kinetic profile as the control group, exhibiting maximal stimulation at 20 min. The stimulatory action of Gen on NO synthesis was time dependent, since statistical differences were detected among each treatment times.
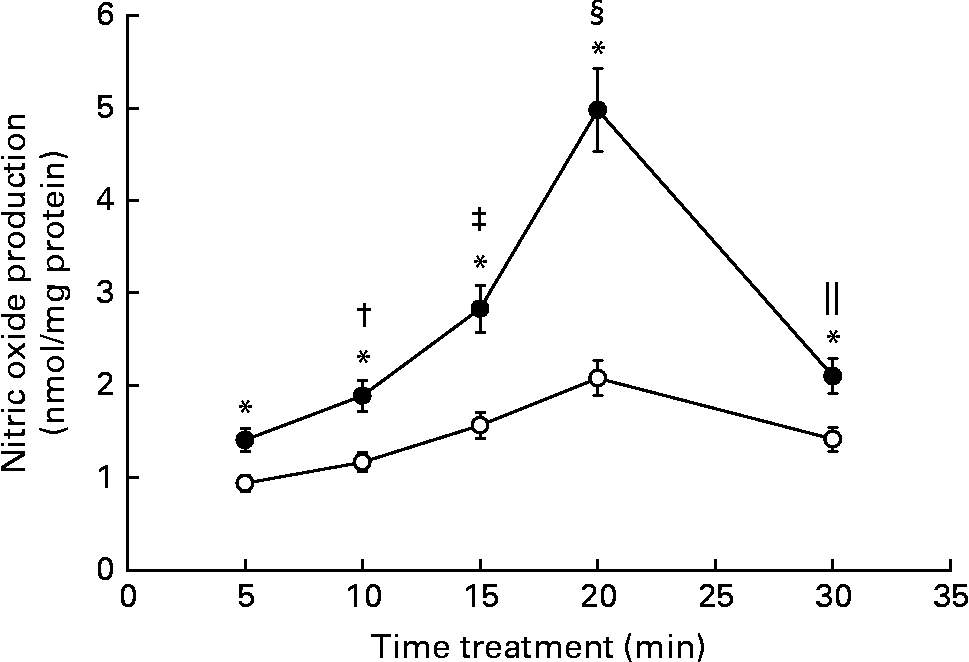
Fig. 2 Effect of genistein (Gen) on nitric oxide production: time–response profile. Starved endothelial cells were treated with 10 nm Gen at the indicated times. Nitric oxide production was measured by Griess reaction as described in the Materials and methods section. Results represent the means and standard deviations of three independent experiments (n 4). Mean values were significantly different: * P < 0·001 with respect to each control value; † P < 0·001 v. Gen 5 min; ‡ P < 0·001 v. Gen 10 min; § P < 0·001 v. Gen 15 min; ∥ P < 0·001 v. Gen 20 min. ●, 10 nm Gen; ○, control.
We tested the hypothesis whether NO pathway participates in the regulation of EC proliferation induced by Gen. To that end, we selected the compound l-nitro-arginine methyl ester as specific NOS inhibitor and measured DNA synthesis in the presence of this compound. Table 1 shows that the preincubation with l-nitro-arginine methyl ester suppressed the proliferative effect elicited by 24 h treatment with 10 nm Gen. Taken together, the results of Fig. 2 and Table 1 suggest that the mitogenic action of the isoflavone involves the participation of ER and NOS pathway.
The effect of Gen on PBM adhesion to EC was measured using cell adhesion assay. Cells were treated with 10 nm Gen for different time intervals, and immediately after that isolated PBM were added. Table 2 shows that isoflavone treatment exhibited lesser monocyte adhesion with respect to the control group. This inhibitory effect was seen after 13–46 h of exposure to the phyto-oestrogen. When EC were exposed to bacterial LPS (1 μg/ml), enhancement of PBM adhesion was observed (Fig. 3 and Table 3). This adhesion was partially attenuated when EC were pretreated with 10 nm Gen for 24 h before bacterial LPS addition (988 (sd 98) v. 1430 (sd 165) cells/μl, Gen+LPS v. Control+LPS). The results from Table 2 and Fig. 3 suggest that Gen treatment not only inhibited leucocyte adhesion in a free proinflammatory agent's microenvironment but also attenuated bacterial LPS-induced monocyte adhesion.
Table 2 Effect of genistein (Gen) on peripheral blood monocyte (PBM) endothelial cells (EC) adhesion at different times of treatment†
(Mean values and standard deviations of three independent experiments (n 4))
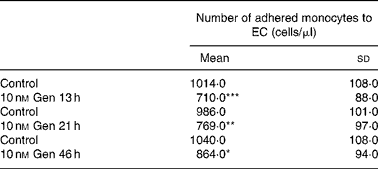
Mean values compared with respective control value: * P < 0·05, ** P < 0·01, *** P < 0·001.
† EC cultures were treated with 10 nM Gen for 13, 21 and 46 h. Subsequently, PBM were seeded on the monolayer and incubated for 2 h. Supernatant containing non-adhered PBM was removed, and non-adhered cells were counted.

Fig. 3 Effect of genistein (Gen) on monocyte/macrophage endothelial cell (EC) adhesion. EC cultures were treated with 10 nm Gen for 46 h in the presence or absence of 1 μg/ml lipopolysaccharide (LPS), which was added 24 h after Gen treatment. Peripheral blood monocyte (PBM) were seeded to cell monolayer for 2 h, and immediately after that the supernatant was removed and the non-adhered PBM were counted. Images show representative fields of each experimental condition ( × 200). The scale bar represents 70 μm.
Table 3 Effect of genistein (Gen) on peripheral blood monocyte (PBM) endothelial cells (EC) adhesion∥
(Mean values and standard deviations of three independent experiments (n 4))

Mean values were significantly different: * P < 0·01 v. control, † P < 0·01 v. control-LPS, ‡ P < 0·01 v. Gen, § P < 0·01 v. control+LPS.
∥ EC cultures were treated with 10 nm Gen for 46 h in the presence or absence of 1 μg/ml LPS, which was added 24 h after Gen treatment. PBM were seeded to the cell monolayer for 2 h, and immediately after that the supernatant was removed and the non-adhered PBM were counted.
Taking in account these observations, we focused our attention on cell adhesion-related genes. Employing RT-PCR technique, we evaluated the Gen regulation on cell mRNA expression of E-selectin, VCAM-1 and P-selectin adhesion molecules. To that end, EC were exposed to the phyto-oestrogen in the presence or absence of LPS. As can be observed in Fig. 4, the mRNA levels of all molecules were marked enhanced after 20 h treatment with bacterial LPS (line D). Gen (10 nm) added alone (line A) down-regulated the mRNA expression of E-selectin, VCAM-1 and P-selectin. Moreover, when Gen was added before bacterial LPS treatment (line B), a partial (P-selectin) or complete (VCAM-1, E-selectin) down-regulation of the enhancement of mRNA expression induced by bacterial LPS was detected.

Fig. 4 Expression of endothelial cell (EC) adhesion molecules mRNA: regulation by genistein (Gen). EC were incubated with 10 nm Gen for 25 h (lane A), 10 nm Gen 5 h+bacterial lipopolysaccharide (LPS) for 20 h (lane B), vehicle alone (lane C) or 1 μg/ml bacterial LPS for 20 h (lane D). RT-PCR was performed as described in the Materials and methods section. PCR amplification products of E-selectin, VCAM-1, P-selectin are shown. The expected bands sizes are indicated. Results represent the means and standard deviations of three independent experiments (n 4). VCAM-1, vascular cell adhesion molecule; GAPDH, glycerldehyde-3-phosphate dehydrogenase.
Since DNA fragmentation is a key feature of cell undergoing apoptosis, we used electrophoretic analysis of DNA to evaluate the role of Gen on EC programmed death. EC were incubated for 24 h with 10 nm Gen. H2O2 (200 μm) was employed as apoptosis inductor. Fig. 5 shows an image of a representative ladder pattern of internucleosomal fragmentation (left) and the quantification of unfragmented DNA (right). Under basal conditions (without H2O2), DNA extracted from control and Gen-treated cells remain unfragmented. H2O2 addition induced a markedly DNA laddering compared with control cells. Preincubation with Gen before H2O2 addition, partially reverses DNA fragmentation elicited by the apoptotic inductor (94 v. 34 % DNA fragmentation, H2O2v. H2O2+Gen, respectively). These results suggest that the phyto-oestrogen attenuated the induction of apoptosis evoked by oxidative stress.

Fig. 5 Effect of genistein (Gen) on DNA fragmentation. Endothelial cell were incubated with 10 nm Gen or vehicle (C) for 24 h in the presence or absence of 200 μm H2O2 added 18 h after Gen treatment. DNA fragmentation was detected as described in the Materials and methods section. A representative image and results of integrated density of unfragmented DNA from three independent experiments are shown. * P < 0·001 v. control; † P < 0·01 v. H2O2; ‡ P < 0·01 v. Gen. ![]() , 10 nm Gen; □, control.
, 10 nm Gen; □, control.
Since cell migration is an important physiological process implicated in vascular tissue healing and reendothelialisation, we investigate the effect of Gen on EC migration. Figure 6 (a) and (b) shows representative microphotography of cell migration assays (0 and 48 h after scraping). The figure shows that the cells have passed the demarcated line and migrated to denuded area after 48 h of treatment with Gen or vehicle alone. As can be seen in Fig. 6(c), a minor number of cells crossed the demarcated line in Gen-treated culture in comparison to control condition (170 (sd 18) v. 65 (sd 6) number of cells/field, control v. 10 nm Gen), suggesting that Gen inhibits EC migration.

Fig. 6 Effect of genistein (Gen) on endothelial cells (EC) migration. Confluent EC cultures were serum starved for 24 h, and cells were removed by scraping. Detached cells were washed with PBS, and the remaining monolayer was treated during 48 h with 10 nm Gen or vehicle. EC migration was recorded at 0 and 48 h after treatment. Dotted lines indicate the boundary between the unscratched and scratched areas. (a) and (b): Images of representative fields of each condition after haematoxylin–eosin staining ( × 40). The scale bar represents 350 μm. (c) Bars show the means and standard deviations of number of migrated cells/field from three separated experiments performed by quadruplicate. * P < 0·001.
Discussion
The present study shows that the isoflavone Gen would exert a protective effect on vascular endothelium, due to its regulatory action on EC proliferation, apoptosis and leucocyte adhesion, events that play a critical role in vascular diseases. This modulatory action was detected either under physiological or inflammatory conditions. The molecular mechanism displayed by the phyto-oestrogen involves the participation of ER and the activation of NO pathway.
Serum concentration of Gen in human consuming dietary soya supplementation varies between 0·74–2·5 μm in Asian(Reference Izumi, Piskula and Osawa33) and 14–20 nm in western population(Reference Bhakta, Higgins and Sevak34). The concentration of Gen employed in the present study (0·1–100 nm) is close to these human plasma circulating levels.
We demonstrated that low concentration of Gen stimulated EC proliferation. At high doses, similarly as reported by other authors(Reference Koroma and de Juan35, Reference Fotsis, Pepper and Adlercreutz36), Gen exhibited an anti-mitogenic effect. Gen exerts both oestrogenic and non-oestrogenic actions that depend on the isoflavone concentration. It is described that Gen acts as protein tyrosine kinase inhibitor at concentrations higher than 10 μm, meanwhile at lower concentration, Gen binds to both isoforms of ER, having high affinity for ER-β. Moreover, it has been reported that, in human umbilical EC, 100 nm Gen strikingly increases ER-β expression(Reference Xu, Zhong and Ghavideldarestani37). We provided evidence that the mechanism of action of Gen involves ER participation, since the ER antagonist ICI 182780 blocked the mitogenic effect of the isoflavone. In agreement with this observation, we have previously reported the involvement of ER in the non-genomic stimulation of vasoactive compounds synthesis induced by Gen.
Endothelial-derived NO is a critical molecule for normal function of vessel, as well as for the progression/reversal of vascular disease. NO represents an important intracellular second messenger with potent vasodilator, anti-inflammatory, anti-atherogenic, anti-thrombotic and anti-apoptotic properties(Reference Moncada38). We have previously demonstrated that, in rat aortic rings, Gen acutely stimulates NO synthesis in a non-genomic manner(Reference Polini, Rauschemberger and Mendiberri17). In the present study, using isolated EC, we confirmed that this stimulatory action was exerted at endothelial level. The kinetic profile exhibited by Gen has a maximal stimulation of NO production at 20 min. This observation is in accordance with other authors(Reference Liu, Homan and Dillon15) who have showed that in bovine and human EC, Gen (1–10 μm) rapidly activates eNOS with maximal effect between 10 and 30 min. It has been reported that Gen stimulates NOS in a protein kinase A-dependent manner(Reference Liu, Homan and Dillon15). Other studies showed that ER modulators non-genomically enhance NOS activity through sequential activation of mitogen-activated protein kinase and phosphatidylinositol-3-kinase/Akt pathways(Reference Simoncini, Varone and Fornari39). Indeed, the natural oestrogen 17-β-oestradiol stimulates NO synthesis via ER/mitogen-activated protein kinase and phosphatidylinositol-3-kinase activation(Reference Meyer, Haas and Prossnitz40). Since we proposed that in our experimental model, the effect of Gen on NO production is mediated by ER, some of these intracellular signal transduction pathways may be involved in Gen action. This fact will be the subject of future studies.
We obtained evidence that shows a cross-talk between genomic and non-genomic effects of the isoflavone. When NO synthesis was suppressed by the presence of the NOS inhibitor l-nitro-arginine methyl ester, the mitogenic action of Gen was abolished. It has been reported that NO mediates the stimulation of EC proliferation and migration induced by IL-33, a member of the IL-1 cytokine family(Reference Choi, Choi and Min41). Some of the mechanisms proposed to be implicated in NO-mediated stimulation of cell proliferation are as follows: (a) the rapid release of NO activates the cytosolic guanylyl cyclase/PKG system which phosphorylates Raf-1 kinase and activates mitogen-activated protein kinase pathway; (b) up-regulation of cyclo-oxygenase-2 via phosphatidylinositol-3-kinase/Akt in a NO-dependent manner; (c) activation of cell cycle machinery enhancing expression of cyclin or decreasing cyclin-dependent kinase inhibitors(Reference Villalobo42).
We showed that Gen reduces EC migration. This inhibitory effect would be relevant to avoid vascular endothelial denudation and to maintain its selective permeable barrier properties by providing a continuous non-thrombogenic lining for the vascular system or for exerting a protective effect against atherosclerosis lesion progression. EC migration in vivo is a complex mechanism regulated by the local balance between pro and inhibitors migratory factors, and the cellular movement depends on coordinating and sensing of cell surroundings(Reference Lamalice, Le Boeuf and Huot43). Since, in our work we measured EC migration using isolated cells under static conditions, at present we could not address the potentially beneficial or deleterious effect of Gen on EC motility.
EC activation by proinflammatory cytokines or bacterial endotoxins predisposes to leucocyte recruitment. Monocytes adhesion is the first critical step in a vascular lesion. EC injury induces synthesis or release of surface adhesion molecules. Since selectins (E and P) are involved in initial leucocyte attachment and rolling, meanwhile, VCAM-1 triggers monocyte adherence and transendothelial migration to the intima(Reference Rao, Yang and García-Cardena4), we choose these CAM as target molecules to study. It has been reported that P- and E-selectins have overlapping and unique functions. P-selectin appears to be able for early leucocyte rolling and initial stages of an inflammatory response, while E-selectin allows slow and stable rolling and more leucocyte adhesion(Reference Jung and Ley44). P-selectin stored within Weibel-Palade bodies may be recruited to the vessel surface as rapidly as few min after inflammatory mediators, reaching its peak after only 10 min, followed by de novo synthesis of this selectin stimulated by the presence of proinflammatory agents such as bacterial LPS(Reference Blann, Nadar and Lip45). E-selectin is expressed constitutively on skin and bone microvessels, and it is induced by de novo synthesis in most organs and EC after inflammatory stimuli(Reference Barthel, Gavino and Descheny46). In the present study, we showed that Gen reduced monocyte adhesion to EC. Indeed, when EC were exposed to a proinflammatory stimulus such as LPS, pretreatment with the isoflavone attenuated the enhancement in leucocyte adhesion induced by LPS. In RT-PCR assays, we demonstrated that, in EC incubated with the isoflavone before bacterial LPS stimulation, the mRNA levels of P-selectin, E-selectin and VCAM-1 were attenuated or blunted compared with bacterial LPS alone. The partial down-regulation of P-selectin observed in our experiments may be due to its kinetic/profile of expression. Pretreatment with Gen could attenuate the stimulation of P-selectin mRNA synthesis after its initial release from EC stores. Moreover, if a lesion is installed, P-selectin blunted synthesis could contribute to prevent its progression. The absence of E-selectin and VCAM-1 mRNA in Gen plus bacterial LPS treatment may be consistent with this proposal. These results may support the hypothesis that Gen treatment would have a protective effect on the endothelium by diminishing initial leucocyte adhesion but also preventing the progression of the lesion in a hostile environment. The activation of EC by bacterial LPS and cytokines conducts to the generation of intracellular reactive oxygen species (mostly hydrogen peroxide) and increases the transcription of adhesion molecules target genes via the NF-κB system(Reference Collins, Read and Neish47). Our results suggest that the inhibitory action of Gen on monocyte adhesion would be due to the regulation of EC adhesion molecules transcription. In agreement with this, in human umbilical vein endothelial cells, red clover extracts containing Gen and daidzein inhibit endothelial expression of ICAM-1 and VCAM-1 induced by bacterial LPS(Reference Simoncini, Garibaldi and Fu48). Moreover, down-regulation of mRNA levels of vascular endothelial-cadherin, integrin α V and multimerin by micromolar concentration of Gen has also been reported(Reference Piao, Mori and Satoh49). Of further interest is the fact that NO exerts a regulatory action on CAM expression. It has been demonstrated that NO inhibits both exocytosis and de novo synthesis of P-selectin(Reference Armstead, Minchenko and Schuhl50, Reference Iafrati, Vitseva and Tanriverdi51). Indeed, in human umbilical vein endothelial cells, Gen inhibits TNFα-induced VCAM-1 expression in an NO-dependent manner(Reference Mukherjee, Nathan and Dinh52). Therefore, the non-genomic stimulatory action of Gen on NO production reported in our study would mediate the inhibitory action of the phyto-oestrogen on CAM expression by affecting either the initial or the later stage of monocyte adhesion. This hypothesis and the intracellular networks involved in Gen inhibitory action on monocytes adhesion will be further investigated in future studies.
EC lesion-prone regions are characterised by increased EC turnover rate, most likely due to enhance apoptosis. Classical risk factors such as oxidised LDL, oxidative stress and angiontensin II stimulate EC apoptosis(Reference Kockx and Herman53–Reference Dimmeler, Rippmann and Weiland55). The apoptotic cells are removed by the blood stream and are replaced by regenerated endothelium formed by neighbouring cells and/or circulating endothelial progenitor cells(Reference Vanhoutte56). Our results show that Gen does not modify basal cell programme death rate, but under oxidative stress conditions, the isoflavone prevents H2O2-induced apoptosis. Gen inhibition of EC apoptosis via Bcl-2/Bax regulation in diabetic EC model has been reported(Reference Xu, Zhong and Ghavideldarestani37).
Taken together, the results obtained in monocytes adhesion assays and DNA laddering may support that Gen would exert a protective effect against oxidative stress or proinflammatory injury in vascular endothelium.
In summary, the present study provides an integral evaluation of the effect of Gen on cellular events that would be altered under vascular injury. The isoflavone might have a beneficial role on EC behaviour prompting vasoactives synthesis and reducing EC motility, apoptosis and monocyte adhesion, either under basal or injury conditions. Whether the administration of this phyto-oestrogen would be helpful to prevent, treat or reverse chronic inflammatory process that underlies many CVD remains to be clarified. Further investigations are required to understand the physiological relevance of our observations.
Acknowledgements
All the authors have seen and approved the contents of the submitted manuscript. All the authors stated that there are no conflicts of interest and all the authors adhere to the Committee on Publication Ethics guidelines on research and publication ethics. This research was supported by grants from the SGCyT, Universidad Nacional del Sur, Argentina (PGI 24/B093); Agencia Nacional de Promoción Científica (PICTO 527, ANPCYT/UNS); Consejo Nacional de Investigaciones Científicas y Técnicas (PIP 5790 CONCET, Argentina). The contribution of each author was as follows: M. J. S., P. H. C. and M. B. R.: doing experiments; M. J. S., P. H. C., M. B. R. and V. L. M.: writing of the manuscript; V. L. M.: designing the study, promotor, investigator and coordinator.









