

Take-Home Points
-
1. The so-called “direct” pathway of the extrapyramidal system is the “go” pathway for motor actions, and is stimulated by dopamine at D1 receptors there.
-
2. The so-called “indirect” pathway of the extrapyramidal system is the “stop” pathway for motor actions, and is inhibited by dopamine at D2 receptors there.
-
3. Tardive dyskinesia theoretically is due to supersensitive D2 dopamine receptors that result from aberrant neuronal plasticity triggered when D2 receptors are chronically blocked. This abnormal neuronal “learning” leads to relative overstimulation of inhibitory dopamine at D2 receptors in the “stop” pathway, resulting in not enough “stop” signaling. This aberrant signaling hypothetically causes the involuntary hyperkinetic movements of tardive dyskinesia.
Introduction
Recognition and treatment of tardive dyskinesia (TD) has attained new importance with the introduction of new treatments.Reference Hauser, Factor and Marder 1 , Reference Fernandez, Factor and Hauser 2 In the modern era of drugs that treat psychosis, TD has become somewhat forgotten—perhaps due to the high degree of attention to cardiometabolic side effects—but has not gone away. Has TD become less common? Less severe? Or just less recognized? There are data suggesting a little bit of truth to all of these.Reference Woods, Morgenstern and Saksa 3 – Reference Carbon, Hsieh, Kane and Correll 5 One thing is for sure: if monitoring for TD has declined especially in outpatients taking D2 antagonists for mood and anxiety disorders, “you can’t find what you aren’t looking for.” Redoubling efforts to look for TD in anyone taking a D2 blocker for any reason may be in order, especially now that treatments are available. To better understand TD and how the new treatments work, we discuss here the pathophysiology of TD as the consequence of drugs for psychosis altering the “traffic signals” coming from the motor striatum and causing not enough “stop.”
Stop and Go Signals for Normal Movement
Movements are coordinated by the extrapyramidal system via two distinct pathways: the “direct” and the “indirect” (Figures 1–3).Reference DeLong and Wichmann 6 – Reference Calabresi, Picconi, Tozzi, Ghiglieu and DiFilippo 9 The so-called direct pathway projects from the motor striatum directly to the globus pallidus interna and is a “go” signal for motor movements (Figure 1; Figures 3A and 3B); the so-called indirect pathway projects indirectly to the globus pallidus interna (via the globus pallidus externa and subthalamic nucleus) to produce a “stop” signal for motor movements (Figure 2; Figures 3A and 3C).
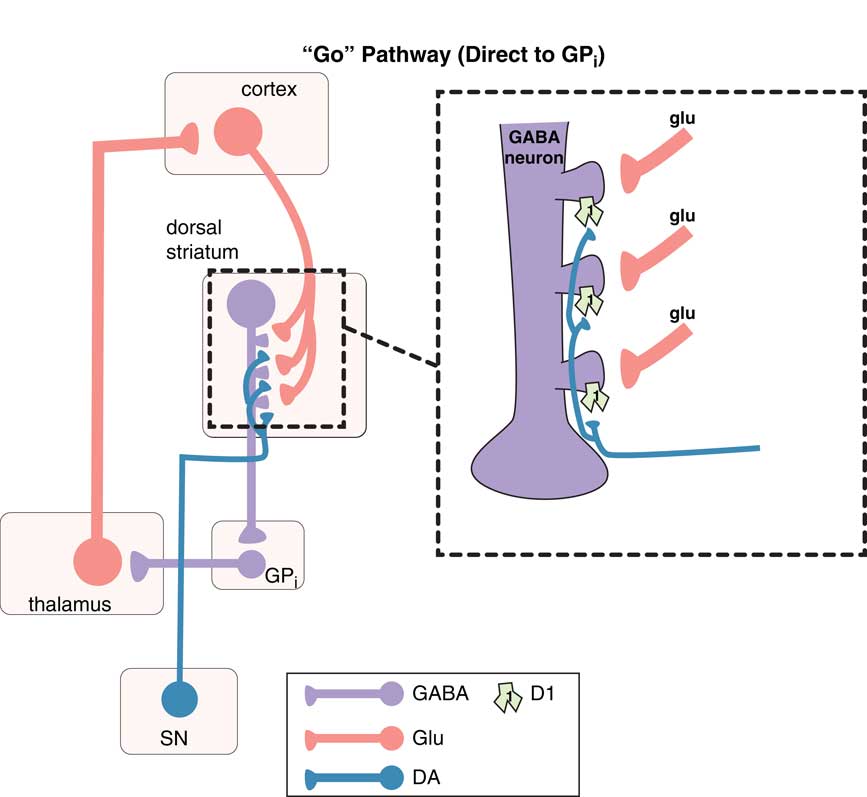
Figure 1 The “go” pathway (the direct pathway of the extrapyramidal system). The so-called direct pathway projects from the motor striatum directly to the globus pallidus interna (GPi) and is a “go” signal for motor movements. Corticostriatal glutamate (glu) inputs synapse onto the heads of dendritic spines of medium spiny GABA neurons in the direct pathway of dorsal (motor) striatum to signal “go,” and dopamine projections from the substantia nigra (SN) synapse on the necks of these same dendritic spines at D1 receptors to amplify the “go” signal (see inset). From GPi, GABA neurons project onto glutamate neurons in the thalamus, which then relay back to the glutamatergic cortical striatal neurons in motor cortex.
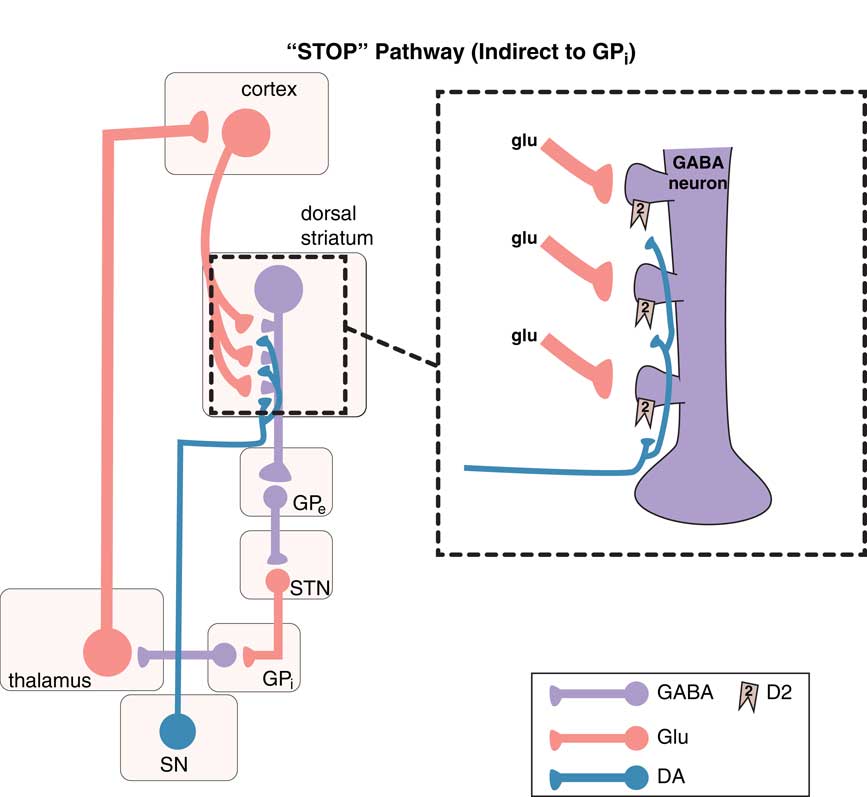
Figure 2 The “stop” pathway (indirect pathway of the extrapyramidal system). The so-called indirect pathway projects indirectly to the globus pallidus interna (GPi) via the globus pallidus externa (GPe) and subthalamic nucleus (STN) to produce a “stop” signal for motor movements. Corticostriatal glutamate (glu) inputs synapse onto the heads of dendritic spines of medium spiny GABA neurons in the indirect pathway of the dorsal (motor) striatum to signal “stop,” and dopamine projections from the substantia nigra (SN) synapse on the necks of these same dendritic spines at D2 receptors to inhibit the “stop” signal (see inset). From GPe, GABA neurons project onto other GABA neurons, which themselves project onto glutamate neurons in the STN that in turn project onto glutamate neurons in the thalamus, which then finally relay back to the glutamatergic cortical striatal neurons in motor cortex.
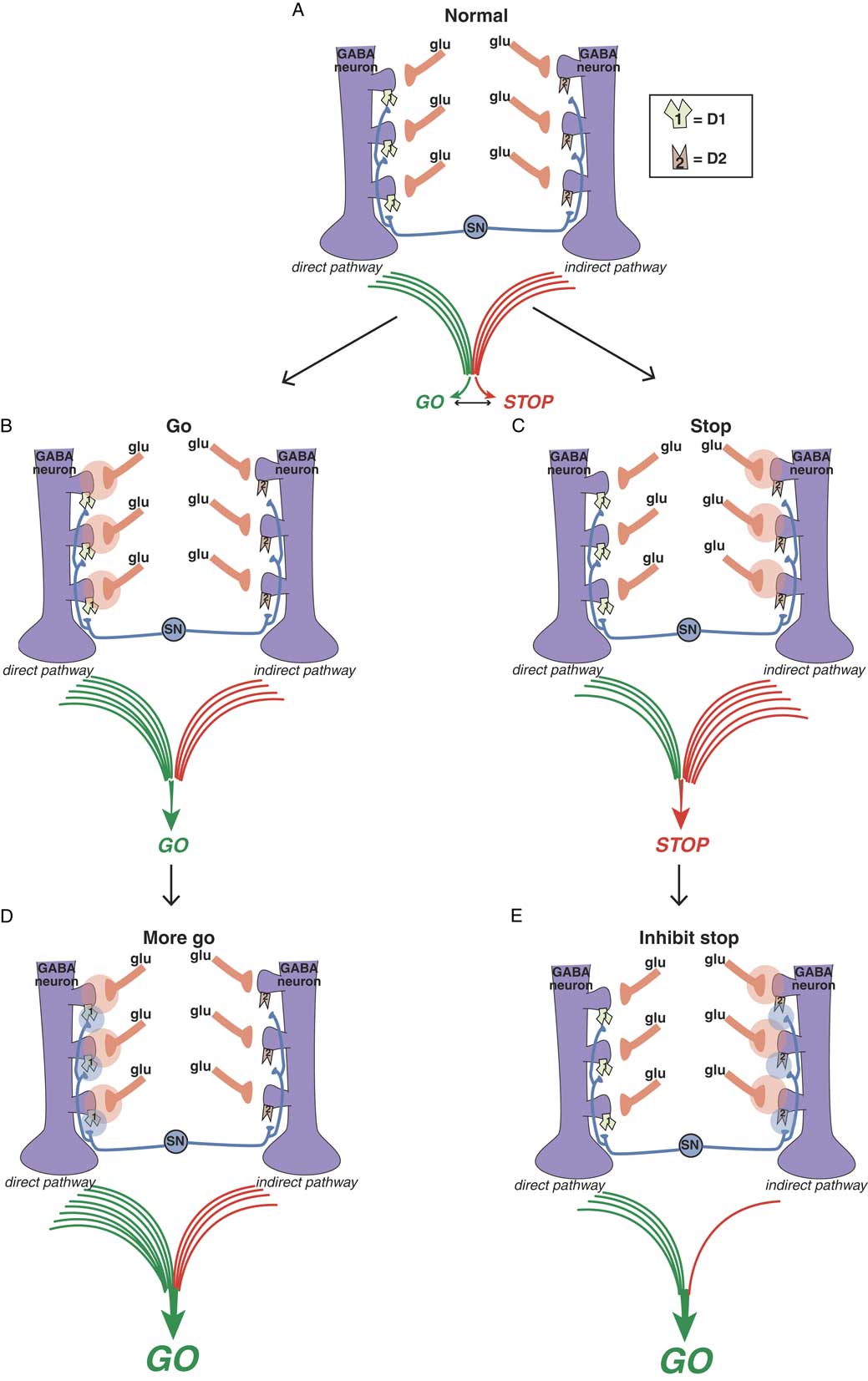
Figure 3A Balance between “go” and “stop” signals in direct and indirect pathways. The direct pathway on the left signals “go” and the indirect pathway on the right signals “stop.” The balance between these two signals determines the coordinated flow of movement. Too much “go” can cause involuntary hyperkinetic dyskinesia, and too much “stop” can cause bradykinesia and rigidity. Purple GABA (gamma amino butyric acid) medium spiny neurons in the motor striatum of both pathways each receive different glutamate (glu) input from the cortex onto the heads of dendritic spines. Regulating this glu input are dopamine terminals arising from the substantia nigra (SN). Dopamine neurons synapse both upon D1 dopamine receptors on the necks of dendritic spines of the direct pathway on the left to boost the glutamate “go” signals there, as well as upon the necks of dendritic spines of the indirect pathway on the right to inhibit the “stop” signals there. Figure 3B. Glutamate in the direct pathway signals “go.” When glutamate is released onto GABA spines of the direct pathway (orange clouds of glutamate neurotransmission on the left), output of the extrapyramidal system favors “go.” Figure 3C. Glutamate in the indirect pathway signals “stop.” When glutamate is released onto GABA spines of the indirect pathway (orange clouds of glutamate neurotransmission on the right), output of the extrapyramidal system favors “stop.” Figure 3D. Dopamine at D1 dopamine receptors potentiates glutamate “go” signals. When dopamine is released onto D1 receptors in the direct pathway (blue clouds of dopamine neurotransmission on the left), the “go” signal arising from glutamate release there is amplified. Figure 3E. Dopamine at D2 dopamine receptors inhibits glutamate “stop” signals. When dopamine is released onto D2 receptors in the indirect pathway (blue clouds of dopamine neurotransmission on the right), the “stop” signal mediated by glutamate release there is inhibited, leading in turn to “less stop” and therefore “more go.”
Dopamine is a sort of traffic cop that regulates “traffic patterns” in both pathways to facilitate motor movements (Figures 3D and 3E).Reference DeLong and Wichmann 6 – Reference Andre, Cepeda and Cummings 16 Dopamine at D1 receptors in the direct pathway interacts with glutamate N-methyl-d-aspartate (NMDA) receptors to facilitate their “go” signals, resulting in “more go” (Figure 3D).Reference Harsing and Zigmond 10 – Reference Andre, Cepeda and Cummings 16 Dopamine at D2 receptors in the indirect pathway interacts with glutamate NMDA receptors to inhibit their stop signals, resulting in “less stop” and, therefore, “more go.”Reference Harsing and Zigmond 10 – Reference Andre, Cepeda and Cummings 16
What’s Wrong with Dopamine and Its Receptors in Schizophrenia?
Before considering what are the effects of acute and chronic D2 receptor blockade, we need to answer the question: what is the cause of dopamine hyperactivity in untreated schizophrenia—too many supersensitive dopamine receptors or too much dopamine release? Some neuroimaging data suggest upregulated D2 receptors in the nucleus accumbens (but not in the motor striatum) of untreated patients with schizophrenia as the cause of their positive symptoms; however, the balance of the evidence currently favors the other explanation, namely, excessive dopamine release in the nucleus accumbens (but not in the motor striatum).Reference Abi-Dargham, Gil and Krystal 17 , Reference Breier, Su and Saunders 18 However, the status of dopamine in untreated schizophrenia is not yet well established, since most studies include patients who had been previously medicated, and even after a few weeks of medication washout, it is not clear that the upregulation of dopamine release was not a compensation due to neuroleptic treatment and not to schizophrenia. Nevertheless, the best evidence now strongly does suggest that the problem in schizophrenia seems to be too much dopamine release but no change in dopamine receptors in the nucleus accumbens, and no documented changes in either dopamine release or dopamine receptors in the motor striatum before drug treatment.
Acute Antipsychotics and Drug-Induced Parkinsonism
Antipsychotic-induced movement disorders are theoretically caused by the effects that D2 antagonism in the motor striatum has on the balance between “stop” and “go” neuronal traffic signals. Positron emission tomography (PET) studies of striatal dopamine binding before and after D2 antagonists suggest that conventional antipsychotics may bind as many receptors in the motor striatum as they do in the nucleus accumbens, meaning that there is no window between antipsychotic actions and drug-induced parkinsonism.Reference Andre, Cepeda and Cummings 16 , Reference Stahl 19 On the other hand, PET studies suggest that D2 blockers with simultaneous 5HT2A blockade create a therapeutic window such that there is less binding of D2 antagonists in the motor striatum. Thus, to a certain extent and within a critical dose range, “you can have your cake and eat it too”: namely, antipsychotic effects (>60% receptor occupancy in the nucleus accumbens) with much less drug-induced parkinsonism (because motor striatum has theoretically <60% receptor occupancy) (see Figure 4C).Reference Stahl 19 , Reference Farde, Nordström, Wiesel, Pauli, Halldin and Sedvall 20
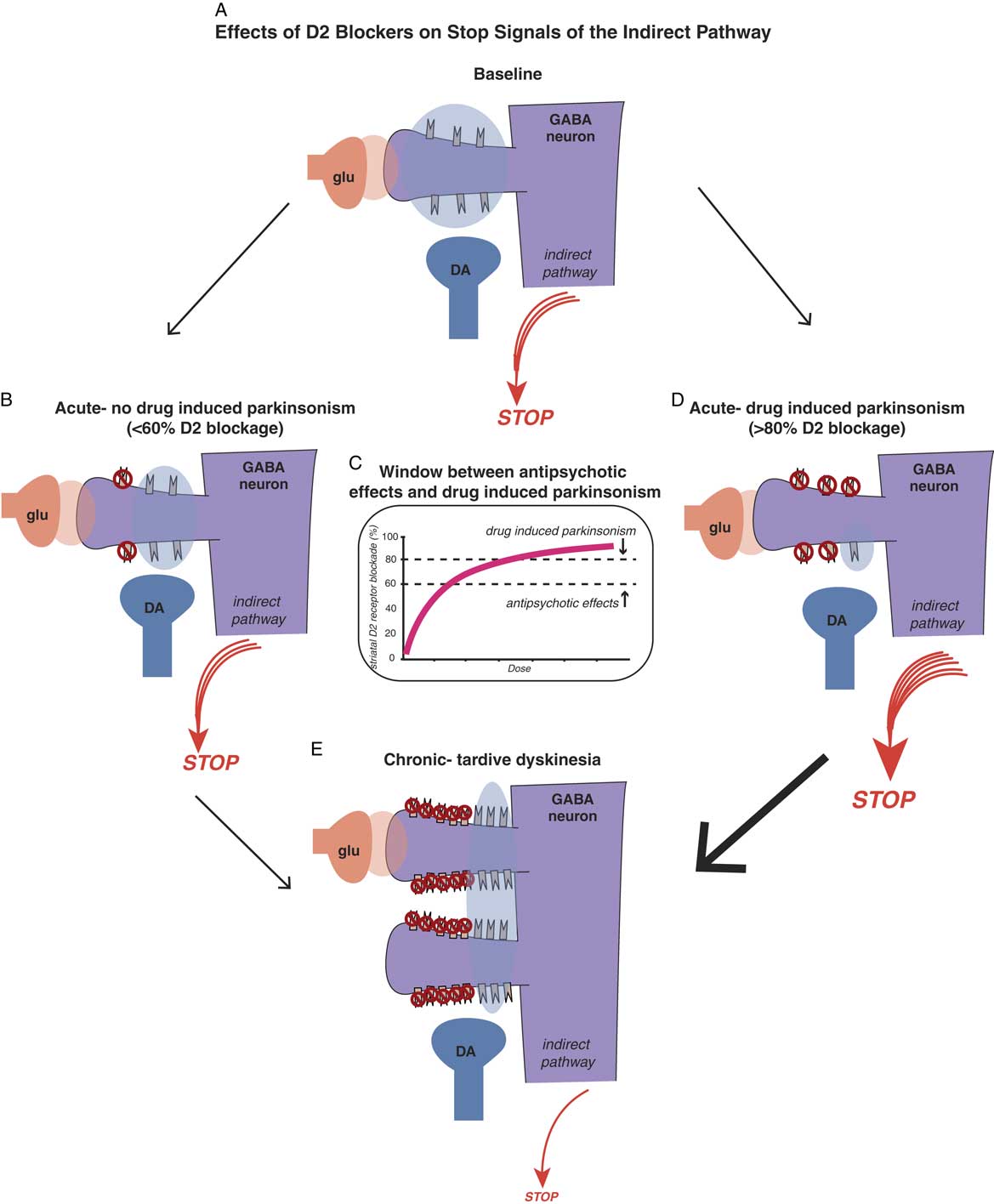
Figure 4A Effects of D2 blockers on “stop” signals of the indirect pathway. Antipsychotic-induced movement disorders are theoretically caused by the effects that D2 antagonism in the motor striatum has on the balance between “stop” and “go” signals. Shown here is a cortical striatal glutamate neuron synapsing upon the head of a GABA medium spiny neuron in the dorsal striatum of the indirect pathway (see orange glutamate cloud), while dopamine D2 receptors receive dopamine input from the substantia nigra to inhibit the glutamate stop signal (see blue dopamine cloud). The balance of glutamate and dopamine shown here is such that a medium sized “stop” signal still emerges from this pathway in the state shown in the illustration. Antipsychotic actions without drug-induced parkinsonism. Acute administration of a dose of a D2 blocker that occupies 60% or fewer D2 receptors in the indirect pathway is theoretically not sufficient to hit the critical threshold for drug-induced parkinsonism (see the stop signal is unchanged from Figure 4A). Figure 4C. There is a therapeutic window for D2 blockers with 5HT2A antagonism, namely a lower threshold for antipsychotic effects (about 60% D2 occupancy in the nucleus accumbens) and a higher threshold for drug-induced parkinsonism (about 80% occupancy of D2 receptors in motor striatum). Doses of drug between these two thresholds will cause antipsychotic actions without motor side effects, as shown in Figure 4B. Doses of drug higher than the threshold for drug-induced parkinsonism will cause motor side effects, such as rigidity and bradykinesia, as well as antipsychotic actions (see Figure 4D). Figure 4D. Antipsychotic actions accompanied by drug-induced parkinsonism. Acute administration of a dose of a D2 blocker that occupies 80% or more D2 receptors in the indirect pathway is theoretically enough to hit the critical threshold for drug-induced parkinsonism and cause motor side effects (see the stop signal is increased compared to Figure 4A). Figure 4E. Chronic administration of D2 blockers causes aberrant synaptic plasticity, such as possible proliferation of spines on GABAergic medium spiny neurons, populated by an increased number of D2 receptors on the necks of these spines. Shown here is what happens when a dose of antipsychotic is given that blocks 60% of these D2 receptors, leaving many extraneous D2 receptors unblocked compared to Figures 4B and 4D, and stimulating many more receptors than shown in Figure 4A. This theoretically causes a dramatic reduction in the stop signal, and thus an increase in the go signal, resulting in the abnormal involuntary hyperkinetic movements of tardive dyskinesia.
These concepts are illustrated in Figure 4, which shows what is theoretically happening at D2 receptors of the indirect pathway in motor striatum when D2 blockers are given. If D2 receptors in the indirect pathway (Figure 4A) are blocked acutely by roughly 60% or less, there may be little or no drug-induced parkinsonism (Figure 4B and 4C). However, if 80% or more of D2 receptors are blocked acutely in the motor striatum, even for drugs with 5HT2A antagonism, drug-induced parkinsonism can be caused (Figure 4C and 4D).
Why are the acute effects of D2 blockade important? The reason is not only because they explain some very troublesome immediate motor side effects of D2 blockade that can interfere with function and cause noncompliance, but also because “as goes drug-induced parkinsonism, so may go TD.” That is, one of the best predictors of who may get TD in the long run could be those patients who exhibit the most drug-induced parkinsonism in the short run, perhaps indicating a particular vulnerability of their motor striatum to the effects of D2 blockade.Reference Kane and Smith 21
Chronic Antipsychotics and Tardive Dyskinesia
What happens to the number and sensitivity of D2 receptors if you block them chronically in the nucleus accumbens? The short answer appears to be: not much. The consensus (although some would disagree) is that chronic D2 blockade does not cause upregulation of D2 receptors in the nucleus accumbens but only in motor striatum.Reference Farde, Nordström, Wiesel, Pauli, Halldin and Sedvall 20 – Reference Blin, Baron and Cambon 24 Evidence for this proposition comes both from PET scans and from the observation that there is little or no supersensitivity psychosis associated with chronic D2 blockade.
What about the motor striatum? These D2 receptors appear to react differently to chronic D2 blockade compared to these same receptors in the nucleus accumbens. Interestingly, D2 receptors in the motor striatum also appear to react in much the same way to chronic stimulation by levodopa in Parkinson’s disease as they do to chronic blockade by D2 antagonists in schizophrenia. Chronic levodopa administration in Parkinson’s disease can lead to levodopa-induced dyskinesias that look very similar to tardive dyskinesia, and may share a similar pathophysiology of aberrant striatal plasticity and abnormal neuronal “learning.”Reference Thiele, Chen and Lo 25 – Reference Cenci and Konradi 27 Perhaps the lesson here is don’t mess with your dopamine receptors in the motor striatum or consequences may ensue!
It has been difficult to prove, but animal models and PET scans in patients with schizophrenia do suggest that chronic D2 blockade in the motor striatum causes upregulated, supersensitive D2 receptors, and this happens to the greatest extent in patients with TD.Reference Kane and Smith 21 – Reference Blin, Baron and Cambon 24 This theory is illustrated in Figure 4E. D2 receptors in the indirect pathway of the motor striatum are upregulated and aberrant neuronal plasticity occurs, perhaps with more dendritic spines and D2 receptors on medium spiny neurons in the indirect pathway. Now, when 60% of D2 receptors are blocked, there are lots of excessive dopamine receptors exposed to overstimulation when dopamine is released. Too much dopamine at D2 receptors in the indirect pathway of the motor striatum translates into excessive inhibition of the “stop” signal; thus, there is “not enough stop” and “too much go,” meaning neuronal traffic without an enforced speed limit, and thus, involuntary hyperkinetic movements of TD.
Conclusion
Understanding how dopamine in the nucleus accumbens acts far differently than it does in the motor striatum, both in treated and untreated patients with schizophrenia, can help explain not only the treatment of positive symptoms of psychosis, but also how acute and chronic treatment with D2 blockers affect each brain area differently. Understanding how dopamine acts in the direct and in the indirect motor pathways via D1 and D2 receptors can help explain how D2 receptor supersensitivity can cause the abnormal neuronal “traffic signals” resulting in the involuntary hyperkinetic movements of TD.




