Elastic fibres are insoluble components of the extracellular matrix (ECM) of extensible connective tissues such as large arteries, skin, lungs, ligaments and auricular cartilage (Ref. Reference Kielty1). They endow these tissues with the mechanical properties necessary to withstand repeated cycles of stretch and recoil through life. They also influence the bioavailability of transforming growth factor β (TGFβ) family growth factors (Ref. Reference Nistala2). Although fibrillin microfibrils arose in early metazoans (Refs Reference Özbek3, Reference Piha-Gossack, Sossin and Reinhardt4) (Fig. 1), elastin and elastic fibres emerged only during vertebrate evolution as an essential requirement to reinforce high-pressure circulatory systems (Ref. Reference Faury5). Elastic fibres comprise an inner core of cross-linked elastin ensheathed within fibrillin microfibrils. These long-lasting structures begin to assemble during mid-gestation, with little adult elastic fibre assembly.

Figure 1. Evolutionary analysis of fibrillin. A list of species from the fibrillin (FBN) sequence alignment was generated, and used to create a common tree using the National Center for Biotechnology Information (NCBI) Taxonomy Browser. The resulting tree was imported into Cytoscape version 2.7, and generated using the organic layout. FBN types were identified using phylogenetic analysis and domain analysis of the FBN multiple sequence alignments and FBN types were mapped onto the common tree. Identified sequences were grouped into 5 categories: vertebrate FBNs 1-3, invertebrate FBN and fibrillin-like epidermal growth factor (EGF) array-containing proteins. The latter sequences had a high identity to FBN sequences but only contained EGF domains organised in arrays. Also indicated are the sequences, from the appearance of chordates that contain the arg-gly-asp (RGD) cell binding motif (branches shown in light brown). By comparison, branches shown in light purple precede chordates and RGD. The first emergence of other key extracellular proteins that interact with FBN is shown (dark blue arrows), along with the evolutionary timescale in million years ago (MYA).
Mature elastic fibres have tissue-specific architectural arrangements that reflect different elastic requirements. Thus, arterial elastic fibres form concentric lamellar layers that support vascular elastic recoil. Dermal elasticity is based on integrated networks of thick reticular elastic fibres and thin fibres in the papillary dermis, and alveolar elastic fibres form fine networks that allow respiratory expansion and contraction (Ref. Reference Kielty1).
Although tropoelastin and fibrillin are the two principal structural components of elastic fibres, several ‘accessory’ molecules contribute to elastic fibre assembly and function. From initial microfibril formation to the deposition and cross-linking of tropoelastin monomers upon microfibrils, these accessory proteins support the progression of elastic fibre formation in a spatially and temporally appropriate manner. For example, small fibulins influence the association of elastin with its cross-linking enzymes, thereby facilitating its stable deposition on microfibrils (see the Assembly of elastic fibres section).
Aberrant elastic fibre formation and/or altered homoeostasis cause many inherited and acquired diseases, with phenotypes ranging from mild (e.g. loose skin) to severe and potentially life-threatening (e.g. vascular defects) (see the Elastic fibre disorders section). Heritable elastic fibre disorders include Marfan syndrome (MFS) caused by fibrillin-1 mutations (Ref. Reference Robinson6) and autosomal dominant cutis laxa (ADCL) caused by elastin mutations (Ref. Reference Sugitani7). Mutations in accessory glycoproteins such as fibulins-4 or -5 cause autosomal recessive cutis laxa (ARCL) (Ref. Reference Berk8), and in a disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin-10 (ADAMTS-10) cause Weill–Marchesani syndrome (WMS) (Ref. Reference Dagoneau9). Acquired elastic fibre disorders arise wherever elastic fibre structure and/or function are compromised; for example, actinic elastosis and vascular degeneration, as well as acquired forms of cutis laxa (CL) and pseudoxanthoma elasticum (PXE).
In this review, we describe the current understanding of microfibril and elastic fibre composition and formation, delineate the diseases associated with elastic fibre defects and summarise the latest therapeutic advances.
Composition of elastic fibres
The structural and associated components of elastic fibres are outlined below; their roles in elastic fibre formation and function are further delineated below (see the Assembly of elastic fibres section).
Structural molecules
Elastin
Elastin is the most abundant protein of elastic fibres, comprising approximately 90% of the mature structure. It is encoded by a gene on chromosome 7q11.2, and secreted as the soluble precursor tropoelastin (60–70 kDa) from elastogenic cells such as fibroblasts, smooth muscle cells and auricular chondrocytes. Tropoelastin has a multi-domain structure comprising alternating hydrophobic and lysine-containing (lysine–alanine and lysine–proline) cross-linking domains. The lysine–alanine-rich domains preferentially participate in desmosine cross-link formation (Ref. Reference Dyksterhuis and Weiss10). The primary transcript can undergo tissue-specific alternative splicing that modifies elastin incorporation into fibres (Ref. Reference Sugitani7).
Tropoelastin is an asymmetric molecule, which has two functionally distinct regions (Ref. Reference Baldock11): an N-terminal elastic coil that endows spring-like properties on tropoelastin, and a C-terminal cell interactive module that facilitates cell adhesion via an association between the αvβ3 integrin and a GRKRK motif (Ref. Reference Bax12). They are separated by a bridge region, within which are domains shown to contain sites of contact for coacervation (Ref. Reference Dyksterhuis13). Destabilisation of this region by mutagenesis has been shown to impair elastogenesis (Ref. Reference Yeo14).
Fibrillins
Fibrillin molecules assemble to form microfibrils (Refs Reference Hubmacher, Tiedemann and Reinhardt15, Reference Ramirez and Sakai16). In man, there are three fibrillin genes, each encoding 350 kDa multi-domain glycoproteins (fibrillins 1–3) (Ref. Reference Piha-Gossack, Sossin and Reinhardt4). Each fibrillin comprises 43 calcium-binding epidermal growth factor-like (cbEGF) domains, five EGF-like domains, seven eight-cysteine-containing (TB) motifs and two hybrid domains with similarities to TB and cbEGF-like domains (Fig. 2). There is a unique proline-rich region in the N-terminal region of fibrillin-1, with corresponding regions of fibrillin-2 and fibrillin-3 being richer in glycine.
Figure 2. Domain structures of elastic fibre molecules. The domain organisations of fibrillins, latent TGFβ-binding protein (LTBPs), fibulins, a disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin (ADAMTS) and ADAMTS-like (ADAMTSL) molecules and tropoelastin are shown, with keys for domain types.
Fibrillin-1 (gene on chromosome 15q21.1) is the most abundant fibrillin isoform; it is expressed throughout life and is required for microfibril homoeostasis, whereas fibrillin-2 (gene on chromosome 5q23-q31) and fibrillin-3 (gene on chromosome 19p3) are expressed predominantly during development (Refs Reference Charbonneau17, Reference Zhang18, Reference Corson19, Reference Sabatier20). Overlapping expression of fibrillins occurs in developing blood vessels, cartilage, bone and lungs, but tissue-specific differences are also apparent, e.g. fibrillin-1 in dermis, fibrillin-2 in peripheral nerves and fibrillin-3 in cartilage, perichondrium and developing bronchi (Ref. Reference Sabatier20). Fibrillin-2 may form the inner core of many fibrillin-1 containing microfibrils (Ref. Reference Charbonneau21). Although rarely detected in adult skin, fibrillin-2 expression can increase during wound healing and in sclerosis (Ref. Reference Brinckmann22). Mouse models confirm the critical importance of fibrillin-1 to vascular development and function. Fibrillin-1 null mice die perinatally from ruptured aortic aneurysm, whereas fibrillin-2 null mice have seemingly normal formation of the aorta, although double-null mice have a much more severe aortic phenotype than fibrillin-1 null mice (Ref. Reference Carta23). Both fibrillins thus perform partially overlapping functions during the development of the aorta and great arteries, with fibrillin-1 able to compensate for loss of fibrillin-2 but not vice versa. The fibrillin-3 gene is disrupted in rodents; so fibrillin-3 is not critical to all mammalian life (Ref. Reference Corson19).
Accessory molecules associated with microfibrils and elastic fibres
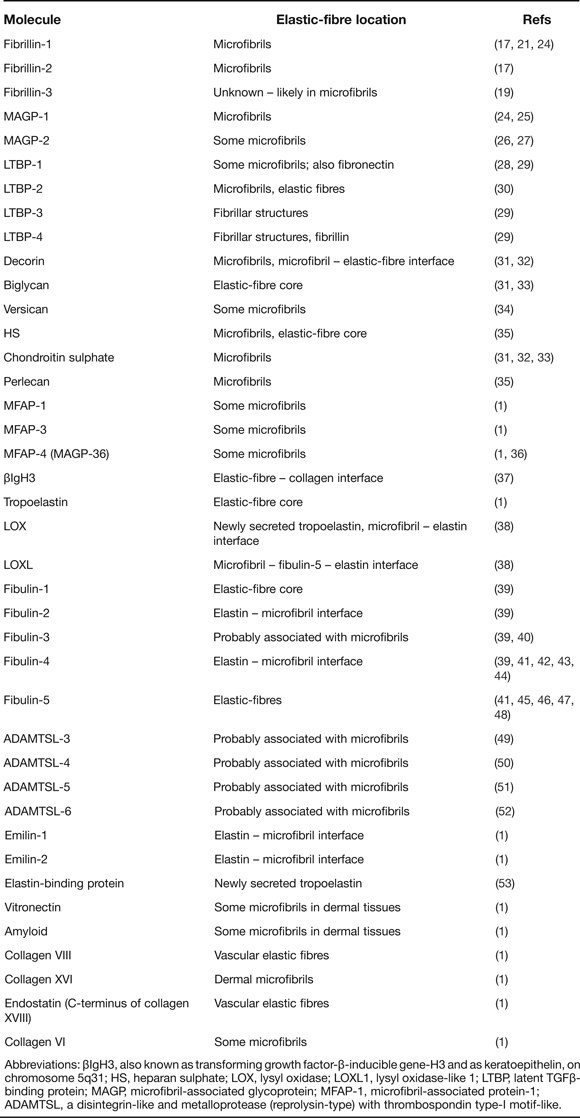
Many microfibril and elastic fibre-associated molecules have been identified by microscopy and immunochemical approaches (reviewed in Ref. Reference Kielty1; updated in Table 1). We summarise here selected molecules shown by functional analysis, mouse models and/or heritable diseases to contribute to elastic fibre formation and function; microfibril-associated molecules include the latent TGFβ binding proteins (LTBPs), ADAMTS isoforms and microfibril-associated glycoprotein (MAGPs) and elastic fibre-associated molecules include the small fibulins 3–5 and lysyl oxidase (LOX).
Table 1. Microfibril and elastic fibre-associated molecules (updated from Ref. Reference Kielty1)
Abbreviations: βIgH3, also known as transforming growth factor-β-inducible gene-H3 and as keratoepithelin, on chromosome 5q31; HS, heparan sulphate; LOX, lysyl oxidase; LOXL1, lysyl oxidase-like 1; LTBP, latent TGFβ-binding protein; MAGP, microfibril-associated glycoprotein; MFAP-1, microfibril-associated protein-1; ADAMTSL, a disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type-I motif-like.
LTBPs: LTBPs 1–4 are large extracellular glycoproteins with a multi-domain structure that has similarities to the fibrillins, comprising cbEGF and TB domains, with an N-terminal cysteine-rich domain. They are expressed in many tissues including heart, placenta, lung, kidney, skeletal muscle and ovary and have cbEGF and TB domain homology with fibrillins (Refs Reference Piha-Gossack, Sossin and Reinhardt4, Reference Todorovic54) (Fig. 2). The third TB domain of LTBPs 1–4 can covalently bind the propeptide (latency-associated peptide, LAP) of the cytokine TGFβ (Refs Reference Gleizes55, Reference Saharinen, Taipale and Keski-Oja56), which is rendered inactive when associated with its propeptide (forming the small latent complex). Association of this complex with LTBP forms the large latent complex, which can be sequestered within the ECM, thereby regulating TGFβ bioavailability (Refs Reference Todorovic54, Reference Gleizes55, Reference Saharinen, Taipale and Keski-Oja56). C-terminal regions of LTBPs 1, 2 and 4 can interact directly with fibrillin-1 through TB4 (Ref. Reference Saharinen, Taipale and Keski-Oja56), thus implicating fibrillin microfibrils in regulating TGFβ activity. LTBP-1 (and thus latent TGFβ) can be deposited in the absence of fibrillins-1 and -2 (Ref. Reference Zilberberg26), although depletion of fibrillin-1 disrupts its pattern of extracellular deposition (Refs Reference Massam-Wu28, Reference Isogai57).
ADAMTS ( a disintegrin-like and metalloprotease (reprolysin-type) with thrombo spondin type-I motif) molecules: Several members of this superfamily, including ADAMTS-10 and ADAMTSL 2–6, have been genetically and/or functionally implicated in microfibril biology (see the Assembly of elastic fibres section). The full superfamily comprises 19 zinc metalloproteases and seven noncatalytic ADAMTS-like (ADAMTSL) proteins, varying greatly in size; many members of this family play roles in morphogenesis, angiogenesis and ovulation and in ECM deposition (Ref. Reference Apte58). Briefly, each ADAMTS protease has an N-terminal protease domain, containing a catalytic module, a disintegrin-like module, a cysteine-rich module and a single thrombospondin type I repeat (TSR) (Fig. 2). The C-terminal (or ancillary) domain is variable between family members, although many contain multiple TSRs and may confer substrate-binding specificity, with the N-terminal domain providing catalytic activity. ADAMTS-10 lacks an optimal furin cleavage site (necessary for activation of ADAMTS enzymes) and is unlikely to be catalytically active in vivo. ADAMTSL family members, including those implicated in microfibril biology, contain multiple TSRs but lack disintegrin-like and catalytic domains. Thus, low/no catalytic activity is a hallmark of ADAMTS family members with identified roles in microfibril biology.
MAGPs: MAGP-1 (a ~31 kDa glycoprotein) was originally detected in microfibril preparations from elastic and nonelastic tissues (Ref. Reference Gibson, Kumaratilake and Cleary59). It has an acidic N-terminal region enriched in proline and glutamine residues, and a cysteine-rich C-terminal portion. It colocalises widely with microfibrils (Ref. Reference Gibson, Kumaratilake and Cleary59), and has been detected in purified microfibrils by mass spectrometry (Ref. Reference Cain60). Surprisingly, mice lacking the MAGP-1 gene show normal microfibrils and elastic fibre assembly (Refs Reference Craft24, Reference Weinbaum61), so it is not essential for elastogenesis. Structurally related glycoprotein MAGP-2 (~25 kDa) has a cysteine-rich central region, is rich in serine and threonine and binds integrins. It colocalises with microfibrils in some tissues (Refs Reference Lemaire27, Reference Gibson62, Reference Penner63, Reference Hanssen64). Both MAGPs can bind fibrillin and microfibrils, and may enhance the deposition of elastin on microfibrils (see the Assembly of elastic fibres section).
Fibulins: Fibulins are extracellular glycoproteins that are classified as Class I or II on the basis of their domain structure and length (Ref. Reference Yanagisawa and Davis39). The closely related Class II (or short) fibulins (fibulins 3–5; 50–60 kDa) each comprise six cbEGF domains, including an atypical N-terminal cbEGF and a C-terminal fibulin (‘FC’) module (Fig. 2) and are widely expressed in developing embryos, particularly cardiovascular and skeletal tissues (Ref. Reference Yanagisawa and Davis39). Fibulins-4 and -5 can bind tropoelastin and LOX or LOX-like (LOXL) cross-linking enzymes, as well as fibrillin-1; by juxtaposing these molecules, they are thought to support the cross-linking of elastin and its deposition on microfibrils.
LOX family: The five members of the LOX family (LOX, LOXL1-4) are secreted as zymogens, and are activated by removal of N-terminal propeptides by bone morphogenetic protein-1 (BMP-1) (Ref. Reference Molnar65). LOX especially, but also its homologue LOXL1 catalyses the oxidative deamination of peptidyl lysine residues in elastin to generate α-aminoadipic-δ-semialdehydes that then spontaneously condense to form covalent desmosine and isodesmosine cross-links, and a very stable insoluble elastin core (Ref. Reference Molnar65). Their C-terminal regions show sequence homology but their N-terminal ‘pro’ regions vary greatly, and may impart specificity to the extracellular targeting of LOX enzymes.
Assembly of elastic fibres
Microfibril formation
Assembly of fibrillin monomers into microfibrils is a complex process. ‘Pro'fibrillin molecules are processed N- and C-terminally by furin/PACE proprotein convertases, either immediately before or upon secretion, thereby facilitating terminal interactions that enable linear and lateral multimerisation (Ref. Reference Marson66). Homotypic N-terminal interactions are enhanced by heparan sulphate (HS) (Ref. Reference Cain67), whereas C-terminal interactions may underpin bead formation (Ref. Reference Hubmacher68). In this way, microfibrils are thought to assemble at the cell surface (Fig. 3a). Microfibril formation is unlikely to be solely a self-assembly process, and there is some evidence for cellular involvement. We reported that, in fibroblasts, microfibril deposition requires fibronectin arg–gly–asp (RGD)-dependent α5β1 integrins (Ref. Reference Kinsey70). Fibrillin-1 can also interact with cells through integrins α5β1 and αvβ3, and αvβ6 in epithelial cells (Refs Reference Bax71, Reference Jovanovic72). It remains to be determined whether direct fibrillin-1 interactions with cells are necessary for microfibril assembly and/or for specific tissue-specific functions of microfibrils (and elastic fibres) such as association with smooth muscle cells in the aorta.
Figure 3. Schematic diagram of microfibril and elastic fibre assembly: elastin assembly. (a) Microfibril assembly occurs pericellularly, and requires fibronectin, integrins and heparan sulphate proteoglycans (HSPG). Fibrillin molecules are secreted and, after processing N- and C-terminally by furin, interact homotypically at N- and C-termini leading to axial and lateral assembly to form microfibrils. Beads may arise from folding of terminal regions. Microfibrils may be stabilised by transglutaminase cross-links. The reason why fibronectin is needed for microfibril deposition is unclear, but it may act as a template for assembly and/or it may stimulate cytoskeletal tension through the α5β1 integrin, thereby facilitating assembly at fibrillar adhesions. Fibrillin-1 also interacts with α5β1, αvβ3 and αvβ6 integrins; however, it is not known whether these interactions are essential for microfibril assembly. Heparin inhibits microfibril assembly, and HSPGs may contribute by facilitating cell surface fibrillin-1 interactions. (b) Elastin assembly occurs pericellularly on ‘microassembly’ and on microfibrils ‘macroaggregates’ (Ref. Reference Wagenseil and Mecham69). Secreted tropoelastin forms globules at the cell surface which become cross-linked by lysyl oxidase; this process may involve αvβ3 integrin interactions with tropoelastin, and integrin interactions with heparan sulphate proteoglycans (HSPGs) which can interact with tropoelastin. Fibulin-4 and fibulin-5 contribute to elastin cross-linking by lysyl oxidase, and probably direct the deposition of elastin globules onto preformed fibrillin microfibrils, to form elastic fibres. Microfibrils and elastic fibres are important matrix storage sites for BMPs and latent TGFβ1.
Assembled microfibrils comprise fibrillin monomers aligned in a head-to-tail arrangement and laterally associated (probably eight molecules in cross-section) (Refs Reference Reinhardt73, Reference Baldock74, Reference Wang, Lu and Baldock75). Fibrillin molecules within tissue microfibrils can be cross-linked by transglutaminase (Ref. Reference Qian and Glanville76). The ‘beads-on-a-string’ appearance of individual microfibrils, their untensioned periodicity (50–60 nm) in tissue sections (Refs Reference Reinhardt73, Reference Baldock74) and their altered structural organisation upon extension (Refs Reference Wang, Lu and Baldock75, Reference Baldock77) has given rise to unstaggered (Refs Reference Baldock74, Reference Wang, Lu and Baldock75) and staggered (Refs Reference Jensen, Robertson and Handford78, Reference Sabatier79) models of fibrillin alignment within microfibrils. The precise alignment of fibrillin molecules within microfibrils awaits further insights, perhaps from approaches such as experimental cross-linking.
Role for fibronectin
In mesenchymal cultures, fibronectin is an essential prerequisite for microfibril formation (Refs Reference Zilberberg26, Reference Kinsey70, Reference Sabatier79). This relationship is surprising because fibrillin microfibrils arose in early metazoans and fibronectin-like molecules only in chordates (Refs Reference Özbek3, Reference Piha-Gossack, Sossin and Reinhardt4, Reference Tucker and Chiquet-Ehrismann80). Fibronectin is able to bind several regions of fibrillin-1, as well as fibrillin multimers, whereas newly deposited microfibrils and fibronectin networks show initial colocalisation (Refs Reference Kinsey70, Reference Sabatier79, Reference Hubmacher81). It has been suggested that fibronectin could act as a template for microfibril deposition (Refs Reference Kinsey70, Reference Sabatier79); however fibrillin microfibrils occur in many lower organisms that lack fibronectin (Ref. Reference Reber-Müller82), and in fibronectin-null cultures (Ref. Reference Dallas83), implying that fibronectin enhances rather than underpins microfibril assembly.
Role for HS
The glycosaminoglycan HS is strongly implicated in microfibril and elastic fibre assembly. It is a component of syndecan and glypican receptors, and of the basement membrane molecule perlecan. Supplementation of cell cultures with heparin effectively ablates microfibril assembly (Refs Reference Tiedemann84, Reference Ritty85). At least six high affinity-binding sites between heparin and fibrillin-1 have been identified (Refs Reference Cain67, Reference Tiedemann84, Reference Ritty85, Reference Cain86), some of which direct N-terminal multimerisation during microfibril assembly (Ref. Reference Cain67) or interactions with cell surface HS (Ref. Reference Bax71). Although it is not known precisely how HS contributes to microfibril deposition, these HS-directed interactions may be essential. Heparin does not disrupt N- and C-terminal fibrillin-1 interactions and so these interactions are compatible with microfibril assembly (Ref. Reference Cain86), and may directly support these homotypic interactions. Murine perlecan (recombinant fragment or molecules isolated from Engelbreth-Holm-Swarm (EHS) sarcoma; Ref. Reference Tiedemann35) or human perlecan expressed by ARPE-19 retinal epithelial cells (Ref. Reference Cain60) binds fibrillin-1 through protein and HS interactions, and directs microfibril-basement membrane interactions. HS competes with MAGP-1 and tropoelastin (Ref. Reference Cain67) to bind fibrillin-1; hence, it may regulate elastin deposition onto microfibrils. HS can also mediate smooth muscle cell interactions with tropoelastin, thus influencing cell phenotype (Ref. Reference Akhtar87). It also directly influences elastin multimerisation (Ref. Reference Gheduzzi88).
Roles for microfibril-associated molecules
LTBP-1 co-localises with microfibrils in some tissues (Ref. Reference Reber-Müller82). This juxtaposition, together with the ability of LTBPs (Refs Reference Kielty1, Reference Özbek3, Reference Piha-Gossack, Sossin and Reinhardt4) to form large latent TGFβ complexes, and to bind fibrillin-1 (Ref. Reference Ono89), implicates microfibrils in TGFβ sequestration. Recently, it was shown that fibrillins are needed for matrix deposition of LTBP-3, but not LTBP-1 (Ref. Reference Zilberberg26). LTBP-1 deposition in the ECM is however, such as fibrillin-1, dependent on fibronectin and LTBP-1 colocalises with microfibrils but not fibronectin over time in culture (Refs Reference Zilberberg26, Reference Reber-Müller82). Fibronectin is also required for LTBP-4 deposition (Ref. Reference Kantola90). However, incorporation of LTBP-2, which does not bind TGFβ, as well as LTBP-3 and LTBP-4, into ECM is dependent on microfibrils (Refs Reference Zilberberg26, Reference Vehvilainen91). LTBP-2 also competes with LTBP-1 to interact with the N-terminal region of fibrillin-1 (Ref. Reference Hirani92). Thus, while none of the LTBPs is necessary for fibrillin microfibril assembly, microfibrils and fibronectin both profoundly influence the extracellular deposition of LTBPs.
Several members of the ADAMTS superfamily are genetically and functionally implicated in microfibril biology (Ref. Reference Hubmacher and Apte93). ADAMTS-10, which causes autosomal recessive WMS (see the Elastic fibre disorders section) may not be physiologically cleaved by furin and is probably not an active enzyme in vivo. ADAMTSL-5 binds both fibrillin-1 and -2, as well as heparin, and co-localises with microfibrils (Ref. Reference Bader51). ADAMTSL-6 binds to the N-terminal region of fibrillin-1, whereas the conditioned medium-containing recombinant ADAMTSL-6 suggested potential to promote fibrillin-1 matrix assembly (Ref. Reference Tsutsui52), and it enhanced microfibrils in an MFS mouse model (Ref. Reference Saito94). ADAMTSL-3 is implicated in a molecular pathway involving fibrillin-1 and ADAMTS-10 (Ref. Reference Sengle49); it can bind fibrillin-1 and may participate in microfibril biogenesis (Ref. Reference Kutz95). Supplementation with recombinant ADAMTSL-4 accelerated fibrillin-1 deposition in culture, and co-localisation with microfibrils (Ref. Reference Gabriel50). It is interesting to speculate that inactive ADAMTS-10 and the ADAMTSL 3–6 molecules could act as ‘decoys’ to protect fibrillins from active ADAMTS enzymes.
MAGP-1 can interact with elastin and may contribute to elastin deposition on microfibrils (Refs Reference Jensen25, Reference Rock96). It strongly binds an N-terminal sequence of fibrillin-1, thereby inhibiting N- and C-terminal fibrillin-1 interactions (Ref. Reference Jensen25), and thus has the potential to fine-tune fibrillin multimerisation. MAGP-1 can also bind TGFβ and BMP-7 (Ref. Reference Weinbaum61), and can activate pSmad2 signalling in culture (Ref. Reference Massam-Wu28), so it may contribute to regulating TGFβ growth factors in association with microfibrils (Ref. Reference Weinbaum61). Intracellular MAGP-1 can modulate expression of versican (Ref. Reference Segade97), which interacts with microfibrils (Ref. Reference Isogai34) and is a regulator of matrix. MAGP-1 can also bind fibronectin (Ref. Reference Werneck98) and could influence fibronectin-mediated microfibril deposition. None of these putative functional contributions to elastic fibre formation can be essential since MAGP-1 null mice have functional elastic fibres (Ref. Reference Craft24). MAGP-2 can interact with fibrillins-1 and -2, has covalent periodic association with isolated microfibrils and co-localises with microfibrils in certain tissues (Refs Reference Lemaire27, Reference Penner63, Reference Hanssen64). MAGP-2 expression peaks during elastic fibre assembly; evidence shows that its overexpression increases elastic fibre formation and that it can stimulate elastic fibre assembly, probably by targeting elastin onto microfibrils (Ref. Reference Lemaire27).
Elastic fibre formation
Tropoelastin has the propensity to self-assemble through a process termed coacervation, which involves rapid molecular association, at increasing temperatures (Ref. Reference Yeo, Keeley and Weiss99). Exposed hydrophobic domains interact in an entropically-driven process, resulting in lysine residues aligning in readiness for LOX-mediated cross-linking. In vitro, the sizes and properties of coacervation ‘droplets’, and the rate at which coacervation and maturation processes proceed, are dependent on tropoelastin concentration, pH, temperature and solution conditions (Ref. Reference Cirulis and Keeley100), as well as the presence of accessory proteins. Fibulin-5 and also fibulin-4, limit aggregation of tropoelastin and slows the maturation phase (Ref. Reference Choi101). The N-terminal region of fibrillin-1 and MAGP-1 both increase maturation velocity and induce clustering of elastin droplets, and the fibrillin-1 fragment induces ‘strings’ of linear droplets (Ref. Reference Cirulis and Keeley100).
In culture and tissues, elastin can self-associate pericellularly into globules that become cross-linked prior to deposition onto microfibrils (Refs Reference Wagenseil and Mecham69, Reference Czirok102, Reference Sato103) (Fig. 3b). This initial stage in elastic fibre assembly, termed ‘microassembly’, may involve interaction of the C-terminal region of tropoelastin with αvβ3 integrin (Ref. Reference Bax12) and HS (Refs Reference Akhtar87, Reference Broekelmann104). Certainly, this region of elastin is critical for elastic fibre assembly (Ref. Reference Sugitani7). In culture, pericellular globule aggregation is coupled with cell motion (Ref. Reference Czirok102), with globule size probably limited by accessory proteins (Ref. Reference Wagenseil and Mecham69) and biophysical constraints (Ref. Reference Cirulis and Keeley100). Once transferred to the microfibril scaffold, the globules coalesce and are stabilised by formation of cross-links facilitated by LOX or LOXL1, probably facilitated by fibulins-4 or -5 (see below), thus forming the insoluble elastin core in a process termed ‘macroassembly’ (Fig. 3b) (Refs Reference Yeo, Keeley and Weiss99, Reference Cirulis and Keeley100).
Roles for fibulins
The homologous small fibulins (Reference Özbek3–Reference Faury5) are critical regulators of elastic fibre assembly (Refs Reference Yanagisawa and Davis39, Reference Kobayashi41). Fibulin-4 colocalises with microfibrils (Ref. Reference Kobayashi41), and binds fibrillin-1 in vitro (Ref. Reference Choudhury42). Fibulin-4 depletion decreases elastic fibre formation but not microfibrillogenesis (Ref. Reference McLaughlin43). Its involvement in the pathogenesis of the disease CL (see the Elastinopathies section) identifies it as an essential element of elastic fibre formation. Fibulin-4 null mice, which die perinatally, have a drastic reduction in desmosine cross-links and grossly impaired elastic fibre formation (Ref. Reference McLaughlin43). Fibulin-4 can bind LOX concurrently with tropoelastin (Ref. Reference Choudhury42) and influence LOX-tropoelastin interactions (Ref. Reference Horiguchi44). Thus, it is considered that fibulin-4 is essential for LOX-mediated elastin cross-linking, probably by forming complexes that juxtapose LOX and elastin (Refs Reference Choudhury42, Reference Horiguchi44). Fibulin-4 may also play a, as yet poorly defined, role in the sequestration of LTBPs (and thus latent TGFβ) within the ECM (Ref. Reference Ono89). Fibulin-5 null mice exhibit a less dramatic elastic fibre phenotype than fibulin-4 null mice, with only 16% decrease in desmosine levels; mice survive well into adulthood but have loose skin (Refs Reference Yanagisawa45, Reference Nakamura46). Fibulin-5 localises at the interface between the elastin core and microfibrils (Ref. Reference Kobayashi41). It can interact with LOXL enzymes, and it enhances tropoelastin self-association and elastic fibre formation (Ref. Reference Hirai47). Fibulin-5 is also capable of integrin-mediated cell attachment, but does not activate α5β1 integrin (Ref. Reference Lomas48). Fibulin-5 may act as a molecular adapter that directs the deposition of elastin microaggregates onto microfibrils (Refs Reference Choudhury42, Reference Wagenseil and Mecham69, Reference El-Hallous105, Reference Zheng106). Indeed, fibulin-5 knockdown in culture-altered elastin aggregates (Ref. Reference Choudhury42). Like its homologues, fibulin-3 null mice exhibit elastic fibre deficiencies in some tissues as well as herniation (Ref. Reference McLaughlin40). A fibulin-3 point mutation (R345W) caused malattia leventinese, a dominantly inherited macular degenerative disease with sub-retinal pigment epithelial deposits (Refs Reference Michaelides107, Reference Marmorstein108) with similarities to age-related macular degeneration (ARMD).
The different severities and tissue defects induced by depletion of fibulins 3–5 may reflect, in part, their tissue-specific expression patterns. It is also likely that some evolutionary diversification of their roles in elastic fibre assembly has occurred, e.g. fibulin-4 preferentially interacts with LOX and fibulin-5 with LOXL enzymes (Refs Reference Choudhury42, Reference Horiguchi44, Reference Hirai47).
Elastic fibre disorders
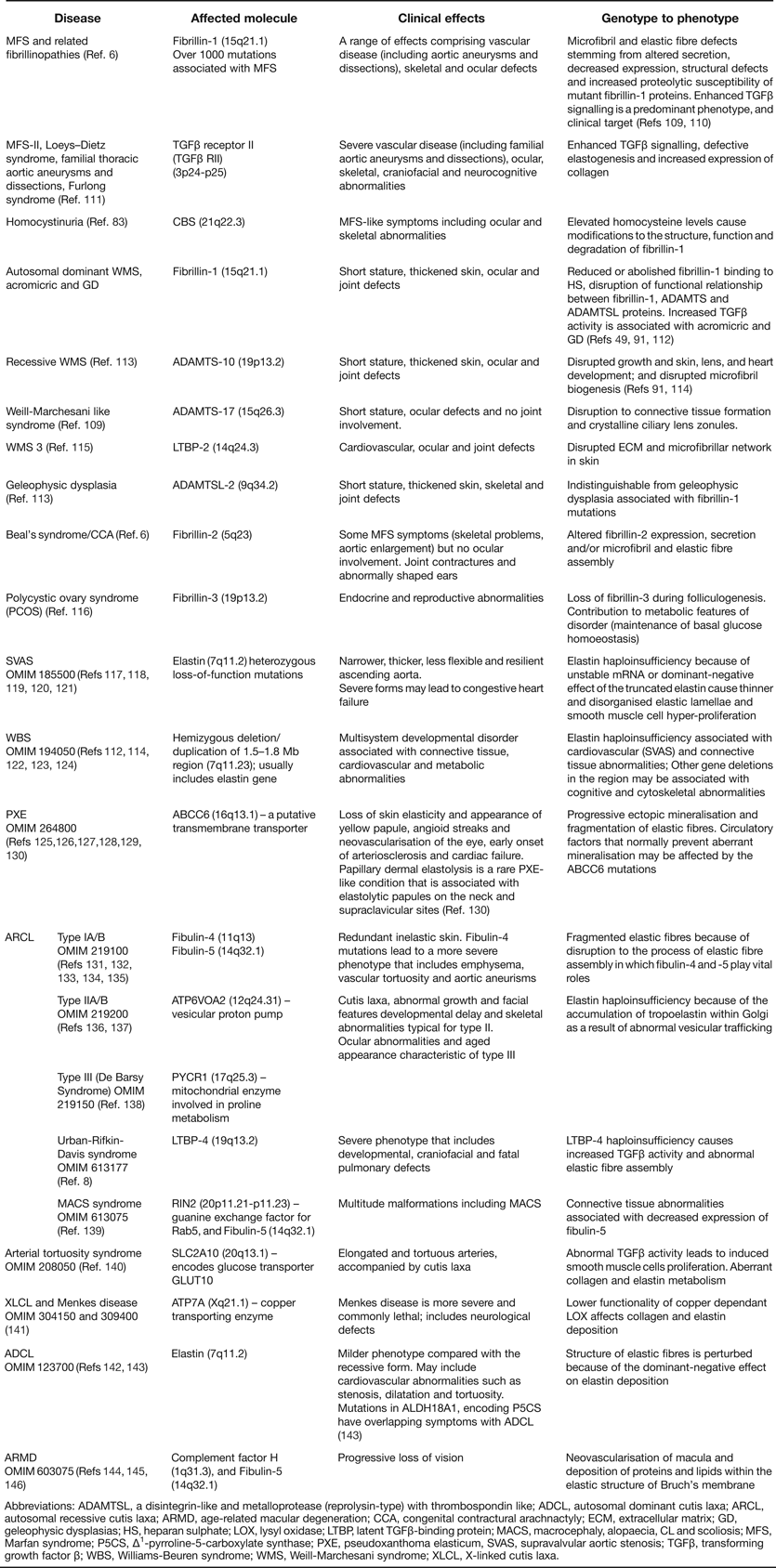
Inherited and acquired diseases of elastic fibres are also reviewed in Ref. Reference Kielty1 (updated in Table 2).
Table 2. Heritable disorders of elastic fibres (updated from Ref. Reference Kielty1)
Abbreviations: ADAMTSL, a disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin like; ADCL, autosomal dominant cutis laxa; ARCL, autosomal recessive cutis laxa; ARMD, age-related macular degeneration; CCA, congenital contractural arachnactyly; ECM, extracellular matrix; GD, geleophysic dysplasias; HS, heparan sulphate; LOX, lysyl oxidase; LTBP, latent TGFβ-binding protein; MACS, macrocephaly, alopaecia, CL and scoliosis; MFS, Marfan syndrome; P5CS, Δ1-pyrroline-5-carboxylate synthase; PXE, pseudoxanthoma elasticum, SVAS, supravalvular aortic stenosis; TGFβ, transforming growth factor β; WBS, Williams-Beuren syndrome; WMS, Weill-Marchesani syndrome; XLCL, X-linked cutis laxa.
Fibrillinopathies
Heritable connective tissue disorders termed fibrillinopathies arise from mutations in fibrillin genes. Fibrillin-1 disorders include MFS, ectopia lentis, MASS (mitral valve prolapse, aortic enlargement, skin and skeletal findings) syndrome, Shprintzen–Goldberg syndrome, Stiff skin syndrome, autosomal dominant WMS and acromicric and geleophysic dysplasias (AD, GD) (Refs Reference Robinson6, Reference Dietz and Pagon109, Reference Loeys116, Reference Faivre115). Fibrillin-1 is also implicated in the pathogenesis of homocystinuria, which exhibits MFS-like symptoms (Ref. Reference Dallas83). Congenital contractural arachnodactyly (CCA; Beal's syndrome) arises from mutations in fibrillin-2, and polycystic ovary syndrome is associated with mutations in fibrillin-3 (Ref. Reference Jordan113). In addition, mutations in LTBP-2 and the ADAMTS family members ADAMTS-10, ADAMTS-17, ADAMTSL-2 and ADAMTSL-4 are associated with the fibrillin-related disorders WMS, a Weill–Marchesani-like syndrome, acromicric and GD and ectopia lentis, demonstrating genetic and functional links between these proteins and fibrillin microfibrils (Refs Reference Dagoneau9, Reference Haji-Seyed-Javadi147, Reference Morales148, Reference Le Goff149).
MFS and related pathologies
MFS (OMIM 154700) is the most prevalent fibrillinopathy, with an incidence of 2–3 per 10,000 people. A complex and phenotypically heterogeneous systemic disorder, MFS is characterised by severe cardiovascular, skeletal and ocular abnormalities, with symptoms ranging from isolated features through progressive classical MFS to severe early onset and rapidly progressive MFS (associated with early childhood mortality). Disease symptoms can reflect both elastic fibre deficiencies and perturbed TGFβ activity (Refs Reference Robinson6, Reference Dietz and Pagon109). A life-threatening complication of MFS is aortic rupture due to dilation of the ascending aorta.
Although commonly inherited as an autosomal dominant trait, 25% of cases arise from de novo mutations; well over 1000 causative fibrillin-1 mutations have been identified. Mutations often affect disulphide bond patterns or calcium-binding consensus sequences within cbEGF domains, resulting in structural alterations that alter assembly and/or integrity of microfibrils and elastic fibres (Ref. Reference Robinson6). There is no single mechanism from fibrillin-1 mutation to disease; mouse models and recombinant fibrillin-1 studies have shown that altered secretion, decreased expression, structural defects and increased proteolytic susceptibility of mutant fibrillin-1 proteins and perturbations in TGFβ activity can all contribute to MFS phenotypes (Refs Reference Robinson6, Reference Dietz and Pagon109, Reference Mellody150, Reference Charbonneau151, Reference Kirschner152, Reference Ashworth153).
Fibrillin-1 is highly susceptible to proteolytic degradation, and increased proteolytic susceptibility appears to play a significant role in the pathogenesis of MFS (Refs Reference Robinson6, Reference Kirschner152, Reference Ashworth153). Murine models of MFS (e.g. C1039 G heterozygote; Ref. Reference Judge154) have revealed gross disruption to the elastic lamellar structure of the aorta (Fig. 4). Matrix metalloproteinases (MMPs) -1, -2, -3 and -9 that can cleave fibrillin-1 are upregulated in thoracic aortas from MFS patients and MMPs-2 and -9 are upregulated in an MFS mouse model (Ref. Reference Chung155). MMP-1 up-regulation has been associated with a fibrillin-1 fragment containing an elastin-binding protein recognition sequence (Ref. Reference Booms156). Recombinant fibrillin-1 fragments harbouring mutations associated with severe neonatal MFS showed enhanced susceptibility to physiological and non-physiological proteases. In addition, cathepsins K and V can cleave fibrillin-1 at multiple sites and thus may play a role in the pathogenesis of MFS (Ref. Reference Kirschner152).
Figure 4. Effects of the fibrillin-1(Fbn)C1039G/+ phenotype, and Smad4 haploinsufficiency (Smad4+/−), on the aortic architecture in mice. Verhoeff-Van Giesen staining revealed architectural abnormalities in the ascending aorta of these mutant mice. Compared with wild-type littermates, FbnC1039G/+ mice had medial thickening and elastic fibre fragmentation. These defects were enhanced in Smad4+/−: FbnC1039G/+ mice. Figure taken from Ref. Reference Holm111 (with permission from the corresponding author and the publisher).
TGFβ is known to control smooth muscle cell differentiation, matrix synthesis and vascular morphogenesis, and perturbed TGFβ signalling has been identified as a primary causative feature of MFS aortic disease progression (Refs Reference Dietz and Pagon109, Reference Jaffe157) (Fig. 4). Mouse studies have shed further light on the complex contribution of TGFβ to disease progression. For example, mice with a fibrillin-1 deletion showed increased TGFβ activity in lung sections compared with wild-type littermates, coupled with impairment in alveolar septation (Reference Neptune158). Increased TGFβ activity also contributed to defective mitral valvulogenesis in a murine model of MFS. Cultured vascular smooth muscle cells from thoracic aortas of fibrillin-1 null mice showed constitutively active Smad2/3 signalling (Ref. Reference Carta159). Non-canonical (non-Smad-mediated) TGFβ signalling also enhances aortic defects in Marfan mice (Ref. Reference Holm111) (Fig. 4).
Enhanced TGFβ activity is associated with other fibrillinopathies including Stiff skin syndrome (Ref. Reference Loeys116), and acromicric and GD that can be caused by fibrillin-1 or ADAMTSL2 mutations (Refs Reference Le Goff149, Reference Le Goff160). These diseases highlight the central role of fibrillin-1 microfibrils in regulating TGFβ bioavailability. Other MFS-like diseases with perturbed TGFβ include MFS2 caused by mutations in the TGFβ receptor II, and Loeys–Dietz syndrome (Refs Reference Dietz and Pagon109, Reference Loeys161), familial aortic aneurysm (Ref. Reference Pannu162) and Furlong syndrome (Ref. Reference Ades163), which are caused by mutations in TGFβ receptors I or II. The rare disorder arterial tortuosity syndrome, which causes arterial elastic fibre fragmentation, might result from altered TGFβ activity as a consequence of mutations in the gene encoding the glucose transporter GLUT10 (Ref. Reference Coucke164). Mutations in the TGFβ repressor SKI cause the Shprintzen–Goldberg syndrome, which has many overlapping features with MFS and Loeys–Dietz syndrome (Ref. Reference Doyle165).
MFS patients are routinely prescribed beta blockers to reduce haemodynamic stress on the aorta (Ref. Reference Dietz and Pagon109). In recent years, uncovering the link between increased TGFβ activity and MFS pathogenesis has led to the development of advanced therapeutic strategies that reduce TGFβ signalling. In particular, treatment with the angiotensin II type I receptor antagonist losartan was shown in MFS mice (Ref. Reference Habashi110) to prevent aortic aneurysm and to improve non-cardiovascular symptoms (such as alveolar septation and muscle repair) (Fig. 5). Several clinical trials have since been assessing the benefits of losartan treatment for MFS patients. In a clinical trial of patients with severe and rapidly progressive MFS, losartan treatment reduced both the rate of aortic root growth to 10% of pretreatment levels (Refs Reference Dietz and Pagon109, Reference Geirsson166). A multicentre losartan trial is in progress (Ref. Reference Brooke117), alongside a trial that studies the combined effects of β-blockers and losartan in MFS patients (Ref. Reference Detaint118). Angiotensin II-receptor blockers are also being assessed for their potential to modulate pathological progression of mitral valve prolapse in MFS patients (Ref. Reference Möberg119). Levels of circulating TGFβ are elevated in MFS mice and patients; thus circulating TGFβ may prove to be a valuable prognostic marker for MFS (Ref. Reference Dietz and Pagon109). Doxycycline has also been shown to delay aneurysm rupture through inhibition of MMPs-2 and -9 (Ref. Reference Xiong120). A recent study has also revealed that treatment of MFS mice with antagonists to the elastin-binding protein motif (GxxPG) can reduce several abnormalities characteristic for MFS, suggesting a potential new method of treatment for cardiovascular manifestations of MFS (Ref. Reference Guo121).
Figure 5. Prevention of aortic aneurysm in mice by losartan. Representative murine ascending aortae (arrowheads), after therapy, are: (a) wild-type mice; (b–f) mice heterozygous for Marfan-causing fibrillin-1 mutation C1039 G (FbnC1039G/+), treated with placebo (b), propanolol (c) or losartan (d–f). Scale bars = 4 mm. Figure taken from Ref. Reference Habashi110 (with permission from the corresponding author and the publisher).
WMS and acromicric and GD
WMS is characterised by short stature, brachydactyly (short digits), eye abnormalities and joint stiffness. Both autosomal (WMS2; OMIM 608328) and recessive (WMS1; OMIM 277600) forms of WMS exist (Ref. Reference Faivre115). Clinical manifestations of the acromicric (AD; OMIM 102370) and GD (OMIM 231050) include short stature, joint defects and thickened skin (Ref. Reference Le Goff160).
Mutations causing autosomal dominant WMS, AD and GD are located within the TB5 domain of fibrillin-1 (Refs Reference Marmorstein108, Reference Jaffe157). This WMS-associated mutation abolishes binding of the TB5 domain to heparin, whereas six tested mutations associated with AD and GD reduce binding (Ref. Reference Cain112). A second N-terminal fibrillin-1 mutation associated with dominant WMS results in an in-frame deletion, abolishing a specific binding site in fibrillin-1 for ADAMTSL-2, -3, -6 and the ADAMTSL papilin (Ref. Reference Sengle49). It was recently demonstrated that ADAMTS-10 can bind fibrillin-1 (Ref. Reference Kutz95), and recessive WMS is associated with ADAMTS-10 mutations (Ref. Reference Dagoneau9). Mutations in ADAMTS-17 and ADAMTSL-2 cause a Weill-Marchesani-like syndrome (WMS-like; OMIM 613195) and GD, respectively (Refs Reference Morales148, Reference Le Goff149). These closely related genetic disorders suggest a functional relationship between ADAMTSL (-2,-4), ADAMTS (-10,-17) and fibrillin-1. It has been hypothesised that ADAMTSL-3, ADAMTS-10 and fibrillin-1 might form a ternary complex and that these interactions may influence integrin interactions with fibrillin-1 because of the proximity of the fibrillin-1 binding sites to an RGD motif (Ref. Reference Sengle49). ADAMTS molecules are also implicated in TGFβ sequestration and activation (Ref. Reference Doyle, Gerber and Dietz114); although the underlying mechanism is unknown, ADAMTS molecules could influence stable deposition of LTBP-containing large latent TGFβ complexes with fibrillin microfibrils. In addition, a mutation in LTBP-2 has recently been associated with WMS (WMS3; OMIM 614819) (Ref. Reference Haji-Seyed-Javadi147).
Elastinopathies
Mutations in elastin and elastic fibre-associated proteins cause disease through loss or gain of function, and increased susceptibility to inflammatory or proteolytic damage of elastic fibres (Refs Reference Wagenseil and Mecham122, Reference Hu123).
Supravalvular aortic stenosis (SVAS; OMIM 185500) occurs in 1 in 20 000 live births, is inherited in an autosomal dominant manner but can also occur sporadically and is characterised by narrowing or obstruction of the ascending aorta. Other defects include obstruction of pulmonary, coronary, carotid and renal arteries. Severe SVAS may lead to dyspnoea, angina, systolic murmur, left ventricular hypertrophy ultimately leading to congestive heart failure. Although congenital non-syndromic forms of SVAS exist, SVAS is typically associated with Williams–Beuren syndrome (WBS), a complex developmental disorder (below).
The spectrum of heterozygous loss-of-function mutations in the elastin gene reflects diverse SVAS phenotypes (Ref. Reference Urban124). Point mutations, translocations and partial deletions of the elastin gene lead to premature stop codons and unstable mRNA. The most widely accepted underlying mechanism of SVAS is elastin haploinsufficiency (Ref. Reference Micale125), where smooth muscle cells of large vessels generate half normal levels of elastin, leading to thinner and disorganised elastic lamellae (Ref. Reference Brooke117). That ELN gene haploinsufficiency is an underlying cause of nonsyndromic SVAS was confirmed by the identification of seven novel elastin gene mutations in 31 SVAS patients with familial and sporadic non-syndromic SVAS (Ref. Reference Urban126). Apart from its structural role, elastin inhibits smooth muscle cell proliferation and promotes organisation of actin filament bundles. In SVAS, smooth muscle cells hyper-proliferate, become hypertrophic and collagen levels increase, leading to thicker, narrower and less flexible and resilient arteries.
Human induced pluripotent stem (iPS) cells from SVAS patients, which exhibit SVAS characteristics, have been generated to study pathogenesis and patient-specific interventions (Ref. Reference Ge127). They reveal that affected smooth muscle cells have fewer organised networks of smooth muscle alpha-actin filament bundles compared with control cells. They also predict therapeutic inhibition of smooth muscle cell overproliferation by decreasing ERK1/2 signalling. Another SVAS therapy by simple sliding aortoplasty has been reported (Ref. Reference Shin128).
Williams–Beuren Syndrome (WBS; OMIM 194050) is a rare multisystem developmental genetic disorder that has a prevalence of 1 in 7500 and usually occurs sporadically (Refs Reference Morris and Mervis129, Reference Pober, Johnson and Urban167). Characteristics associated with WBS include intellectual deficit, connective tissue defects, cardiovascular abnormalities (SVAS, also peripheral pulmonary aortic stenosis and arterial hypertension), dimorphic facial features, short stature and metabolic defects (infantile hypocalcaemia and abnormal glucose intolerance). Mitral valve disease is also associated with WBS, with a prevalence of approximately 40%.
WBS results from a hemizygous deletion (1.5–1.8 megabase), and less frequently duplication, of 26–28 genes, including elastin, that localise to the Williams syndrome region of the chromosome 7q11.23 (Ref. Reference Urban131). These deletions arise as a consequence of meiotic misalignment of repetitive sequences that flank the WBS chromosome region (Ref. Reference Schubert132). Deletions cause the phenotypes outlined above, whereas duplications typically result in milder phenotypes (Refs Reference Urban131, Reference Schubert132).
Deletion of one copy of elastin gene is the most common and most recognised genetic rearrangement known in WBS, as it occurs in over 96% of WBS patients, and elastin haploinsufficiency underlies the cardiovascular manifestations in WBS, most prominently SVAS. Smooth muscle cells of WBS patients produce reduced levels of elastin (Ref. Reference Urban124). Treatments for the vascular symptoms of WBS commonly involve reparative surgery of aortic stenosis and individualised hypertension treatment (Ref. Reference Pober133).
Pseudoxanthoma elasticum (PXE; OMIM 264800) is a heritable autosomal recessive disorder with a prevalence of approximately 1 in 25 000–100 000 (Refs Reference Chassaing134, Reference Uitto135). PXE is characterised by progressive ectopic mineralisation and fragmentation of elastic fibres that leads to ocular, cutaneous and cardiovascular abnormalities. PXE becomes initially evident when the accumulation of calcium/phosphate complexes in the skin causes the appearance of yellow papule and loss of elasticity. In the eyes, these complexes cause angioid streaks and neovascularisation, leading to gradual loss of vision. Early onset of arteriosclerosis and cardiac failure are the most serious complication. PXE is caused by loss-of-function mutations in the ABCC6 gene, which encodes a putative transmembrane transporter, especially abundant in the liver (Ref. Reference Bergen136). The dysfunctional transporter may affect levels or activity of circulatory factors, such as fetuin-A and matrix Gla-protein, responsible for prevention of aberrant mineralisation in normal conditions (Ref. Reference Uitto, Li and Jiang137). Therapies for the ocular symptoms of PXE focus on treating neovascularisation of the eye by intravitreal injections of vascular endothelial growth factor (VEGF) inhibitors (Ref. Reference Georgalas138). Other approaches such as supplementation of diet with magnesium, introduction of anti-mineralising factors, regeneration of the liver and stem-cell therapy are being assessed.
Cutis laxa (CL) comprises inherited (Ref. Reference Berk8) and acquired disorders that are characterised by abnormal elastic fibres and loose sagging skin that often gives an aged appearance. CL can be inherited in ADCL, ARCL or X-linked forms (XLCL). Based on different mutations and phenotypic manifestations, autosomal recessive types can be further divided into type I (A and B), II (A and B) and III (De Barsy syndrome), Urban-Rifkin-Davis syndrome, macrocephaly, alopaecia, CL and scoliosis (MACS) syndrome and arterial tortuosity syndrome. Recessive forms of CL tend to be more severe, often resulting in childhood mortality (Ref. Reference Berk8).
ARCL type I (OMIM 219100) is caused by mutations in the genes encoding fibulin-4 (subtype A) or fibulin-5 (subtype B) (Refs Reference Hucthagowder139, Reference Renard140, Reference Loeys141, Reference Claus142). Fibulin-4 mutations usually cause more severe and often lethal phenotypes with emphysema, tortuosity, aortic aneurysms and joint laxity. Fibulin-4 mutations were previously thought to be rare, but recently, in a small population, a novel fibulin-4 mutation has been identified in 22 infants suffering from severe arteriopathy syndrome (Ref. Reference Kappanayil168).
The two subtypes of ARCL type II (OMIM 219200) are both associated with loss-of-function mutation in the ATP6VOA2 gene, which encodes a vesicular proton pump, thus affecting vesicular trafficking. The mutation indirectly leads to tropoelastin accumulation within the Golgi and thereby significantly reduced deposition of mature elastin (Ref. Reference Hucthagowder143). Mutations in the PYCR1 gene, which encodes for a mitochondrial enzyme involved in proline biosynthesis (pyrroline-5-carboxylate reductase 1), have also been associated with this form of CL (Ref. Reference Reversade169).
The genetic cause of the De Barsy syndrome/CL-corneal clouding-mental retardation syndrome (OMIM 219150) is currently unknown; however, because of the significant overlap with the phenotypic characteristics of type II ARCL, mutations in ATPVOA2 and PYCR1 seem to be the likely underlying cause of this syndrome (Ref. Reference Leao-Teles170).
Urban-Rifkin-Davis syndrome (OMIM 613177) is a newly characterised type of ARCL that is caused by mutations in LTBP-4 (Ref. Reference Urban171). A severe phenotype including developmental, craniofacial and fatal pulmonary abnormalities is common. Haploinsufficiency of LTBP-4 is likely to cause increased TGFβ activity and abnormal elastic fibre assembly.
MACS syndrome (OMIM 613075) is a multitude malformation syndrome that includes macrocephaly, alopaecia, CL and scoliosis. It is caused by mutations in RIN2, which is a guanine exchange factor for Rab5, which in turn controls endocytic trafficking. It is unlikely that these mutations underlie the connective tissue abnormalities, which more probably are attributable to fibulin-5 deficiency, which MACS patients exhibit (Ref. Reference Basel-Vanagaite172).
Arterial tortuosity syndrome (OMIM 208050) is characterised by elongated and tortuous arteries, along with CL. Mutations in the SLC2A10 gene that encodes the glucose transporter GLUT10 are associated with this syndrome. Although previously thought that the mutation may lead to abnormal TGFβ activity, stimulating smooth muscle cell proliferation, it has also been suggested that the loss of GLUT10 rather results in aberrant collagen and elastin metabolism (Ref. Reference Segade173).
XLCL (OMIM 304150) and Menkes disease (OMIM 309400) are both caused by mutations in the ATP7A gene that encodes for a copper-transporting enzyme. Defects in this enzyme reduce functionality of copper-dependent enzymes such as LOX, affecting elastin and collagen deposition. Although both disease forms have overlapping phenotypic manifestations, XLCL tends to be milder. Severe and commonly lethal Menkes disease is associated with neurological defects. Although neonatal diagnosis and early treatment with copper can improve survival, some residual activity of ATP7A is essential (Ref. Reference Berk8). Recently, gene replacement therapy has been suggested as a therapeutic approach to alleviate Menkes disease (Ref. Reference Kaler130).
ADCL (OMIM 123700) is generally defined as a milder form of the inherited CL spectrum, typically characterised by aged facial appearance. Systemic manifestations are less common and may involve gastrointestinal abnormalities, hernias, pulmonary aortic stenosis, tortuosity, emphysema and aortic root dilatation (Ref. Reference Callewaert174). ADCL-causing mutations do not lead to elastin haploinsufficiency, in contrast to SVAS, but rather have a dominant-negative effect on elastin deposition. Indeed, mutant elastin has abnormal binding to fibrillin-1 and enhanced self-association of elastin, which caused decreased mature elastin deposition (Ref. Reference Callewaert174). Increased endoplasmic reticulum stress, induced apoptosis and increased TGFβ signalling are also reported (Ref. Reference Shifren and Mecham144). Mutations in ALDH18A1, encoding Δ1-pyrroline-5-carboxylate synthase (P5CS) have overlapping symptoms with ADCL, with altered elastic fibre ultrastructure (Ref. Reference Skidmore145).
Buschke–Ollendorff syndrome (OMIM 166700) is a rare autosomal dominant condition, which includes skin lesions containing elastic fibres, and osteopoikilosis, and is caused by mutations in the LEMD3 gene, which encodes a protein involved in bone and connective tissue morphogenesis (Ref. Reference Schena146). The encoded protein can antagonise TGFβ signalling at the inner nuclear membrane.
Acquired elastic fibre disorders
Acquired elastic fibre disorders arise wherever elastic fibre structure and function are compromised, including tissues such as skin, blood vessels and lungs. Some examples are highlighted below.
Acquired CL usually develops in adulthood. Characteristic secondary destruction of elastic fibres occurs because of dermal inflammation caused by other medical conditions or reactions to medicines. Systemic manifestations, which often lead to fatal outcomes, may include emphysema, hernias and vascular dilatation (Ref. Reference Hu175).
Marshall syndrome is a type of acquired CL typically affecting children after suffering neutrophilic dermatosis (Sweet's syndrome) (Ref. Reference Timmer176).
Solar/actinic elastosis is a result of a prolonged exposure to sunlight, leading to degeneration of the elastic tissue of the dermis and consequently to premature ageing of the skin. Deposition of the elastotic material and decreased collagen synthesis lead to rough, inelastic, wrinkled and hyperpigmented skin. Current therapy of choice is a skin-rejuvenating photodynamic therapy in which photosensitisers and diverse light sources have been successfully used to reverse the effects of photodamage (Ref. Reference Karrer177).
Acquired PXE is characterised by cutaneous mineralisation and fragmentation of elastic fibres leading to lax inelastic skin. Unlike the heritable PXE, the acquired PXE is non-systemic and is limited to the skin only. Exposure to chemicals such as calcium–ammonium–nitrate salts, mechanical stress and abnormal calcium–phosphate metabolism has been implicated in the development of this disease (Ref. Reference Lewis178). There are no generally accepted therapies for the acquired PXE. Papillary dermal elastolysis is a rare condition, similar to PXE, which usually affects elderly women, and is associated with elastolytic papules on the neck and supraclavicular sites (Ref. Reference Alves179).
Ageing changes to blood vessels and lungs: loss of elastic fibre integrity and elastic recoil are common features of ageing changes to blood vessels especially the elastic arteries, whereas pulmonary emphysema is associated with loss of elasticity and pathological alveolar remodelling (Ref. Reference Pasquali-Ronchetti and Baccarani-Contri180).
ARMD (OMIM 603075) is one of the most frequent causes of loss of vision and is characterised by progressive deposition of extracellular aggregates and neovascularisation in the macula region of the eye. A multitude of genes have been associated with this disease, with mutations in complement factor H being the most common cause of the disease (Ref. Reference Klein181). Mutations in fibulins, and especially fibulin-5, have also been implicated in ARMD. Fibulin-5 mutations lead to haploinsufficiency which may cause the observed phenotype by altering the structure of the elastic Bruch's membrane of the macula (Ref. Reference Lotery182). Anti-VEGF-A therapy has been successfully used to alleviate neovascularisation in the ARMD. A recent review has highlighted other possible therapeutic approaches, and stressed the importance of supporting the coping mechanisms of the retinal pigment cells of the macula (Ref. Reference Ambati and Fowler183).
Elastosis perforans serpiginosa (EPS; OMIM 130100) is a rare degenerative skin disease, which exhibits loss of elastic fibres in the upper dermis, following extended treatment with D-penicillamine that chelates copper and inhibits cross-linking of elastin by copper-dependent lysyl oxidase (Ref. Reference Atzori184).
Research in progress and outstanding research questions
Recent progress in treating MFS and related diseases with losartan and other therapies that target TGFβ activity have led to improved patient outcomes. However, further understanding of the biology of elastic fibres and their assembly is urgently needed to advance the therapeutic prospects for elastic fibre diseases. Current research on elastic fibres aims to define how cells and their integrin and heparan sulphate proteoglycans (HSPG) receptors direct the assembly of microfibrils and elastic fibres, how fibronectin and the ADAMTS and ADAMTSL molecules influence microfibril deposition and how pericellular processing (by furin/PACE enzymes) and cross-linking (by LOX and tissue transglutaminase enzymes) regulate elastic fibre deposition. New insights into how LTBPs are deposited in the matrix, and how active TGFβ and BMPs are released from these complexes are needed to resolve precisely how TGFβ growth factor bioavailability is controlled. All this information is critical for elucidating how defects in microfibrils and elastic fibres cause distinct genetic and acquired diseases by perturbing different stages in their assembly, their structural integrity or their susceptibity to proteolytic attack. We also need to understand much better how inflammatory enzymes cause elastic fibre remodelling and loss of both microfibril and elastic fibre functional integrity in disease and ageing. There is a need to identify biomarkers of elastic fibre damage to enable early diagnosis and treatment. Much recent progress has been made in the engineering of vascular constructs based on elastin and elastic fibre components, and this approach offers great promise for the repair of damaged elastic tissues. Such advances will provide mechanistic targets to prevent elastic fibre degeneration and to encourage de novo elastic fibre formation in regenerating tissues.
Acknowledgements and funding
Current research on microfibrils and elastic fibres in our laboratory is funded by the Medical Research Council (UK) and Wellcome Trust.