



Introduction
Parkinson’s disease (PD) has become the second most prevalent neurodegenerative disease. However, despite significant research efforts, there is still no disease-modifying therapy. PD varies widely in its clinical presentation and progression, and this represents a challenge for clinical trials aimed at investigating targeted therapies in disease subgroups sharing similar clinical characteristics.Reference Espay, Schwarzschild and Tanner1 Unfortunately, the utility of clinical features to define subgroups of patients with similar disease trajectories is limited, as most patients transition between these subtypes during the disease course.Reference Tropea and Chen-Plotkin2,Reference Alves, Larsen, Emre, Wentzel-Larsen and Aarsland3 Instead, genetic, imaging and biochemical biomarker-based subtyping may be a more objective and reliable alternative for classifying PD.Reference Tropea and Chen-Plotkin2 This approach may provide deeper insights into the pathophysiology of PD, which is critical for identifying targets for effective disease-modifying treatments.Reference Frasier, Fiske and Sherer4 It may also delineate biologically homogeneous subgroups of patients who have distinctive clinical phenotypes and disease trajectories, facilitating better monitoring and potentially leading to the prediction of disease progression and therapeutic responses.Reference Espay, Schwarzschild and Tanner1,Reference Marras, Chaudhuri, Titova and Mestre5 Here, we review studies that have applied the biomarker-phenotype approach and described new PD subtypes. We highlight how biomarker-based phenotyping can guide clinical research and precision medicine, in which disease-modifying treatments and tailored symptomatic treatments can be personalized for each patient.
Genetics
PD susceptibility is undoubtedly influenced by genetic factors, which are likely to contribute to some extent to almost all PD cases.Reference Ye, Robak, Yu, Cykowski and Shulman6 Only 5%–10% of PD cases are caused by high-penetrance mendelian alleles, and these cases often present with different phenotypes compared to sporadic cases.Reference Ye, Robak, Yu, Cykowski and Shulman6 Describing these phenotypes has potential benefits for both clinical and research settings. Yet, different phenotypes can conceal similar genetic mechanisms. Thus, it is also important to understand the fundamental biology and its role in disease development and propagation. Multiple mechanisms have been proposed to explain how genetic factors contribute to PD pathogenesis, namely, synaptic, lysosomal, mitochondrial and immune dysfunction.Reference Ye, Robak, Yu, Cykowski and Shulman6 Here, we briefly discuss the underlying pathophysiology and phenotypes of four major PD-related genes.
SNCA
The aggregation of α-synuclein (αSyn) is thought to play a role in the pathophysiology of PD, although conclusive evidence is lacking regarding whether it is causative or compensatory. Many postmortem studies have revealed αSyn aggregates in the brains of PD patients, forming Lewy bodies (LB).Reference Vijiaratnam, Simuni, Bandmann, Morris and Foltynie7 SNCA mutation carriers have diffuse and severe LB pathology in the brainstem and cortex.Reference Schneider and Alcalay8 SNCA mutations, which are autosomal dominant, are hypothesized to lead to a gain of function that promotes αSyn accumulation.Reference Koros, Simitsi and Stefanis9 This accumulation is thought to disrupt multiple cellular pathways, leading to impairment of protein degradation and clearance, thereby creating a vicious cycle of αSyn aggregation. When toxic αSyn accumulates in presynaptic terminals, it causes synaptic dysfunction, neurodegeneration and cell death, thereby contributing to the clinical presentation of PD. Different types of SNCA mutations induce various molecular effects.Reference Serratos, Hernández-Pérez, Campos, Aschner and Santamaría11
Point mutations and gene multiplications have been reported in SNCA-PD, with the most common point mutation being p.A53T.Reference Koros, Simitsi and Stefanis9 Despite marked familial variability, some phenotypic resemblances are observed among p.A53T carriers.Reference Ricciardi, Petrucci and Di Giuda12 With 90% penetrance, the disease tends to be more aggressive than idiopathic Parkinson’s disease (iPD), featuring an average onset at 46 years of age, classic iPD motor symptoms that are levodopa-responsive, less common resting tremor and early motor complications.Reference Papadimitriou, Antonelou and Miligkos13,Reference Bostantjopoulou, Katsarou, Papadimitriou, Veletza, Hatzigeorgiou and Lees14 Clinicians must assess the premotor phase, as nonmotor features such as olfactory dysfunction and severe orthostatic hypotension (OH) are prominent.Reference Papadimitriou, Antonelou and Miligkos13 Dementia typically occurs within 5–7 years of disease progression.Reference Bostantjopoulou, Katsarou, Papadimitriou, Veletza, Hatzigeorgiou and Lees14 On dopamine imaging, p.A53T mutation carriers show symmetrical loss of radioligand uptake, distinguishing them from iPD patients.Reference Bostantjopoulou, Katsarou, Gerasimou, Costa and Gotzamani-Psarrakou15
Gene multiplications include triplications and, more commonly, duplications.Reference Trinh, Zeldenrust and Huang16 Patients with triplications exhibit 100% penetrance and experience disease onset around 40 years of age, with important nonmotor symptoms, early dementia as a hallmark feature and death approximately 7 years after disease onset.Reference Koros, Simitsi and Stefanis9,Reference Ferese, Modugno and Campopiano17 Imaging shows frontoparietal atrophy and severe striatal dopaminergic deficit.Reference Olgiati, Thomas and Quadri18 Duplication carriers exhibit highly heterogeneous phenotypes with milder disease and 40%–50% penetrance.Reference Koros, Simitsi and Stefanis9 The reduced penetrance and high phenotypic variability can cause one’s clinical presentation to resemble iPD, while another can mimic triplication disease.Reference Koros, Simitsi and Stefanis9 Gene multiplications respond adequately to levodopa initially.Reference Jia, Fellner and Kumar19
LRRK2
The LRRK2 gene encodes leucine-rich repeat kinase 2, a synaptic protein involved in vesicular trafficking and endocytosis.Reference Piccoli, Condliffe and Bauer20 Mutations in LRRK2 are associated with both autosomal dominant PD and iPD, resulting in increased kinase activity of the LRRK2 protein.Reference Jia, Fellner and Kumar19,Reference Di Maio, Hoffman and Rocha21 This toxic gain of function is thought to be associated with neurotoxicity.Reference Ye, Robak, Yu, Cykowski and Shulman6 Pathological findings are heterogeneous, including synucleinopathy, tauopathy and pure nigrostriatal degeneration, with the latter being the only consistent feature.Reference Koros, Simitsi and Stefanis9
Overall, LRRK2-PD is the most similar to iPD among the genetic forms of PD, though some differences remain.Reference Khan, Jain and Lynch22 LRRK2-PD has a later age of onset (after 50) compared with other genetic forms of PD, with mild, early motor symptoms and a slower disease progression in terms of nonmotor symptoms.Reference Koros, Simitsi and Stefanis9,Reference Ahamadi, Mehrotra and Hanan23 One study identified a predominance of lower extremity involvement and a higher prevalence of postural instability and gait impairment.Reference Alcalay, Mirelman and Saunders-Pullman24 Patients often maintain their cognitive function for many years and have a lower risk of developing dementia.Reference Tolosa, Vila, Klein and Rascol25,Reference Healy, Falchi and O’Sullivan26 However, the most common mutation, Gly2019Ser, is frequently associated with diffuse LB pathology, OH and dementia.Reference Kalia, Lang and Hazrati10
In terms of nonmotor symptoms, olfactory impairment, depression and rapid eye movement (REM) sleep behavior disorder (RBD) are less common.Reference Healy, Falchi and O’Sullivan26,Reference Ehrminger, Leu-Semenescu and Cormier27 Autonomic dysfunction is similar to that seen in iPD.Reference Gaig, Vilas and Infante28 Due to the low penetrance (25%–80% for the Gly2019Ser mutation)Reference Koros, Simitsi and Stefanis9 and less prominent prodromal nonmotor features, assessing the premotor phase can be challenging. However, this phase is critical for developing disease-modifying treatments, as it may represent a window for preventing the disease’s pathophysiological progression. Therefore, further research is needed to identify nonclinical biomarkers for prodromal LRRK2-PD. These patients typically respond similarly to iPD to symptomatic treatments, such as levodopa or deep brain stimulation.Reference Tolosa, Vila, Klein and Rascol25
GBA
Mutations in the GBA gene are also a well-known risk factor for PD. It encodes β-glucocerebrosidase (GCase), a lysosomal protein involved in the degradation of sphingolipids. In human-induced pluripotent stem cells, reduced GCase activity leads to the accumulation of sphingolipid substrates and facilitates αSyn accumulation, leading to deleterious effects on neuronal cells. In turn, αSyn accumulation reduces GCase activity, perpetuating lysosomal dysfunction.Reference Mazzulli, Xu and Sun29 GCase activity is decreased in PD patients, both with and without GBA mutations, although it is lower in those with GBA mutations.Reference Mullin, Smith and Lee30 Furthermore, reduced GCase activity is thought to hinder mitochondrial energy production and increase oxidative stress.Reference Cleeter, Chau and Gluck31 In addition, neuroinflammation and microglial activation have been found in brain regions susceptible to LB in carriers of GBA mutations without PD.Reference Mullin, Stokholm and Hughes32 However, it remains unclear whether elevated levels of cytokines in the serum and CSF of PD patients are also present in GBA-PD patients and whether they contribute to disease development. Nonetheless, pathophysiological mechanisms may differ between GBA variants, suggesting that patients could benefit from treatments tailored to their specific variant.Reference Senkevich, Rudakou and Gan-Or33
GBA mutations have typically been associated with Gaucher disease (GD). GBA variants are classified as severe or mild, based on the severity of GD they cause.Reference Senkevich, Rudakou and Gan-Or33 Both are associated with a higher risk of PD, with severe variants conferring a higher risk.Reference Gan-Or, Amshalom and Kilarski34 In addition, the GBA risk variants p.E326K and p.T369M do not lead to GD but increase the risk of developing PD.Reference Senkevich, Rudakou and Gan-Or33 GBA-PD patients have an earlier age of disease onset by 1.7–6 years compared to noncarriers,Reference Koros, Simitsi and Stefanis9 with a 10%–30% penetrance.Reference Senkevich, Rudakou and Gan-Or33 Despite phenotypic variability, some clinical features can help distinguish mild from severe variant carriers.
Carriers of the severe GBA variant have worse OFF motor symptoms and are more likely to exhibit psychotic symptoms, apathy, OH and severe hyposmia. They also have a higher risk of dementia and death compared to noncarriers. Mild GBA variant carriers are primarily distinguished from the severe variants by a lower risk of dementia, although still higher than in noncarriers. Studies with higher statistical power may find other differences between severe and mild variants.Reference Cilia, Tunesi and Marotta35 Compared to noncarriers, p.E326K risk variant carriers show faster motor progression and more prevalent cognitive impairment with faster cognitive decline as measured by MoCA scores, while p.T369M variant carriers show faster disease progression to the third stage of the Hoehn and Yahr scale.Reference Senkevich, Rudakou and Gan-Or33
Although GBA-PD is characterized by more prevalent or severe nonmotor features, motor symptoms are less defined. Some studies show rapid motor progressionReference Brockmann, Srulijes and Pflederer36 and more fluctuations,Reference Senkevich, Rudakou and Gan-Or37 while others suggest that motor progression is similar to noncarriers.Reference Sidransky, Nalls and Aasly38 Interestingly, carriers of both GBA and LRRK2 variants tend to have a milder phenotype than those with GBA variants alone, suggesting that LRRK2 variants may offer a protective effect over GBA variants.Reference Senkevich, Rudakou and Gan-Or33 GBA-PD patients generally respond well to levodopa.Reference Koros, Simitsi and Stefanis9
PRKN
Mutations in the PRKN gene can lead to early-onset PD in homozygous or compound heterozygous carriers.Reference Koros, Simitsi and Stefanis9 It has an autosomal recessive inheritance pattern, unlike the three previously described genes.Reference Senkevich, Rudakou and Gan-Or37 PRKN, along with the PINK1 gene, is responsible for degrading dysfunctional mitochondria in neurons.Reference Narendra and Youle39 Mutations in PRKN prevent normal parkin-mediated mitophagy. Because this process is normally initiated by high levels of reactive oxygen species, its dysfunction could lead to an excessive accumulation of these free radicals, resulting in toxicity for dopaminergic neurons.Reference Senkevich, Rudakou and Gan-Or37 PRKN also plays a role in innate immunity, and when impaired, neuroinflammatory processes can contribute to dopaminergic neuron death.Reference Senkevich, Rudakou and Gan-Or37 Unlike SNCA, LRRK2 and GBA-related PD, neuropathology studies have shown that αSyn accumulation and LB are absent in PRKN-PD. The disease is also thought to be more specific to the substantia nigra and locus coeruleus.Reference Senkevich, Rudakou and Gan-Or37
As with other genetically linked forms of PD, there is marked phenotypic variability in PRKN-PD. Penetrance can be incomplete.Reference Koros, Simitsi and Stefanis9 The median age of onset is 31 years, and patients usually present with milder disease.Reference Ye, Robak, Yu, Cykowski and Shulman6 Motor features include dystonia in the lower extremities, early gait and balance problems and common motor fluctuations and dyskinesias.Reference Khan, Graham and Critchley40 Nonmotor symptoms are less prominent than in iPD, except for psychiatric manifestations such as anxiety, panic attacks, depression and psychosis, which are more frequent. Olfaction and cognition are usually preserved, even after years of disease progression. Patients respond well to levodopa or anticholinergics, even more so than iPD patients.Reference Koros, Simitsi and Stefanis9
Genetic testing is typically reserved for patients with a family history of PD, early disease onset or ethnic risk factors.Reference Jia, Fellner and Kumar19 However, this does not align with patients’ and families’ interest in understanding their genetic background.Reference Ye, Robak, Yu, Cykowski and Shulman6 A less restrictive approach to genetic testing could help reconcile this gap and expand the pool of patients eligible for clinical trials exploring targeted and disease-modifying treatments. Clinicians must explain the goals, benefits and risks of these tests based on the best available evidence.Reference Jia, Fellner and Kumar19 Expanding genetic testing also poses ethical challenges, such as informing healthy individuals that they are carriers of mutations for less-understood diseases with low penetrance.Reference Tolosa, Vila, Klein and Rascol25
Although no medication currently modifies the progression of PD, clinicians can provide tailored symptomatic treatments to alleviate some of the most debilitating symptoms (Table 1).Reference Marras, Chaudhuri, Titova and Mestre5,Reference Koros, Simitsi and Stefanis9,Reference Jia, Fellner and Kumar19 Awareness of a patient’s genetic profile can aid in early symptom detection and timely, appropriate care.
Table 1. Symptomatic treatment can be tailored to the genetic subtypes of PD and their respective phenotypes. Adapted from Marras et al. (2020)Reference Marras, Chaudhuri, Titova and Mestre5
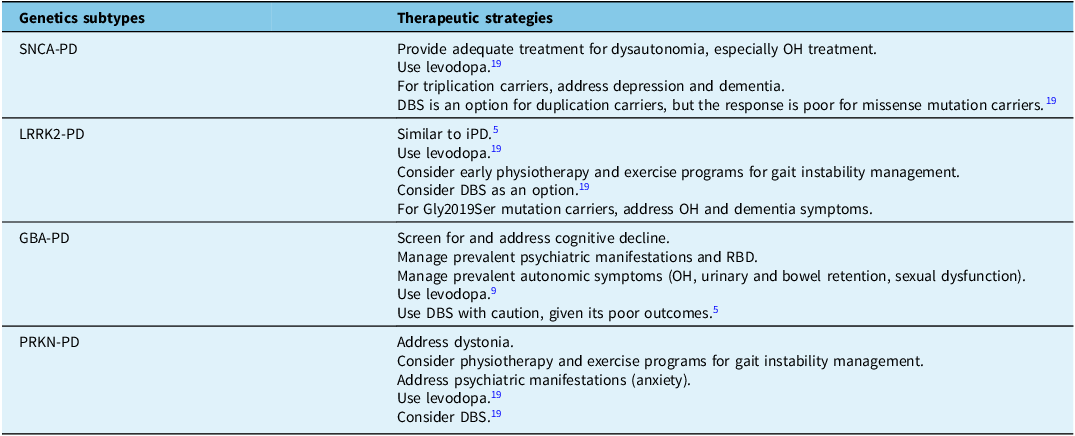
PD = Parkinson’s disease; OH = orthostatic hypotension; DBS = deep brain stimulation; iPD = idiopathic Parkinson’s disease; RBD = REM sleep behavior disorder.
Imaging
Neuroimaging is an essential tool for improving our understanding of the pathophysiology of PD and exploring how these mechanisms vary from patient to patient. It can help identify biomarker-based PD subtypes and could aid in describing, monitoring and possibly predicting disease progression.
MRI
MRI enables us to investigate specific features in the brains of PD patients and is easily accessible. T1-weighted structural MRI is used to measure cortical and subcortical volumetric changes and atrophy,Reference Mitchell, Lehéricy, Chiu, Strafella, Stoessl and Vaillancourt41 which represent axonal degeneration and neuronal cell death found in PD. Interestingly, a relationship has been found between brain connectivity, clinical features and the progression of atrophy in PD.Reference Pan, Jiang, Zhang, Zhang, Wang and Cheng42 Many studies have described MRI-based subtypes of PD and found that patterns of cortical atrophy can underpin distinct disease courses (Table 2).Reference Pan, Jiang, Zhang, Zhang, Wang and Cheng42–Reference Uribe, Segura and Baggio47 For example, progressive posterior parietal and temporal thinning could be related to semantic fluency deterioration.Reference Uribe, Segura and Baggio46 Despite the variations in imaging protocols and methodologies that complicate direct comparisons, widespread cortical thinning on MRI has been associated with more severe cognitive and motor symptoms.Reference Pan, Jiang, Zhang, Zhang, Wang and Cheng42–Reference Wang, Cheng and Rolls44,Reference Uribe, Segura and Baggio46,Reference Cao, Pang and Yu48 However, some patients maintain higher function despite brain atrophy, reflecting the brain’s compensatory capacity (brain reserve). Physical activity, known to increase brain volume in older adults, could help patients build brain reserve as a preventive measure.Reference Wang, Cheng and Rolls44
Table 2. T1-weighted MRI is used to identify PD subtypes based on brain atrophy and its relation to clinical phenotype and disease progression
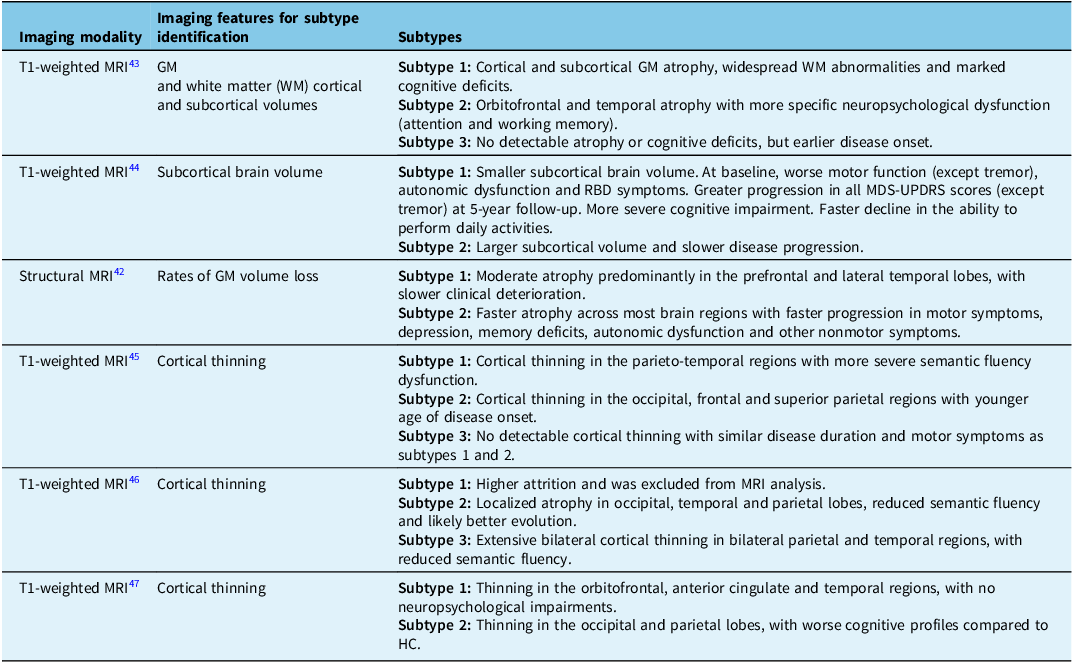
HC = healthy controls; PD = Parkinson’s disease; RBD = REM sleep behavior disorder.
Specific MRI techniques can be used in clinical trials as outcome measures.Reference Mitchell, Lehéricy, Chiu, Strafella, Stoessl and Vaillancourt41 For instance, free-water diffusion MRI, which reflects neurodegeneration and neuroinflammation,Reference Mitchell, Lehéricy, Chiu, Strafella, Stoessl and Vaillancourt41 is associated with 4-year disease progression on the Hoehn and Yahr scale.Reference Burciu, Ofori and Archer49 Moreover, neuromelanin-sensitive MRI and iron-sensitive MRI can assess specific dopaminergic neuron populations in the substantia nigra and are potential disease progression biomarkers.Reference Mitchell, Lehéricy, Chiu, Strafella, Stoessl and Vaillancourt41 Finally, functional MRI, which maps brain connectivity, can help identify patterns of neurodegeneration. Abnormal sensorimotor functional connectivity, found in the supplementary motor area of drug-naïve PD patients and carriers of LRRK2 mutations at the prodromal stage, is partially corrected by levodopa therapy. If these connectivity changes correlate with corticostriatal functional disruption, functional MRI could serve as a tool for predicting symptom development and treatment response.Reference Sasikumar and Strafella50 This imaging technique has been used in a multimodal MRI approach to identify new PD subtypes.Reference Cao, Pang and Yu48 A diffuse-malignant PD subtype, characterized by reduced spontaneous neuronal activity in the visual cortex and diffuse gray matter (GM) atrophy, showed more severe motor symptoms and cognitive dysfunction compared with the mild subtype. In contrast, the latter presented increased neuronal activity in the frontal, temporal lobes and sensorimotor cortex, mild GM atrophy and less severe motor and cognitive impairment. Given PD’s heterogeneous pathophysiology and clinical presentation, a single imaging modality cannot fully reflect the disease. Thus, objective PD subtyping is likely to benefit from multimodal imaging, as well as genetic and biochemical biomarkers.
Positron emission tomography and single-photon emission computed tomography
Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) can be used in PD with tracers that target different neurotransmitters. Reduced uptake of serotonin-specific PET tracers reflects presynaptic serotonergic disruption in cortical and subcortical regions and is associated with PD progression.Reference Sasikumar and Strafella50 Serotonergic degeneration has been associated with the severity of several neuropsychiatric symptoms, including apathy, depression and anxiety.Reference Maillet, Krack and Lhommée51 Other imaging markers may provide insights into prodromal PD,Reference Sasikumar and Strafella50 as patients with idiopathic RBD present with reduced cholinergic markersReference Gersel Stokholm, Iranzo and Østergaard52 and increased microglial activation.Reference Stokholm, Iranzo and Østergaard53 Moreover, brain glucose metabolism imaging has been correlated with disease severityReference Zhang, Li and Yuan54 and can help discriminate PD from atypical parkinsonism.Reference Sasikumar and Strafella50,Reference Tripathi, Tang and Feigin55
Dopaminergic PET and SPECT imaging identify striatal presynaptic dopaminergic deficits. Many studies have found a poor correlation between dopaminergic imaging and clinical progression.Reference Mitchell, Lehéricy, Chiu, Strafella, Stoessl and Vaillancourt41 Nevertheless, a study described three PD subtypes with differing cognitive prognoses based on cerebral perfusion patterns evaluated by 18F-FP-CIT PET, an imaging marker for nigrostriatal integrity.Reference Chung, Kim and Park56 Subtype 1 had preserved cortical uptake, young age at disease onset and better cognitive function. Subtype 2 had decreased uptake in frontal, temporal and parietal regions, with a higher risk of dementia compared to subtype 1. Subtype 3 had extensive decreased uptake, including in the occipital region, with older age at disease onset, poorer cognitive function and risk of dementia higher than subtype 1 but similar to that in subtype 2. Additionally, another study found robust PD subtypes using a combination of clinical, MRI dopamine transporter scan and radiomics imaging features.Reference Salmanpour, Shamsaei, Saberi, Hajianfar, Soltanian-Zadeh and Rahmim57 It identified mild, intermediate and severe subtypes in terms of dopaminergic deficit, which correlated both for motor and nonmotor domains, although the intermediate subtype had worse tremors overall.
Phosphodiesterase 10A is an enzyme traceable with 11C-IMA107 PET that modulates dopaminergic striatal pathways. Although not used for subtyping, reduced levels have been linked to longer disease duration and more severe symptoms in PD.Reference Sasikumar and Strafella50,Reference Niccolini, Foltynie and Reis Marques58 Consequently, phosphodiesterase 10A may represent a potential target for novel therapies.
Cardiac meta-iodobenzylguanidine scintigraphy
Cardiac meta-iodobenzylguanidine (MIBG) scintigraphy has provided new insights into the pathophysiology of PD. Reduced uptake of myocardial MIBG, associated with cardiac sympathetic denervation, has been documented in PD patients and is thought to reflect the degeneration of the cardiac plexus driven by peripheral LB pathology, possibly via the vagus nerve.Reference Pitton Rissardo and Fornari Caprara59 This may point to a distinction between peripheral and central LB deposition. Supporting this distinction is the observation that, in early-stage PD, plasma αSyn levels correlate with cardiac denervation but not with degeneration of nigrostriatal pathways.Reference Pitton Rissardo and Fornari Caprara59 MIBG scintigraphy is also used for PD subtyping. Researchers have described three subtypes based on this imaging technique: one that had initial cardiac sympathetic denervation, one with preserved innervation at the initial and follow-up imaging and a converter subtype whose imaging was initially normal but later showed cardiac denervation.Reference Yoo, Ryu and Oh60 An increasing degree of asymmetry in nigrostriatal degeneration was found among the groups, with the initially denervated subtype having the most severe and symmetric nigrostriatal degeneration. The authors concluded that these subtypes could reflect distinct origins and patterns of PD pathobiology spread – peripheral, central or converging midway. Furthermore, it was found that converters had preserved memory and that it could be the result of non-dopaminergic compensatory mechanisms such as serotonergic or noradrenergic circuits.Reference Yoo, Oh and Ryu61 Moreover, other researchers have found that central serotonergic pathways are linked to cardiac sympathetic innervation, suggesting that these pathways may play a role in cardiac sympathetic dysfunction in PD.Reference Pitton Rissardo and Fornari Caprara59
MIBG scintigraphy can also predict PD phenotypes, mainly in the context of nonmotor symptoms. Regarding autonomic dysfunction, OH was associated with cardiac denervation on MIBG scintigraphy in patients with early and mild disease.Reference Kim, Park and Oh62 Other studies have found no association between heart rate variability, sympathetic or parasympathetic function and MIBG scintigraphy in PD.Reference Pitton Rissardo and Fornari Caprara59 This imaging technique may be used for risk assessment of syncope, monitoring disease burden and predicting disease progression.Reference Pitton Rissardo and Fornari Caprara59 Indeed, subtypes of PD with normal or mild cardiac denervation have a more benign disease course, with less severe cortical atrophy and nigrostriatal damage.Reference Tsujikawa, Hasegawa and Yokoi63 Relationships have also been found between cardiac denervation on MIBG scintigraphy and cognitive dysfunction, dysphagia, hyposmia, depression, anxiety and RBD. Interestingly, abnormal cardiac MIBG scintigraphy has been shown to correlate with the incidence of falls and the progression of rigidity and axial motor symptoms.Reference Pitton Rissardo and Fornari Caprara59
Biochemical
Blood, CSF and urine are additional sources of biomarkers that could help us better understand the neurodegenerative processes underlying PD, predict disease phenotype and progression, gauge treatment response and guide researchers in developing targeted therapies. It is important to consider that blood samples are more accessible, less invasive and often more acceptable to patients than a lumbar puncture for CSF collection.
As discussed earlier, αSyn is an important marker of PD pathogenesis. Total αSyn levels in plasma or in extracellular vesicles (EV) derived from neurons have prognostic implications (Table 3).Reference Chan, Chung, Hsieh, Wu and Hong64–Reference Hirayama, Nakamura and Watanabe79 Some studies reported that EV αSyn levels were higher and associated with worse motor progression,Reference Niu, Li and Li66,Reference Shi, Liu and Cook67 while another showed reduced levels of total αSyn in PD patients.Reference Chung, Chan, Chen, Hung and Hong68 Lower EV αSyn could result from aggregation of αSyn within neurons, thereby reducing its transport in EVs across the blood-brain barrier, which would also help explain the low CSF αSyn levels observed in these PD patients.Reference Chung, Chan, Chen, Hung and Hong68 These contradictory findings about EV levels of αSyn in PD patients highlight the need for further research.Reference Chung, Chan, Chen, Hung and Hong68
Table 3. Blood, CSF and urine biomarkers are related to clinical PD phenotypes
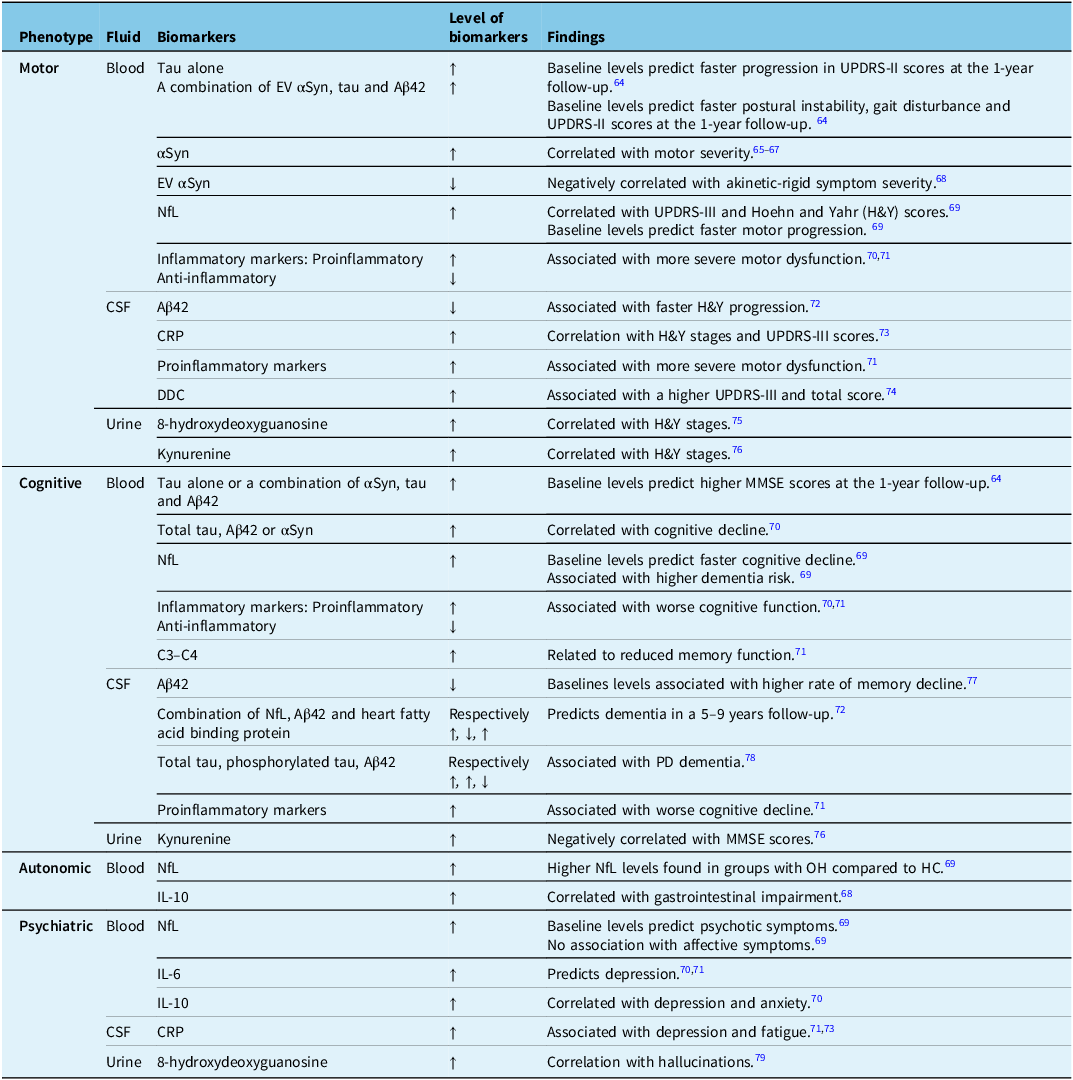
CRP = C-reactive protein.
Biomarkers of Alzheimer’s disease, including amyloid beta (Aβ) and tau protein, are also relevant to PD. Amyloid plaques and neurofibrillary tangles have been detected in the brains of PD patients, along with LB pathology, and they correlate with faster cognitive decline.Reference Tönges, Buhmann and Klebe70,Reference Irwin, Lee and Trojanowski80 Lower CSF Aβ42, an isoform of Aβ, has been found in PD patients with dementia, and this finding can be explained by increased Aβ deposition on PET imaging.Reference Hu, Yang and Gong78 Furthermore, Aβ and tau accumulation promotes αSyn aggregation, and vice versa.Reference Clinton, Blurton-Jones, Myczek, Trojanowski and LaFerla81 Elevated tau levels in PD patients with dementia suggest that tauopathy may play a role in PD pathophysiology.Reference Hu, Yang and Gong78
Neurofilament light chain (NfL) is a marker of neuronal damage in many neurological diseases, reflecting the rate of progression at a specific point in time rather than cumulative damage.Reference Buhmann, Magnus and Choe69 It also reflects nigrostriatal degeneration.Reference Diekämper, Brix and Stöcker82 Thus, NfL could be a promising candidate for monitoring PD progression and characterizing its phenotype. Moreover, since blood and CSF NfL levels correlate,Reference Hansson, Janelidze and Hall83 blood samples may be prioritized.
Inflammatory biomarkers are detectable in the blood and CSF of PD patients. Although it remains unclear whether inflammation is a cause or an effect in PD,Reference Tönges, Buhmann and Klebe70 it is believed to contribute to PD pathogenesis. In fact, αSyn activates microglia, leading to neuroinflammation and the release of cytokines and chemokines. αSyn also stimulates peripheral cytokine production and cytotoxic T-cell responses.Reference Zimmermann and Brockmann71
Dopa decarboxylase (DDC) is crucial for the synthesis of neurotransmitters, particularly dopamine. It has been hypothesized that brain neurons may produce more DDC in response to dopaminergic neurodegeneration.Reference Pereira, Kumar and Hall84 Elevated CSF DDC levels have been observed in prodromal PD patients with RBD and hyposmia, suggesting that DDC upregulation may begin early in the disease. Higher CSF DDC levels also correlate with motor symptom severity.Reference Rutledge, Lehallier and Zarifkar74 Another explanation for elevated CSF DDC could be that it first increases in the periphery and is then transported into the CSF.Reference Pereira, Kumar and Hall84
Two potential urinary biomarkers for PD are 8-hydroxydeoxyguanosine and kynurenine. 8-hydroxydeoxyguanosine is a product of DNA base modification caused by oxidative stress – a process implicated in dopaminergic neuron degeneration. Kynurenine is also associated with oxidative stress, and the kynurenine pathway may contribute to PD pathophysiology.Reference Gopar-Cuevas, Duarte-Jurado and Diaz-Perez85
Table 3 shows that some biochemical biomarkers can help predict clinical progression. For instance, a patient with higher blood IL-6 and NfL could face an increased risk of depression and psychotic symptoms. Therefore, the clinician could screen for worrisome symptoms, ensure closer follow-up, provide prevention strategies and refer the patient to mental health specialists as needed. These biological signatures, combined with imaging and genetic data, may help create homogeneous cohorts with similar disease trajectories for clinical trials. In the clinic, they might also aid follow-up and complement clinical tools like the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), which have their own limitations.Reference Regnault, Boroojerdi, Meunier, Bani, Morel and Cano86
Clinical research
Highlighting biomarker-based phenotypes is essential for improving clinical trial design. For example, carriers of different GBA variants and carriers of both GBA and LRRK2 variants may experience different disease trajectories and phenotypes. Considering these differences could help improve stratified randomizationReference Marras, Fereshtehnejad and Berg87 and power calculations and help predict motor and nonmotor complications, as well as mortality and attrition during these studies.Reference Marras, Chaudhuri, Titova and Mestre5 Additionally, patients with slower disease progression, such as those with less subcortical atrophyReference Wang, Cheng and Rolls44 or preserved MIBG uptake,Reference Totsune, Baba and Sugimura88 may require longer study durations.Reference Tolosa, Vila, Klein and Rascol25
Describing new PD subtypes can also help researchers better anticipate challenges when recruiting patients or defining inclusion criteria. For instance, GBA-PD patients tend to show faster motor progression and a shorter premotor phase. Therefore, recruitment for studies investigating targeted therapies in the premotor phase for this population could be more challenging. In contrast, this could also be beneficial, since a faster disease progression could potentially show the effect of treatments more quickly or with a smaller sample size.Reference Senkevich, Rudakou and Gan-Or37 Expanding genetic testing would make it easier to identify potential candidates.
Subtyping can also aid in participant selection. In trials investigating dementia prevention, we could include SNCA-PD patients, since they generally develop dementia more rapidly, after 5–7 years of disease. This concept can be extended to other subtypes at high risk of dementia described in this review, such as the carriers of the GBA E326K risk variant, subtypes defined by 18F-FP-CIT PET and MRI,Reference Chung, Kim and Park56 or those identified by the combination of CSF NfL, Aβ42 and heart fatty acid binding protein.Reference Bäckström, Eriksson Domellöf and Linder72 Also, because LRRK2 mutations increase the risk of both autosomal dominant forms of PD and iPD, the latter group could be included in studies investigating LRRK2-targeted therapies, thereby enlarging the pool of participants for trials.Reference Tolosa, Vila, Klein and Rascol25
In early-phase clinical trials investigating disease-modifying treatments, the primary outcome measures should focus more on imaging and biochemical biomarkers.Reference Vijiaratnam, Simuni, Bandmann, Morris and Foltynie7 These measures are objective and more likely to change rapidly than clinical features, especially in patient groups with a lower prevalence of prodromal symptoms, such as those with LRRK2-PD. These outcome measures should be specific to the subtypes investigatedReference Wang, Cheng and Rolls44 and will require clinical correlation in later phases.
Building on the importance of biomarkers, genetic biomarkers hold promise for clinical trials, as expanding knowledge of the pathophysiology behind genetic forms of PD reveals potential targets for disease-modifying treatments. Many clinical trials are currently investigating molecules that target specific pathophysiological mechanisms of genetic subtypes of PD. In SNCA-PD (and iPD), trials attempt to halt αSyn accumulation through various strategies or target already accumulated αSyn with passive and active immunization. These approaches aim to limit cellular pathway disruption and the resulting neurodegeneration. Also, because LRRK2 mutations increase kinase activity, current trials focus on developing kinase inhibitors.Reference Tolosa, Vila, Klein and Rascol25 For GBA mutations, the goal is to increase GCase activity with GCase chaperonesReference Colucci, Avenali and De Micco89 or gene therapy,Reference Abeliovich, Hefti and Sevigny90 thereby limiting lysosomal dysfunction and αSyn accumulation. Active immunization targeting accumulated αSyn is also under study for GBA-PD. To date, research aimed at identifying molecules that slow the progression of PRKN-PD remains in the preclinical stage.
Conclusion
This review presents available data on biomarker-based phenotyping and explores the description of more objective and reliable PD subtypes based on biomarkers rather than clinical measures. Genetic, neuroimaging and biochemical markers can help better describe the various clinical presentations of PD patients and subgroups of patients who follow similar disease trajectories. Not only are they promising tools for clinicians to predict disease course and tailor symptomatic treatments, but they could also help develop better clinical trials and provide a pathophysiological foundation for the development of disease-modifying treatments.
Further validation of these subtypes is needed. In addition, we must consider ethical concerns related to expanding genetic testing, as well as the costs and burden of frequent lab and imaging tests. Collaboration among neurologists, geneticists, imaging specialists and other health professionals will be essential for integrating these tools into clinical practice, ultimately advancing care for patients and their families.
Acknowledgments
The authors have no acknowledgments to disclose.
Author contributions
CB contributed to the conceptualization of the manuscript, conducted the literature review and wrote the manuscript. MB contributed to the conceptualization of the manuscript and wrote and revised the manuscript.
Funding statement
None.
Competing interests
None.




 Open access
Open access