


Citrus fruits contain an ample amount of vitamin C and are an important food group in our diet. In addition to this essential nutrient, they also contain phytochemicals that may improve our health. Naringenin is the major bioflavonoid isolated from citrus fruits. Animal studies have demonstrated that the administration of naringenin can deter or protect against 7,12-dimethylbenz(α)anthracene (DMBA)-induced mammary tumorigenesis(Reference So, Guthrie and Chambers1), azoxymethane-induced colon carcinogenesis(Reference Leonardi, Vanamala and Taddeo2), DMBA-induced oral carcinogenesis(Reference Miller, Peacock and Bourland3) and N-methyl-N-nitro-N-nitroso-guanidine-induced gastric carcinogenesis(Reference Ekambaram, Rajendran and Magesh4).
Polycyclic aromatic hydrocarbons (PAH) are chemical carcinogens that can be found in industrial emissions, char-broiled meat, tobacco smoke and overheated cooking oil(5, Reference Agency6). Upon exposure, our body metabolises these compounds into DNA-attacking species through cytochrome p450 (CYP) 1A1 and 1B1 catalysis(Reference Gonzalez and Gelboin7). On the other hand, PAH are ligands to the aryl hydrocarbon receptor (AhR). After binding to PAH, the AhR migrates to the nucleus and dimerises with the AhR nuclear translocator. The dimer interacts with xenobiotic-responsive element (XRE) and stimulates the transcription of genes coding for CYP1 family enzymes(Reference Dertinger, Lantum and Silverstone8–Reference Safe10). The observations that PAH fails to induce tumours in AhR(Reference Shimizu, Nakatsuru and Ichinose11) or CYP1B1(Reference Buters, Sakai and Richter12) knockout mice illustrate the importance of the respective proteins in the process of carcinogenesis.
The significance of CYP1B1 in human carcinogenesis is still unclear. CYP1B1 polymorphisms with increased activity are associated with breast cancer risk in Asian women(Reference Zheng, Xie and Jin13). Increased CYP1B1 activity or expression can also be affected by other gene expression. Polymorphism of AhR resulting in lower mRNA expression may also lower the mRNA expressions of ARNT and CYP1B1 (Reference Helmig, Seelinger and Dohrel14). However, meta-analyses on the association of CYP1B1 and breast cancer risk display mixed results(Reference Paracchini, Raimondi and Gram15, Reference Economopoulos and Sergentanis16).
Many PAH have been isolated in the diet(Reference Phillips17), and DMBA is a model PAH commonly used in experiments. The effect of DMBA on gene expression should parallel those of dietary PAH. The present study was designed to examine the inhibitory effect of naringenin on CYP1B1 expression and enzyme activity. As aforementioned that CYP1B1 is an important enzyme in PAH-induced carcinogenesis, the present study might elucidate the underlying mechanism of the chemoprevention of diets rich in fruit and vegetables. In the promoter region of CYP1B1, researchers have located eight XRE and one oestrogen response element (ERE) with verified functionalities(Reference Tsuchiya, Nakajima and Yokoi18, Reference Tsuchiya, Nakajima and Kyo19). Since naringenin is a weak agonist to the oestrogen receptor(Reference Bovee, Schoonen and Hamers20) and AhR(Reference Wang, Yeh and Iwamoto21), we hypothesised that the citrus flavonoid suppresses CYP1B1 expression through antagonising ERE or XRE DNA binding. Concentrations of naringenin covering the physiological as well as pharmacological intake were administered.
Materials and methods
Chemicals
Naringenin, ethoxyresorufin and DMBA were purchased from Sigma Chemicals. All other chemicals, if not stated, were acquired from Sigma Chemicals.
Cell culture
MCF-7 cells (American Type Culture Collection) were cultured in Roswell Park Memorial Institute-1640 phenol red-free media (Sigma Chemicals) with 10 % fetal bovine serum (Invitrogen Life Technology) and incubated at 37°C and 5 % CO2. These cells were routinely subcultured when reaching 80 % of confluency. Cell density in each experiment was maintained at 5 × 102 cells/mm2.
Ethoxyresorufin-O-deethylase activities in intact MCF-7 cells
The ethoxyresorufin-O-deethylase (EROD) activity assay method was performed as described previously(Reference Ciolino and Yeh22). In brief, MCF-7 cells in ninety-six-well plates were treated with 1 μm-DMBA and 0·1, 1 or 5 μm-naringenin. The medium was then removed and the cells were washed twice by 100 μl PBS. Then, 50 μl ethoxyresorufin (5 μm) and salicyclamide (1·5 mm) dissolved in PBS were added in each well, and incubated at 37°C for 15 min. The reaction was stopped by 50 μl of ice-cold methanol, and the resorufin generated was measured by a FLUOstar Galaxy microplate reader (BMG Labtechnologies) with an excitation wavelength at 544 and 590 nm as the emission wavelength. The activities were quantified against resorufin standards.
Enzyme inhibition assays
Recombinant human CYP1A1 and CYP1B1 proteins expressed in insect cells (Supersomes®) were purchased from Gentest Corporation. In brief, 2 pmol of protein were incubated in 100 μl PBS, pH 7·2 with 400 nm-ethoxyresorufin and naringenin in different concentrations. The reaction was initiated by adding 500 μm-NADPH, and terminated by adding 100 μl of ice-cold methanol after 20 min of incubation. Fluorescence was measured as described earlier.
Quantitative real-time RT-PCR assay
MCF-7 cells were seeded in six-well Costar plates and underwent various treatments. After 24 h, total RNA was extracted from the cells using TRIzol reagent (Invitrogen). The RNA's concentration and purity were determined by its absorbance at 260/280 nm. First DNA strands were synthesised from 3 μg of total RNA using oligo-dT primers and Moloney murine leukaemia virus reverse transcriptase (USB Corporation). Target fragments were quantified by real-time PCR and an ABI Prism 7700 Sequence Detection System (Applied Biosystems) was employed for these assays. Taqman®/VIC® MGB probes and primers for CYP1B1 and GAPDH (Assay-on-Demand™) and Real-time PCR Taqman Universal PCR Master Mix were all obtained from Applied Biosystems. PCR were set up as described in the manual, which were validated by the company. Signals obtained for glyceraldehyde-3-phosphate dehydrogenase served as a reference to normalise the amount of RNA amplified in each reaction. Relative gene expression was analysed using the 2ΔΔCt method(Reference Livak and Schmittgen23).
Western blot analysis
Cells were washed once with PBS (pH 7·4) and harvested into a 1·5 ml microtube with 0·5 ml lysis buffer (PBS, 1 % NP40, 0·5 % sodium deoxycholate and 0·1 % SDS). The lysis buffer contained protease inhibitors (40 mg phenylmethylsulfonyl fluoride/l, 0·5 mg aprotinin/l, 0·5 mg leupeptin/l, 1·1 mm-EDTA and 0·7 mg pepstatin/l) and phosphatase inhibitor cocktail (PhosphoSTOP tablet; Roche). The harvested cells were then lysed with a cell disruptor (Branson Ultrasonics Corporation) on ice for 30 s. The protein concentration of cell lysate was determined by DC protein assay (BioRad). Then, 50 μg of lysate protein were separated on 10 % SDS–PAGE and transferred onto an Immobilon PVDF membrane (Millipore). Anti-CYP1B1 (Abcam) and anti-actin primary (Sigma Chemicals) and secondary antibodies conjugated with horseradish peroxidase (Santa Cruz Biotechnology) were used for protein detection. An Enhanced Chemiluminescence Detection Kit (Amersham) provided the chemiluminescence substrate for HRP, and the targeted protein was visualised by autoradiography.
Luciferase reporter gene assay
Construction of the CYP1B1 truncation reporter gene plasmid
The 5′-flanking region upstream ( − 2299 to +25) from human CYP1B1 was cloned in the pGL3-promoter plasmid. Different truncation plasmids were constructed from the region as listed in Fig. 1. The detailed procedure has been described previously(Reference Tsuchiya, Nakajima and Yokoi18).
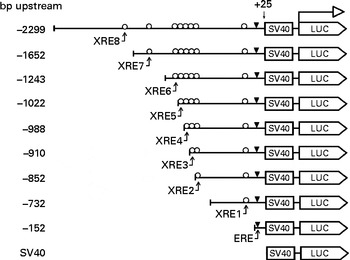
Fig. 1 Design of the CYP1B1 truncation reporter gene plasmids. Xenobiotic-responsive elements (XRE) within the CYP1B1 promoter were sequentially truncated. Locations of the response elements are oestrogen response element (ERE) ( − 63 to − 49), XRE1 ( − 268 to − 264), XRE2 ( − 838 to − 834), XRE3 ( − 853 to − 849), XRE4 ( − 944 to − 940), XRE5 ( − 993 to − 989), XRE6 ( − 1028 to − 1024), XRE7 ( − 1494 to − 1490) and XRE8 ( − 1679 to − 1675). The design facilitated the identification of a specific XRE interaction. SV40, Simian Virus 40 sequence; LUC, firefly luciferase sequence.
Construction of xenobiotic-responsive element-driven reporter gene plasmids
XRE-driven reporter plasmids were constructed as described previously(Reference Chan, Wang and Leung24). The synthesised XRE-containing fragment was digested with SmaI and BamHI and subcloned into a firefly luciferase reporter vector pTA-Luc (Clontech). ERE-luciferase reporter plasmids were gifts from Dr D McDonnell (Duke University, NC, USA). These two plasmids were known to be responsive to XRE or ERE activation only.
Dual luciferase assays
MCF-7 cells were seeded at 105 cells/well in twenty-four-well plates. After 24 h, transient transfection was performed using 1 μl Lipofectamine and 2 μl Plus reagent (Invitrogen Life Technologies) per well. Reporter plasmids (0·25 μg, containing DNA fragments derived from the CYP1B1 promoter or XRE) were transfected into cells in serum-free medium. The renilla luciferase vector pRL-CMV (Promega Corporation) was co-transfected as an internal correction for transfection efficiency. After 6 h incubation, cells were cultured in phenol red-free medium supplemented with 10 % charcoal dextran-treated FBS. The medium was removed 16 h later and the cells were treated with 1 μm-DMBA and 0·1, 1 or 10 μm-naringenin for 24 h. The cells were lysed, and the activities of the luciferases were determined using the Dual-Luciferase Assay Kit (Promega Corporation). Luciferase bioluminescence was quantified by using a FLUOstar Galaxy plate reader. Transactivation activities represented by firefly luciferase light units were then normalised with that of renilla luciferase.
Electrophoretic mobility shift assay
Nuclear protein extract was isolated by using the NucBuster™ protein extraction kit (Novagen®; EMD Biosciences, Inc.). In brief, cells were co-treated with DMBA and naringenin. After 24 h of incubation, the cells were washed, trypsinised and centrifuged at 500 g at 4°C. Reagent 1 was added to the packed cells. Nuclear extract was isolated from the cell suspension by vortexing and centrifugation. The nuclear protein was stored at − 80°C until assayed. XRE oligonucleotide, as designed below, was synthesised and labelled by the DIG Gel Shift Kit, 2nd Generation (Roche Diagnostics GmbH):
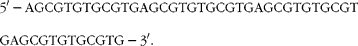 $$\begin{eqnarray} 5\prime -AGCGTGTGCGTGAGCGTGTGCGTGAGCGTGTGCGTGAGCGTGTGCGTG-3\prime . \end{eqnarray}$$
$$\begin{eqnarray} 5\prime -AGCGTGTGCGTGAGCGTGTGCGTGAGCGTGTGCGTGAGCGTGTGCGTG-3\prime . \end{eqnarray}$$
The nuclear protein was incubated with the labelled probe, sonicated salmon sperm DNA, poly(deoxyinosinic-deoxycytidylic) and binding buffer (400 mm-KCl, 80 mm-HEPES, 2 mm-dithiothreitol, 0·8 mm-EDTA and 80 % glycerol, pH 8) provided in an electrophoretic mobility shift assay accessory kit (Novagen) for 30 min at room temperature. The reaction mix was then separated on a 4–6 % non-denaturing gel in 0·5 × Tris–borate–EDTA at 100 V. The labelled oligonucleotide was electrotransferred to a Nylon membrane, fixed by UV light, blocked and washed. The shifted oligonucleotide was detected by the anti-Digoxigenin-AP conjugate and the chemiluminescent substrate CSPD® provided in the kit.
Statistical methods
A Prism®5.0 (GraphPad Software, Inc.) software package was used for statistical analysis. Results were analysed by one-way ANOVA followed by the post hoc ranking test and the significant level was set at P< 0·05.
Results
Effect of naringenin on ethoxyresorufin-O-deethylase activity in MCF-7 cells
EROD activity was determined in intact MCF-7 cells treated with DMBA and naringenin. The results indicated that naringenin was able to impede DMBA-induced EROD activity at or above 0·1 μm (Fig. 2). The IC50 value was estimated to be about 0·5 μm-naringenin. EROD activity presented was normalised by absorbance obtained from the 3-(4,5-dimethylthiazol-2-yl)-2,5-dipheryltetrozolium bromide (MTT) assay.
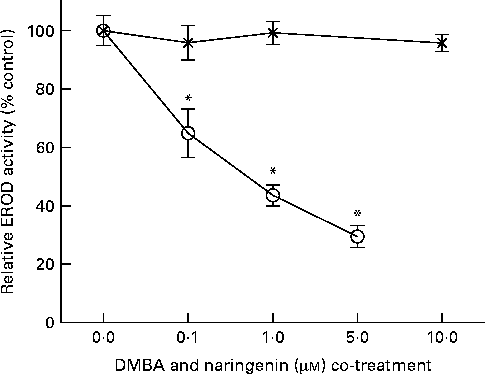
Fig. 2 Naringenin (![]() ) inhibited ethoxyresorufin-O-deethylase (EROD) activity in 7,12-dimethylbenz(α)anthracene (DMBA)-induced MCF-7 cells. MCF-7 cells were seeded in ninety-six-well culture plates and treated with naringenin or co-treated with 1 μm-DMBA and naringenin (
) inhibited ethoxyresorufin-O-deethylase (EROD) activity in 7,12-dimethylbenz(α)anthracene (DMBA)-induced MCF-7 cells. MCF-7 cells were seeded in ninety-six-well culture plates and treated with naringenin or co-treated with 1 μm-DMBA and naringenin (![]() ). After 24 h of treatment, the cells were assayed for EROD activity, and cell viability was determined on a separate set of cultures with equivalent treatment. Values are means (n 4), with their standard errors represented by vertical bars. * Mean values were significantly lower than the cultures treated with DMBA alone (P< 0·05).
). After 24 h of treatment, the cells were assayed for EROD activity, and cell viability was determined on a separate set of cultures with equivalent treatment. Values are means (n 4), with their standard errors represented by vertical bars. * Mean values were significantly lower than the cultures treated with DMBA alone (P< 0·05).
Analysis on the inhibition of CYP1 enzymes
EROD activity was decreased in cultures co-treated with naringenin, and the results could be contributed by CYP1A1 and CYP1B1 inhibition. Therefore, specific enzyme inhibition assays were carried out on recombinant human CYP1A1 and CYP1B1 enzymes. The results showed that naringenin inhibited CYP1B1 but not CYP1A1 in the concentration range tested (Fig. 3), and the IC50 value was about 5 μm.
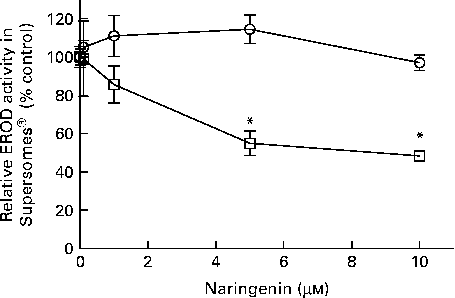
Fig. 3 Enzyme inhibition assay of naringenin on recombinant CYP1A1 and CYP1B1 proteins. Ethoxyresorufin-O-deethylase (EROD) assay was performed on human recombinant CYP1A1 (![]() ) and CYP1B1 (
) and CYP1B1 (![]() ) in the presence of the indicated concentrations of naringenin and ethoxyresorufin. Values are means (n 3), with their standard errors represented by vertical bars. * Mean values were significantly lower than samples treated with 7,12-dimethylbenz(α)anthracene alone (P< 0·05).
) in the presence of the indicated concentrations of naringenin and ethoxyresorufin. Values are means (n 3), with their standard errors represented by vertical bars. * Mean values were significantly lower than samples treated with 7,12-dimethylbenz(α)anthracene alone (P< 0·05).
CYP1B1 mRNA expression
Real-time RT-PCR showed that the DMBA treatment significantly (P< 0·05) increased the levels of CYP1B1 mRNA by 2·5-fold. The induced expression of CYP1B1 mRNA was significantly (P< 0·05) reduced by naringenin at concentrations ranging from 1 to 10 μm (Fig. 4(A)). However, naringenin given alone had no effect on the expression (Fig. 4(B)). The results indicated that naringenin by itself has no effect on CYP1B1 transcription.
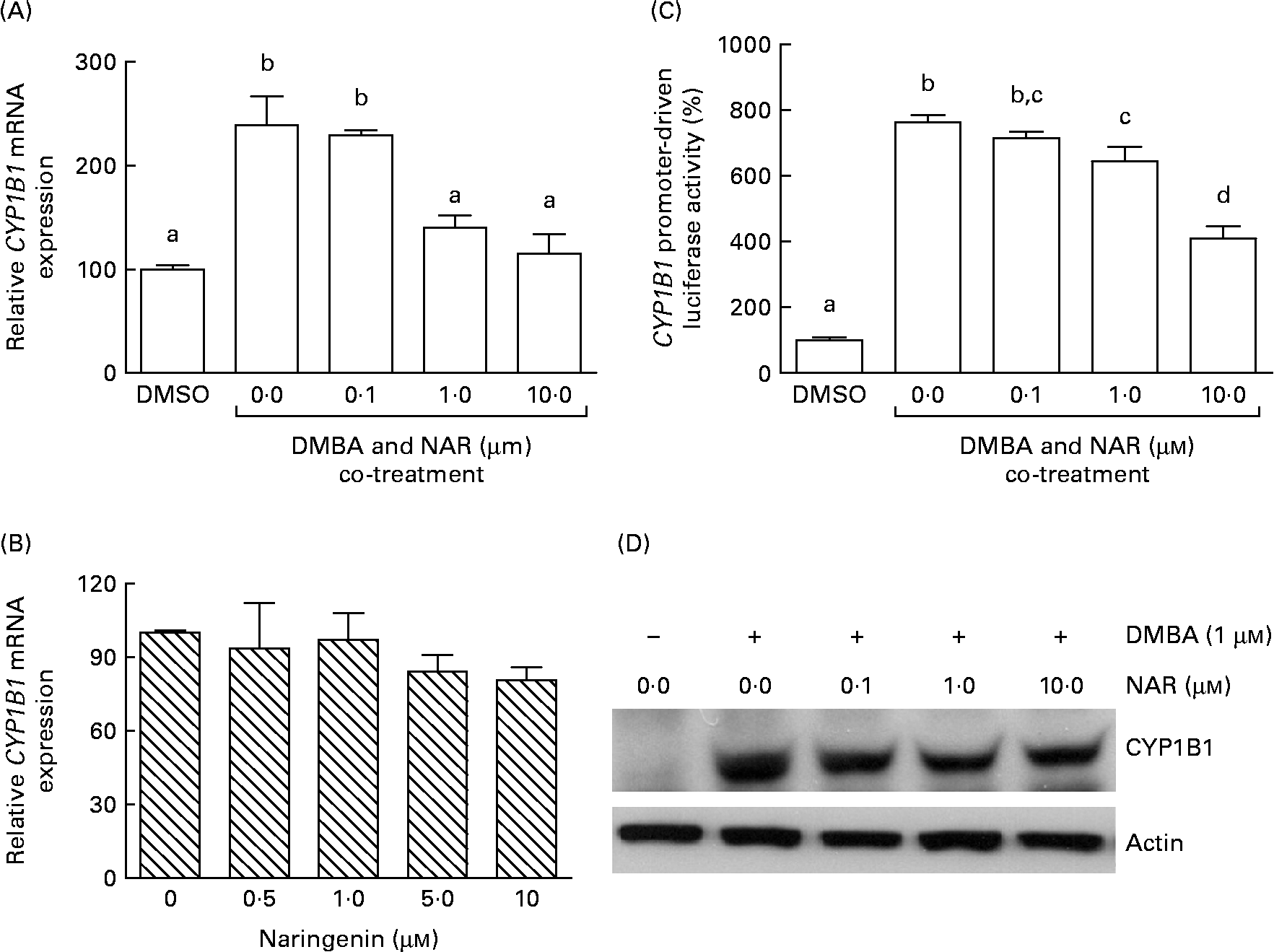
Fig. 4 Effect of naringenin (NAR) on CYP1B1 expression. MCF-7 cells were (A) co-treated with 1 μm-7,12-dimethylbenz(α)anthracene (DMBA) and NAR or (B) treated with NAR alone, and cultured for 24 h. mRNA expression of CYP1B1 was quantified by real-time RT-PCR. Values are means (n 3), with their standard errors represented by vertical bars. Mean values were significantly different (P< 0·05) among the treatments and the order is b>a. (C) Reporter gene assay was performed to verify the transcriptional regulation. In this assay, MCF-7 cells were seeded in twenty-four-well plates and were transfected with CYP1B1 reporter plasmid ( − 2299) and treated with NAR. Luciferase activities were determined in the cell lysate. Values are means (n 3), with their standard errors represented by vertical bars. Mean values with unlike letters were significantly different (P< 0·05), and the order is b>c>d>a. (D) The immunoblot image of the CYP1B1 protein, which is a representation of two independent experiments with similar results. DMSO, dimethyl sulfoxide.
Reporter gene assay on CYP1B1 promoter
As the DMBA-induced CYP1B1 mRNA expression was suppressed by naringenin, CYP1B1 promoter-driven luciferase reporter assay was performed to verify whether the gene transactivation was repressed by naringenin. The results demonstrated that naringenin suppressed the promoter transactivation at or above 1 μm (Fig. 4(C)) and was consistent with the mRNA results.
Immunoblot of CYP1B1
The Western blot results (Fig. 4(D)) were similar to the trend of mRNA. The results implicated that the expression of CYP1B1 under the naringenin treatment was probably regulated at the transcriptional level.
Oestrogen response element-driven luciferase activities
These reporter assays were carried out because ERE and XRE motifs have been located in the promoter region of CYP1B1. Naringenin by itself could induce ERE-driven luciferase activity (Fig. 5(A)) and the activity of the truncation reporter plasmid ( − 152) was induced in the co-treatment with DMBA (Fig. 5(B)). As plasmid ( − 152) contained an ERE, these data did not support the hypothesis that naringenin suppressed CYP1B1 mRNA expression by antagonising oestrogen receptor binding.
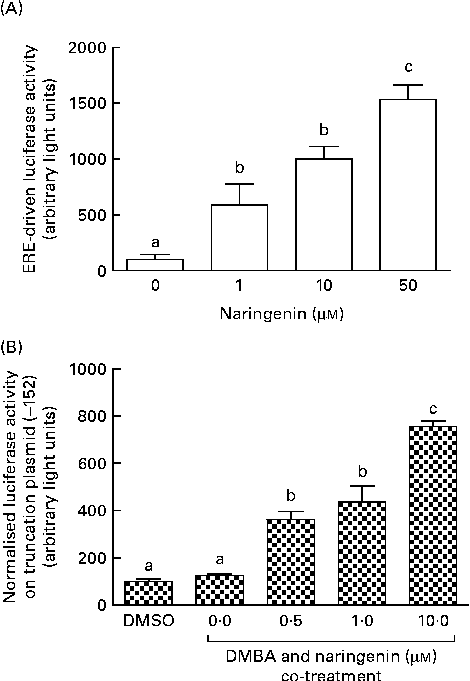
Fig. 5 Naringenin activated oestrogen response element (ERE). MCF-7 cells were seeded in forty-eight-well plates and maintained in Roswell Park Memorial Institute-1640 medium. The cells were switched to serum-free medium for 6 h and transfected with (A) ERE-driven reporter gene plasmid or (B) CYP1B1 ( − 152) truncation reporter plasmid. The cells were then treated with naringenin and assayed for luciferase activities. Values are means (n 3), with their standard errors represented by vertical bars. Mean values with unlike letters were significantly different (P< 0·05), and the order is b>c>a. DMSO, dimethyl sulfoxide.
Xenobiotic-responsive element-driven luciferase activities
On the other hand, DMBA induced the XRE-driven luciferase activity by about 3·5-fold, and this induction was significantly repressed by co-treating with naringenin at concentrations as low as 0·1 μm (Fig. 6(A)). Further study by using the electrophoretic mobility shift assay indicated a similar trend in the DMBA-induced XRE binding and the repression by naringenin (Fig. 6(B)).
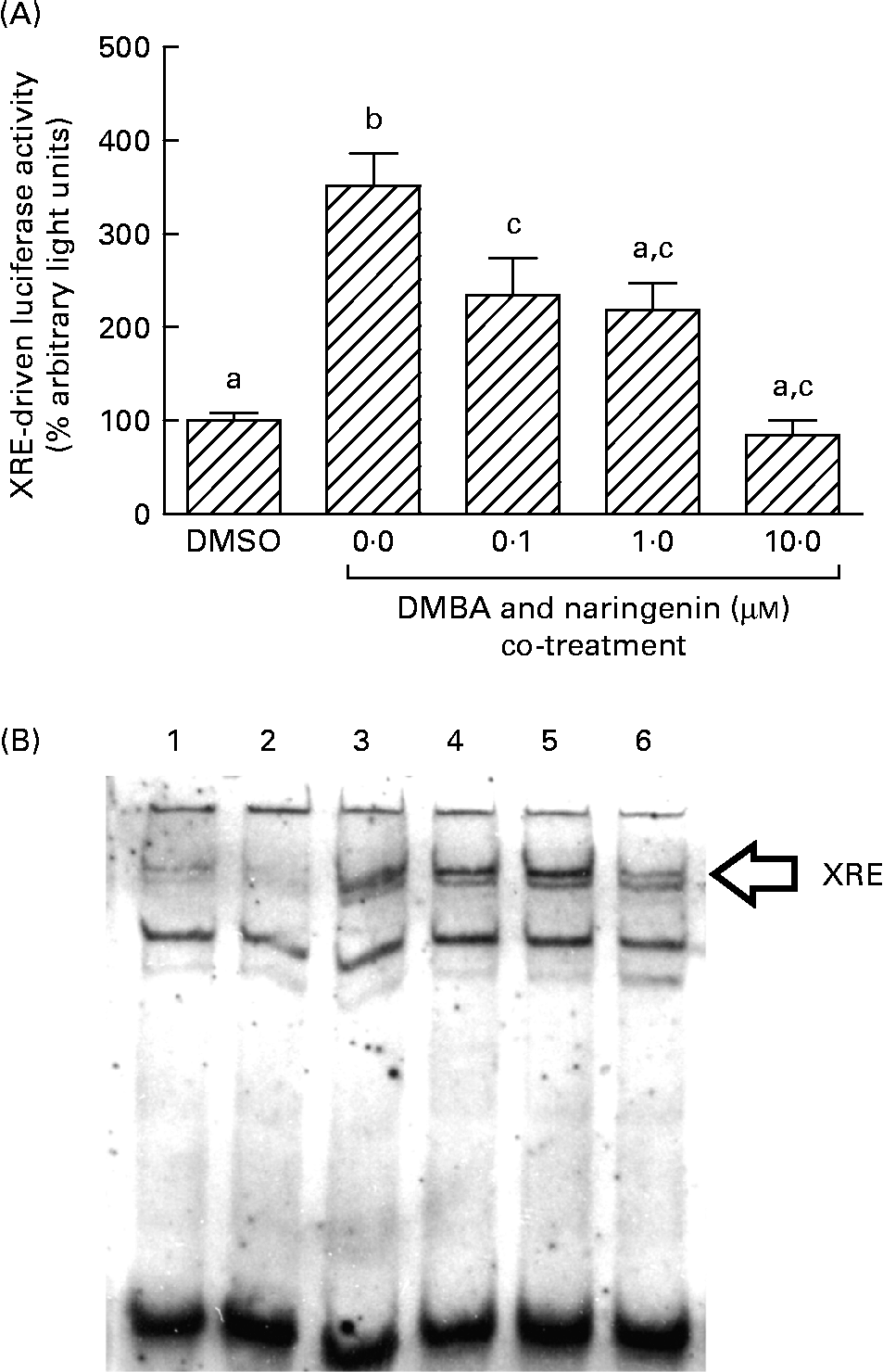
Fig. 6 Gene transactivation with respect to xenobiotic-responsive element (XRE). MCF-7 cells were seeded in forty-eight-well plates and maintained in Roswell Park Memorial Institute-1640 medium. The cells were switched to serum-free medium for 6 h and transfected with XRE-driven reporter gene plasmids. (A) The cells were then treated with naringenin and assayed for luciferase activities. Values are means (n 3), with their standard errors represented by vertical bars. Mean values with unlike letters were significantly different (P< 0·05), and the order is b>c>a. Subsequently, nuclear extract samples were prepared from the 7,12-dimethylbenz(α)anthracene (DMBA) and naringenin co-treated MCF-7 cells and electrophoretic mobility shift assay was performed. (B) Lanes labelled with 3–6 are samples incubated with the respective naringenin concentrations of 0, 0·1, 1·0 and 10·0 μm under DMBA co-treatment; lane 1 is the sample extracted from DMBA treatment and incubated with Ah receptor antibody; lane 2 is the sample treated with the carrier solvent dimethyl sulfoxide (DMSO) only. Data represent one of two independent experiments with comparable results.
Reporter gene assay on truncated CYP1B1 promoter
In order to identify the specific XRE was antagonised by naringenin, a series of truncation reporter plasmids were constructed. Among all reporter signals from the XRE-containing truncation plasmids, only those from plasmids ( − 988) and ( − 2299) were significantly induced by DMBA (Fig. 7). However, naringenin appeared to be only counteracting XRE(Reference Dertinger, Lantum and Silverstone8) in plasmid ( − 2299). Naringenin co-administered with DMBA induced luciferase activity in plasmid ( − 152), but the induction was not caused by DMBA. As demonstrated in Fig. 5, naringenin should be responsible for the induction through ERE binding. However, the induction was not carried over beyond ( − 152). Some silencing factors could be present from − 152 to − 910 and from − 988 to − 1652 since luciferase activity progressively decreased in this 5′-flanking region.

Fig. 7 Identification of the xenobiotic-responsive element-specific transactivation domain by using the truncation reporter gene assay. MCF-7 cells were seeded in forty-eight-well plates and maintained in Roswell Park Memorial Institute-1640 medium. The cells were switched to serum-free medium for 6 h and transfected with a series of CYP1B1 truncation reporter plasmids. The cells were then treated with 10 μm-naringenin (NAR) and assayed for luciferase activities. Values are means (n 3), with their standard errors represented by horizontal bars. Mean values with unlike letters were significantly different (P< 0·05) under the same truncation plasmid, and the order is b>c>a. DMBA, 7,12-dimethylbenz[α]anthracene; DMSO, dimethyl sulfoxide; SV40, Simian virus 40.
Discussion
Similar to previous findings(Reference Doostdar, Burke and Mayer25), naringenin inhibited recombinant CYP1B1 but not CYP1A1 in the present investigation. As the inhibition on cellular EROD activity was more potent than that in the recombinant protein, we also evaluated the phytochemical's effect on the transcriptional control of CYP1B1. The present results demonstrated that naringenin suppressed DMBA-induced CYP1B1 mRNA expression by antagonising XRE binding. Although the CYP1B1 promoter segment encompassing ERE was activated by naringenin, the response element did not take part in the gene transactivation. On the other hand, naringenin was not demonstrated to be a ligand of AhR in the present study. It could interact with the AhR at a non-ligand binding region and induced conformational changes in the ligand-binding domain or the ARNT-interacting domain. The altered structure of AhR prevented the receptor from binding to PAH or ARNT. A previous study has shown that inhibitors of heat shock protein 90(Reference Hughes, Guttenplan and Marcus26) may deter CYP1B1 transcription. Alternatively, the flavonoid could inhibit heat shock protein 90 and prevented the nuclear translocation of AhR.
CYP1B1 contains eight XRE in its promoter region. The position of specific XRE activation and/or suppression could be important in regulating CYP1B1 transactivation. A previous study has shown that XRE interactions at − 834 and − 853 are essential for the transcriptional control under 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment(Reference Tsuchiya, Nakajima and Yokoi18). By contrast, XRE transactivations at − 940 and − 1675 were induced by DMBA in the present study. However, only the latter XRE appeared to be suppressed by naringenin. Another possible interacting sequence was ERE. Since naringenin is a weak agonist to the oestrogen receptor and a functional ERE site is located in the 5′-flanking region of CYP1B1, the phytochemical could exert agonist/antagonist activity to the oestrogen receptor and block the transcriptional activity. However, the present data suggested that the interaction at ERE ( − 152) was augmented rather than suppressed in cells co-treated with DMBA and naringenin. The induction signal was silenced as the length of the promoter progressively increased. The biological significance of the antagonism of naringenin on the specific XRE is not known; however, we speculate that this could facilitate an additive effect on CYP1B1 suppression. Other dietary compounds may antagonise XRE in the other segments of the CYP1B1 promoter, and the combined suppression effect could be magnified.
On the other hand, Sp1-binding sites have also been identified in the 5′-flanking region of CYP1B1 (Reference Tang, Wo and Stewart27, Reference Sun, Boverhof and Burgoon28). The importance of Sp1 sites is not clear. Sp1 binding may initiate repressor activity(Reference Kaczynski, Conley and Fernandez Zapico29), while others have shown that the binding may confer enhancer activities(Reference Wo, Stewart and Greenlee30). We have found that the citrus phytochemical also suppressed CYP1A2 expression (data not shown). Since the promoter region of CYP1A2 contains no Sp1, it is unlikely that Sp1 was a factor in the present study.
Protein kinase C(Reference Delescluse, Lemaire and de Sousa31) and tyrosine and serine/threonine kinases(Reference Kikuchi, Hossain and Yoshida32) are potential signalling pathways mediating XRE transactivation. Nevertheless, the present results did not demonstrate any changes in protein kinase C in cultures treated with naringenin and/or DMBA (data not shown).
The promotive effect of CYP1B1 on breast carcinogenesis is 2-fold. It may initiate mutation by metabolising PAH into DNA-damaging species. In addition, CYP1B1 introduces an –OH group to oestrogen at the C-4 position to form 4-OH catechol oestrogen(Reference Liehr33). The hydroxylated oestrogen is genotoxic while maintaining the oestrogenic properties of its parent compound(Reference Liehr33). A previous study has shown that naringenin and citrus juices are protective against DMBA-induced mammary carcinogenesis in Sprague–Dawley rats(Reference So, Guthrie and Chambers1). The present study suggest that CYP1B1 inhibition could be the underlying mechanism.
CYP1B1 is expressed in the heart, kidney, prostate, ovary and mammary gland(Reference Shimada, Hayes and Yamazaki34) and can be a potential target for chemoprevention in the tissues(Reference Peter Guengerich, Chun and Kim35). Naringenin, as shown in the present study, can be an agent for chemoprotection. The physiological relevance of naringenin dosage in the present study is not known. Vallejo et al. (Reference Vallejo, Larrosa and Escudero36) have shown that consuming 400 ml of a commercial orange juice may produce a plasma C max of 0·44 μm-naringenin in adults. The lowest effective dose observed in the present study is still achievable from oral consumption.
In conclusion, the present study illustrated that naringenin inhibited the enzyme activity and repressed CYP1B1 mRNA expression with specific XRE antagonism. These properties might be important in nutraceutical development.
Acknowledgements
This study was supported by the Chinese University of Hong Kong. C. H. P. performed most of the experimental work; T. Y. W. and Y. W. contributed to the EROD enzyme assay and ERE transfection work, respectively. Y. T., M. N. and T. Y. cloned and constructed the CYP1B1 truncation reporter plasmids. L. K. L. designed, coordinated and oversaw the execution of this project. The authors declare that there are no conflicts of interest with respect to this study.







