Introduction
Tardive dyskinesia (TD) is a delayed and often irreversible motor complication resulting from long-term exposure to centrally acting dopamine D2 receptor blocking agents. Documented over 60 years ago, many consider it a legacy condition of an era when only potent (first generation) antipsychotic drugs were available. This attitude contributed in part to a stunning increase in label and off-label use of second-generation (so-called atypical) antipsychotic drugs with combined dopamine and serotonin (D2/5-HT2A) blocking properties. Recent studies from different countries suggest that the TD landscape has changed appreciably since the advent of the atypical drugs, but long-term prevalence rates over 20% are still being reported. Thus, one can reasonably expect TD to remain on the radar screen in the foreseeable future. The enduring knowledge gap in pathophysiology greatly impeded the development of effective preventative and curative measures against TD, and the synthesis of novel antipsychotic drugs that would not trigger the molecular signaling defects leading to TD. Still, progress is being made in clarifying the modern epidemiology of TD, the neural circuitry involved, and the contribution of dopamine signaling confirmed by newer effective vesicular monoamine transporter type 2 (VMAT2) function inhibitors, which rekindled the interest about TD and enriched a practical management algorithm for the condition. This brief update will focus on these aspects. Further reading on the definition and recognition of the different tardive syndromes can be found elsewhere. Reference Friedman1
Epidemiology and Functional Impairment
Recent studies have contributed to shed light on the challenge of TD and its impact on quality of life. The reputed anosognosia of patients about TD and its lack of impact on quality of life have been called into question. In an online survey of a selected sample of patients with various psychiatric diagnoses, Reference McEvoy, Gandhi and Rizio2 TD subjects had worse figures for health-related quality-of-life scores and social withdrawal than those without TD, and this was particularly true of those with schizophrenia, a surprising finding given that approximately 50% of subjects with schizophrenia have apparently no or low awareness of TD. Reference Emsley, Niehaus, Oosthuizen, Koen, Chiliza and Fincham3 In a cohort of 204 patients living with a schizophrenia spectrum disorder (54%) or another psychiatric condition and TD, over 40% reported that their involuntary movements had at least some impact on their quality of life, and three-quarters felt self-conscious or embarrassed about these movements. Reference Caroff, Yeomans and Lenderking4 In 110/204 patients with TD, an overlap was documented between the clinician-rated assessment of TD severity and the patient self-assessment. A recent review revealed a regrettable paucity of studies on the functional impairment and physical capacity limits related to the body distribution of TD. Reference Strassnig, Rosenfeld and Harvey5 Impaired dexterity may compromise professional and domestic activities. Gait pattern and balance control are affected in up to 59% of those affected, in addition to speech and swallowing mechanisms. Severe oral pain and problems with teeth and/or dentures had also been reported previously. Reference Blanchet, Popovici, Guitard, Rompré, Lamarche and Lavigne6 Severe TD is associated with social stigma and increased suicide rates. A bidirectional unexplained relationship has long been recognized between TD and cognitive impairment, Reference Strassnig, Rosenfeld and Harvey5 perhaps suggesting that the signaling alterations and plastic changes underlying TD extend beyond the sensorimotor territory of the subcorticofrontal circuitry.
Contemporary epidemiological data generated from long-term prospective studies on the differential risk of TD with atypical antipsychotic drugs are beginning to emerge, not devoid of selection biases. They often recruit TD-free subjects not drug-naïve at baseline, who have been typically exposed to several typical antipsychotics previously. These individuals may be considered in a sensitized state promoting TD induction, or in a compensatory state effectively fending off drug sensitization. Within these limits, a large prospective study (N = 352) conducted in Connecticut between 2000 and 2005 concluded that the incidence of TD with atypical drugs was similar to findings generated locally two decades ago during the era of conventional antipsychotic drug therapy. Reference Woods, Morgenstern and Saksa7 A recent meta-analysis cumulating 41 studies constituted a reality check for clinicians claiming that TD had vanished with modern drug therapy, reporting an overall mean prevalence of TD of 25.3%, lower in those under current second-generation antipsychotic drugs (20%) relative to those under first-generation drugs (30%). Reference Carbon, Hsieh, Kane and Correll8 In Caroff et al. [Reference Caroff, Yeomans and Lenderking4], studies reporting on 352 participants never exposed to first-generation antipsychotics, including 80 children, prevalence of definite TD was determined to be 7.2%, or 9.6% among the adult population, a favorable outcome influenced by the mean age (under 40) of the participants and overrepresentation (41.7%) of patients on clozapine, a less TD-prone treatment. In an Indian cohort that matched patients for age, gender, and duration of therapy, an insignificant difference in risk was found between those on first-generation versus second-generation drugs, but this time risperidone was overrepresented in the latter class. Reference Achalia, Chaturvedi, Desai, Rao and Prakash9 Risperidone is a strong D2 antagonist of equipotent and even superior antipsychotic efficacy compared to haloperidol. Reference Tarsy, Baldessarini and Tarazi10,Reference Csernansky, Mahmoud and Brenner11 Its use has been associated with a TD risk greater than other atypical agents, similar to typical (oral and depot) antipsychotic agents. Reference Novick, Haro, Bertsch and Haddad12 A retrospective, population-based cohort study that interrogated three different databases in Ontario on the incidence of TD in >20,000 senior citizens with dementia found no difference between those exposed to typical or atypical antipsychotics, but once again risperidone was overrerepresented. Reference Lee, Sykora and Gill13 Reported figures continue to be influenced by the average age of the cohort under scrutiny, and one French study conducted in patients under age 40 found a prevalence rate of TD of 8.3%. Reference Misdrahi, Tessier and Daubigney14 Any reduction in TD prevalence is worth mentioning, but recent findings are no reason to celebrate.
Reappraisal of the Dopamine Receptor Supersensitivity Hypothesis
Several studies have attempted to shed light on the genetic risk and induction mechanisms for TD. Many different pharmacokinetic (linked to drug metabolism) and pharmacodynamic (linked to drug response) genetic markers of interest were examined by candidate gene investigations and genome-wide association studies, with promising leads identified for CYP2D6, the dopamine receptor D2 and D3 genes, and the VMAT2 gene SLC18A2. Reference Zai, Maes, Tiwari, Zai, Remington and Kennedy15 Although a predictive multigene TD panel to determine the individual risk in a personalized medicine approach is not within reach, these studies indirectly support the dopamine hypothesis of TD.
The early concept of TD as a dopamine supersensitivity condition stemmed from observations that its intensity is aggravated or attenuated following the abrupt withdrawal or dosage increase of the offending drug, respectively, and enhanced following challenge with dopaminomimetics. In addition, an imbalance in striatal dopamine receptors was found following chronic antipsychotic drug exposure in experimental rats with upregulation in the density of striatal D2, not D1, receptors. Reference See, Aravagiri and Ellison16 This result was confirmed regardless of the presence or absence of TD-like vacuous chewing movements (VCM). Reference Knable, Hyde and Herman17 The D2 receptor upregulation was confirmed in vitro in addition to increased oxidative stress indices, Reference Deslauriers, Lefrancois, Larouche, Sarret and Grignon18 both prevented by lipoic acid, but this antioxidant failed to improve TD-like movements in a rat model. Reference Lister, Andreazza and Navaid19 In another rat experiment, long-term exposure to haloperidol increased striatal D2 binding density, but no electrophysiological support for the development of supersensitive neuronal responses to selective D1 or D2 agonists was documented. Reference Jiang, Kasser, Altar and Wang20 Human positron emission tomography examining D2-like (D2/3) receptor binding with the radioligand bromospiperone, Reference Blin, Baron and Cambon21 or methylspiperone, Reference Andersson, Eckernäs and Hartvig22 did not find evidence for D2/3 receptor upregulation in subjects with TD examined off-drug. In another report, [11C]raclopride binding was found to be increased by 30–40% in nine subjects under long-term antipsychotic drug treatment with or without TD, but a positive correlation was observed between D2/3 binding level and TD intensity. Reference Silvestri, Seeman and Negrete23 Postmortem analysis of [3 H]spiperone binding to D2 receptors in the putamen of subjects medicated for schizophrenia and controls (N = 27/group) showed no difference between those with or without TD, Reference Kornhuber, Riederer and Reynolds24 but such studies are inevitably conducted long after TD induction in senescent nigrostriatal pathways.
Given the inconsistencies in published data, the dopamine receptor supersensitivity hypothesis was revisited in an experimental nonhuman primate model following long-term exposure to haloperidol or clozapine chosen as recognized prototypical drugs for conventional and atypical antipsychotics, respectively. Reference Blanchet, Parent, Rompré and Lévesque25 According to the National Institute of Mental Health Psychoactive Drug Screening Program, these drugs display equivalent high in vitro affinity for dopamine D2 and D3 receptors. In rat tissue ex vivo, Reference Davoodi, te Riele and Langlois26 both haloperidol and clozapine display preferential D2 receptor occupancy, of greatest potency with haloperidol, but dose-dependent D3 receptor occupancy is always observed. At clinically relevant doses, D3 receptor occupancy is greater for clozapine (60%) than for haloperidol (16%). A nonhuman primate positron emission tomography imaging study using [11C]-(+)-PHNO confirmed D2 and D3 receptor occupancy with both drugs, with calculated D2:D3 selectivity values of 2.38 and 5.25 for haloperidol and clozapine, respectively. Reference Girgis, Xu and Miyake27 Unexpectedly, striatal D2-labeled [125I]iodosulpride binding level was found comparable between unmedicated and chronically medicated monkeys under long-term haloperidol exposure, regardless of TD status. Reference Mahmoudi, Lévesque and Blanchet28 Strong upregulation in [125I]iodosulpride binding was observed following haloperidol exposure in the internal globus pallidus, of equal magnitude between TD-expressing and TD-free animals. In contrast to the rat model, long-term haloperidol exposure caused a robust upregulation in D3-labeled [125I]7-OH-PIPAT binding in all regions examined by standard receptor autoradiography, to a greater extent in the anterior striatal areas of TD-expressing monkeys. Using a double in situ hybridization procedure, a 3-fold increase in the fraction of preprotachykinin (PPT)-positive neurons co-expressing D3 mRNA was found in the anterior putamen, compared to PPT-negative cells, indicating that this upregulation mainly took place in direct striatal projection neurons. This increase correlated with TD intensity in the lateral putamen. Clozapine exposure spared D3 receptors in the basal ganglia. It is still unclear how striatal dopamine D3 receptor upregulation in direct striatal output neurons contributes to TD at the cellular or circuit level, but D3 receptors have been associated with behavioral sensitization and levodopa-induced dyskinesia along the direct pathway, Reference Bézard, Ferry and Mach29 possibly suggesting common mechanisms. The finding warrants confirmation in human studies. The foregoing results also raise the possibility of fundamental inter-species differences in terms of D3 receptor biology and suggest that caution should be used when interpreting experimental TD data generated in rodents.
The revival of the dopamine hypothesis of TD through striatal D3, not D2, receptor upregulation sets the stage for exploring different strategies designed to attenuate D3 receptor availability or signaling. In view of their high affinity for dopamine (20-fold higher than D2 receptors), Reference Sokoloff, Giros, Martres, Bouthenet and Schwartz30 small changes in density or function may greatly impact on synaptic transmission. Further research will need to determine whether striatal monomeric or heteromeric D3 receptors are upregulated in a distinct way. In rats, half of the medium spiny neurons at origin of the direct striatal efferent pathway expressing dopamine D1 receptor mRNA co-express D3 receptor mRNA, Reference Surmeier, Song and Yan31 and strong experimental evidence suggests the existence of D1–D3 receptor heterodimers in the striatum, where the D3 partner may potentiate D1 receptor-mediated responses spared by antipsychotic drugs and contribute to an overactive direct pathway. Reference Marcellino, Ferré and Casadó32 Thus, experimental drugs dampening neural activity along the direct pathway would be worth testing to improve motor behavior.
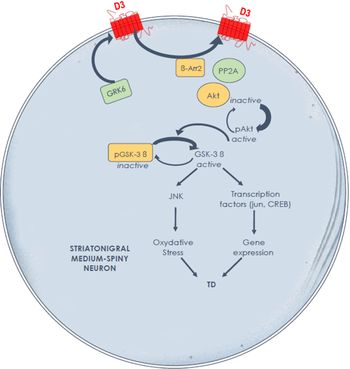
The experimental evidence generated in a relevant experimental TD model suggests key molecular mechanisms in the striatum to guide future development of preventive and palliative antidyskinetic strategies. At the signaling level, data suggest that cyclic adenosine monophosphate (cAMP)-independent mechanisms may compound the foregoing alterations in striatal D3 receptor membrane density and that the glycogen synthase kinase-3 beta (GSK-3β) is a key enzyme. In contrast to the rodent model, Reference Emamian, Hall and Birnbaum33 our primate results indirectly point to a post-translational modification of striatal GSK-3β with a major shift in enzymatic activity toward its unphosphorylated (active) form in TD-expressing animals, with the potential to distinctly alter numerous targets, physiological processes, and neuroinflammation (Figure 1). Reference Hernandez, Mahmoudi, Cyr, Diaz, Blanchet and Lévesque34,Reference Golpich, Amini and Hemmati35 This cascade of events in TD-expressing monkeys could contribute to overactive striatal dopamine signaling. Indeed, GSK-3β overactivity has been reported in a knockout mouse model lacking the dopamine transporter, producing a phenotype of motor hyperactivity and stereotypies. Reference Beaulieu, Sotnikova and Yao36 Of note, GSK-3β inhibition has been shown to decrease dopamine D1 receptor activation and membrane density, Reference Wang, Sun, Ren, Meltzer and Zhen37 with the potential to modulate direct pathway activity and mitigate hyperkinetic conditions. The GSK-3β inhibitors SB 216763, as well as two histamine H3 receptor agonists, provided benefit in a mouse model of motor hyperactivity or stereotypies resulting from dopamine D1 agonist exposure, Reference Miller, Tallarida and Unterwald38,Reference Rapanelli, Frick and Horn39 and the antidyskinetic benefit afforded by TDZD8 (GSK-3β inhibitor) administration in a rat model of levodopa-induced dyskinesia was blocked by pretreatment with a selective dopamine D1 agonist. Reference Xie, Lin and Wang40 Brain-penetrant, small molecules providing GSK-3β inhibition would certainly be worth testing in an experimental TD model.
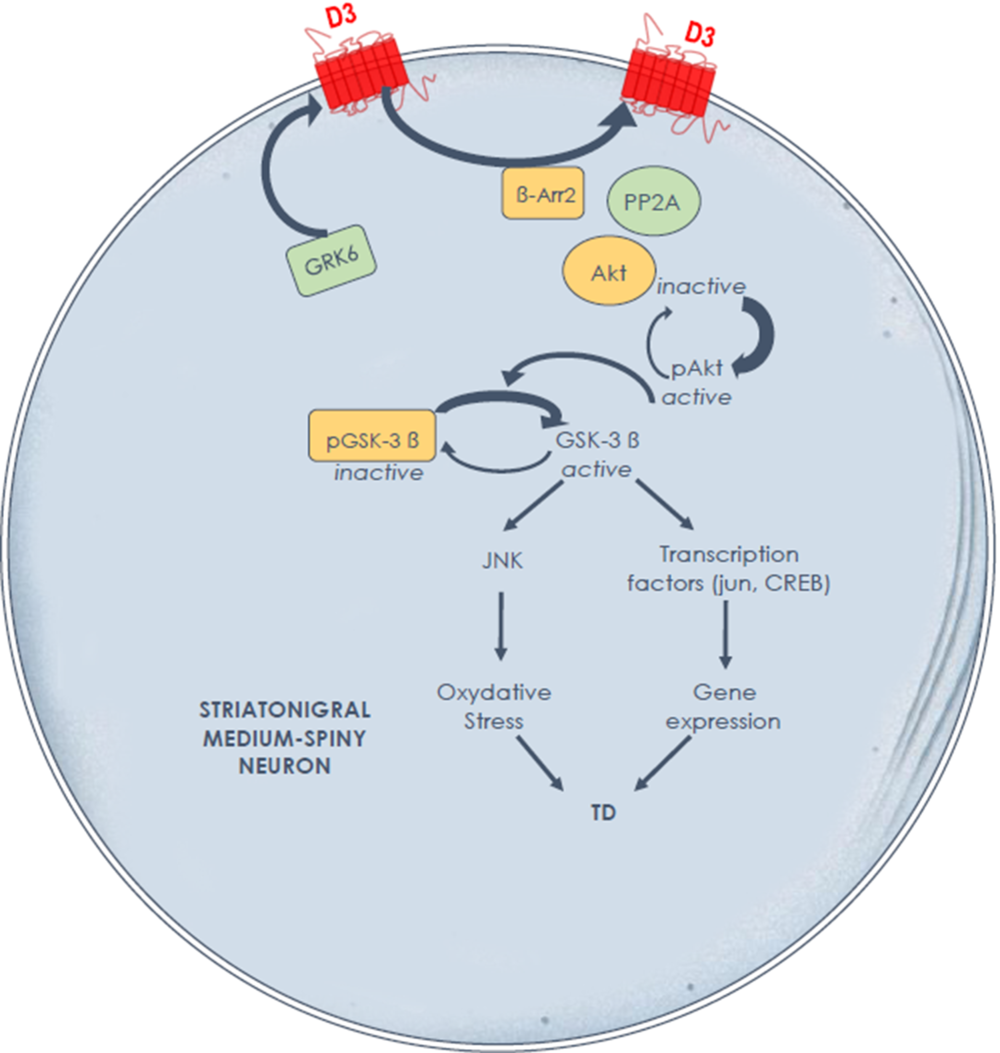
Figure 1: Hypothesis of overactive dopamine signaling Along the direct striatal output pathway in experimental tardive dyskinesia, through dysregulated D3 receptors and GSK-3β signalingReference Mahmoudi, Lévesque and Blanchet28,Reference Hernandez, Mahmoudi, Cyr, Diaz, Blanchet and Lévesque34 the D3 receptor not only displays high affinity for dopamine, but also has the potential to amplify D1 receptor-mediated responses. A high fraction of GSK-3β is maintained in its nonphosphorylated (active) form in nonhuman primates with tardive dyskinesia. Further studies are needed to determine the causal link between these signaling alterations and tardive dyskinesia induction and maintenance.
Chemically diverse GSK-3 inhibitors, such as zinc, lithium, valproic acid, tetrahydrocannabinol, histamine H3 receptor agonists, cholinergic agents, glutamate N-methyl-D-aspartate (NMDA) antagonists, and others, have been tested in various animal models (see review Reference Chico, Van Eldik and Watterson41 ). For instance, both lithium and valproate increased (inactive) phospho[Ser9]GSK-3β levels in the rat frontal cortex. Reference Kozlovsky, Amar, Belmaker and Agam42 Chronic lithium use has been reported to protect and attenuate TD in a population of schizophrenic patients, Reference van Harten, Hoek, Matroos and van Os43 whereas short-term effects were disappointing. Reference MacKay, Sheppard and Saha44 Valproic acid improved the VCM response in the reserpine rat model, Reference Peixoto, Abílio, Silva and Frussa-Filho45 but no clinical benefit was documented. Reference Fisk and York46 On the other hand, the antioxidant ginkgo biloba extract EGb761 has been shown to inhibit (Ser9) phosphorylation of GSK-3β Reference Kwon, Lee and Cho47 but somehow reduced TD in a single placebo-controlled clinical trial. Reference Zhang, Tan and Zhang48
Important knowledge gaps need to be filled in order to improve TD management. Several non-dopaminergic small-molecule candidates could eventually find their way to the clinic in order to prevent or alleviate TD. The most promising targets appear those able to dampen neural activity along the direct striatal output pathway. The ideal approach would be to design a novel class of antipsychotic drugs avoiding the detrimental neurochemical events underlying TD.
Clinical Efficacy of New VMAT2 Inhibitors
Blockade of VMAT2 function has been used as a strategy to deplete monoamine stores and manage different hyperkinetic conditions including troublesome TD. For decades, tetrabenazine and reserpine were the only drugs available in that class, with preference to the former because of its selectivity for VMAT2 over VMAT1 binding, reversibility, and shorter duration of response, decreasing adverse effects. The pharmacokinetic and pharmacodynamic profiles of tetrabenazine have complicated its use. Reference Guay49 It is a mixture of enantiomers undergoing liver metabolism by the cytochrome P450 enzymes CYP2D6 with some contribution by CYP1A2. The metabolites of the β-isomers display low-affinity antagonism for dopamine D2 receptors, which may become problematic if the drug accumulates. Metabolites have short half-lives between 2 and 8 hours, resulting in plasma drug fluctuations attended by repeated dosing during the day. A careful slow titration is required, up to an approximate daily dose of 100 mg if necessary. The Food and Drug Administration (FDA) has recommended proceeding to CYP2D6 genotyping in individuals taking over 50 mg daily, but the advantages of genotyping analysis in clinical practice remain uncertain. Expectedly, poor metabolizers tend to display more adverse effects than others. Reference Mehanna, Hunter, Davidson, Jimenez-Shahed and Jankovic50 The efficacy of tetrabenazine has been tested in various hyperkinetic movement disorders, mainly in small open-label studies. Reference Leung and Breden51 Sustained improvement in TD ratings was observed in over 80% of a group of 149 patients according to a retrospective study that allowed a daily dose up to 300 mg. Reference Kenney, Hunter and Jankovic52 Dose-dependent adverse effects such as somnolence (27%), depression (15%), parkinsonism (12%), and akathisia (9%) were documented. Reference Kenney, Hunter, Mejia and Jankovic53 Depression occurred more commonly in those with a pre-existing history (50/78 cases). Other uncommon potentially serious adverse effects include neuroleptic malignant syndrome, dysphagia, pneumonia, corrected QT interval prolongation, and suicide attempts. Co-administration of strong cytochrome P450 2D6 inhibitors (e.g., fluoxetine, paroxetine, bupropion, quinidine, and terbinafine) is liable to raise the risk of adverse events and warrants caution.
It is against these limitations and lack of FDA-approved drug treatment in TD that two novel VMAT2 inhibitors, valbenazine and deutetrabenazine, were approved in the USA as add-on therapy following rigorous protocols involving central video rating. Valbenazine is a highly selective reversible VMAT2 inhibitor converted by hydrolysis of the valine ester to (+)-α-dihydrotetrabenazine, one of the main metabolites of tetrabenazine. Its half-life of 20 hrs allows single daily dosing. It is slowly metabolized by CYP3A4/5, with some contribution by CYP2D6, offering narrowed off-target receptor binding and side-effects profiles. In 2013, a phase 2, randomized, placebo-controlled, 6-week flexible dose study (NBI-98854-1202 trial or KINECT Phase 2 study, N = 102) was completed in subjects with moderate to severe TD that allowed a daily dose range of valbenazine between 25 and 75 mg. Reference O’Brien, Jimenez, Hauser, Factor, Burke, Mandri and Castro-Gayol54 Ten participants (5 in each arm) discontinued the trial. After titration, 76% of patients in the active drug arm reached the maximal dose. The active drug improved baseline TD ratings, and 49% of participants showed ≥50% improvement in TD rating compared to 18% for the placebo group. The 6-week improvement on the rating scale was corroborated by the Clinical Global Impression of Change-TD (CGI-TD) score. No serious adverse events occurred, and fatigue and headache (10%) were the most common side effects compared to placebo (4%). In the 6-week KINECT Phase 3 study (N = 234), Reference Hauser, Factor and Marder55 fixed doses of 40 mg and 80 mg improved blinded TD ratings and CGI-TD score in a sustained manner, and the proportion of participants showing ≥50% improvement in TD rating was 40% (80 mg) and 24% (40 mg) compared to 9% for placebo. These figures were confirmed in the blinded 1-year extension KINECT Phase 3 study (N = 198, all on active drug), completed by 61% of the enrolled participants. Reference Factor, Remington and Comella56 The discontinuation rate due to adverse effects was 15.7%. The most common adverse effects included headache (7.1%), urinary tract infection (6.6%), diarrhea and dizziness (5.6%), suicidal ideation (5.1%), and depression (4%). The drug did not induce or worsen akathisia or parkinsonism. Three participants experienced syncope, but no clinically relevant electrocardiogram (ECG) alterations were documented. Thus, ECG monitoring is advised only for those known for corrected QT prolongation or arrhythmias. Long-term safety for up to 48 weeks was also assessed in the open-label KINECT Phase 4 study (N = 167 at entry). Reference Marder, Singer and Lindenmayer57 Of note, 27% of participants were taking an anticholinergic agent, perhaps influencing the detection of parkinsonism. In the first 4 weeks, 22% of participants experienced at least one treatment-emergent adverse event, considered mild to moderate in 97.5% of cases, similar to those previously reported. After week 4, serious treatment-emergent adverse events, or treatment-emergent adverse events leading to discontinuation, were recorded in 13.7% and 11.8% of participants, respectively. Dose reduction to 40 mg/d was required in 11 (6.7%) participants. Dose reduction should be considered for those on potent CYP2D6 or CYP3A4 inhibitors, and coadministration with a potent CYP3A4 inducer (e.g., rifampin, phenytoin, carbamazepine, St. John’s wort) is not recommended.
For the synthesis of deutetrabenazine, nontoxic atoms of deuterium replaced hydrogen in the molecule and strengthened bonds with carbon, thus stabilizing its metabolism and smoothing fluctuations. This allows twice-daily dosing within a daily dose range of 12–48 mg much lower than for tetrabenazine, thereby reducing the adverse effects profile and potential for dopamine D2 receptor occupancy. In two 12-week placebo-controlled studies (Aim to Reduce Movements in Tardive Dyskinesia [ARM-TD], Addressing Involuntary Movements in Tardive Dyskinesia [AIM-TD]) Reference Fernandez, Factor and Hauser58,Reference Anderson, Stamler and Davis59 and their open-label extension study of up to 2 years, Reference Fernandez, Stamler and Davis60 active treatment was well tolerated and produced rapid and sustained reduction in mean TD rating along the Abnormal Involuntary Movements Scale. In the AIM-TD study (N = 298) that included three fixed active treatment arms of 12, 24, and 36 mg, significance in TD rating improvement was achieved for the 24- and 36-mg arms. Reference Anderson, Stamler and Davis59 The proportion of patients showing ≥50% improvement in TD rating was greater for the 36-mg (33%) and 24-mg (35%) arms than in the placebo arm (12%). The incidence of adverse effects was similar to placebo, including suicidality (2.7%). During an extended open-label study (N = 343), Reference Fernandez, Stamler and Davis60 76% of participants received at least 54 weeks of treatment and 35% of participants reached the highest daily dose of 48 mg by week 80. Adverse effects were deemed mild to moderate in most cases, including somnolence and anxiety (8.5%), and depression (7.9%).
Post-marketing clinical use of the different VMAT2 inhibitors for various hyperkinetic indications and treatment durations has generally been favorable, with preference for the newer agents in view of their better tolerability. Reference Niemann and Jankovic61 Nonetheless, cases of valbenazine-induced parkinsonism have been reported in subjects over age 40, Reference Akbar, Kim and Friedman62 raising the importance to carefully monitor patients particularly during the first months of treatment. The demonstrated tolerability and safety profile of the new VMAT2 inhibitors will hopefully promote their approval by other national regulatory agencies. These efficacy data extend the practical experience gathered from the clinical use of tetrabenazine and fuel the dopamine hypothesis of TD.
Management Algorithm
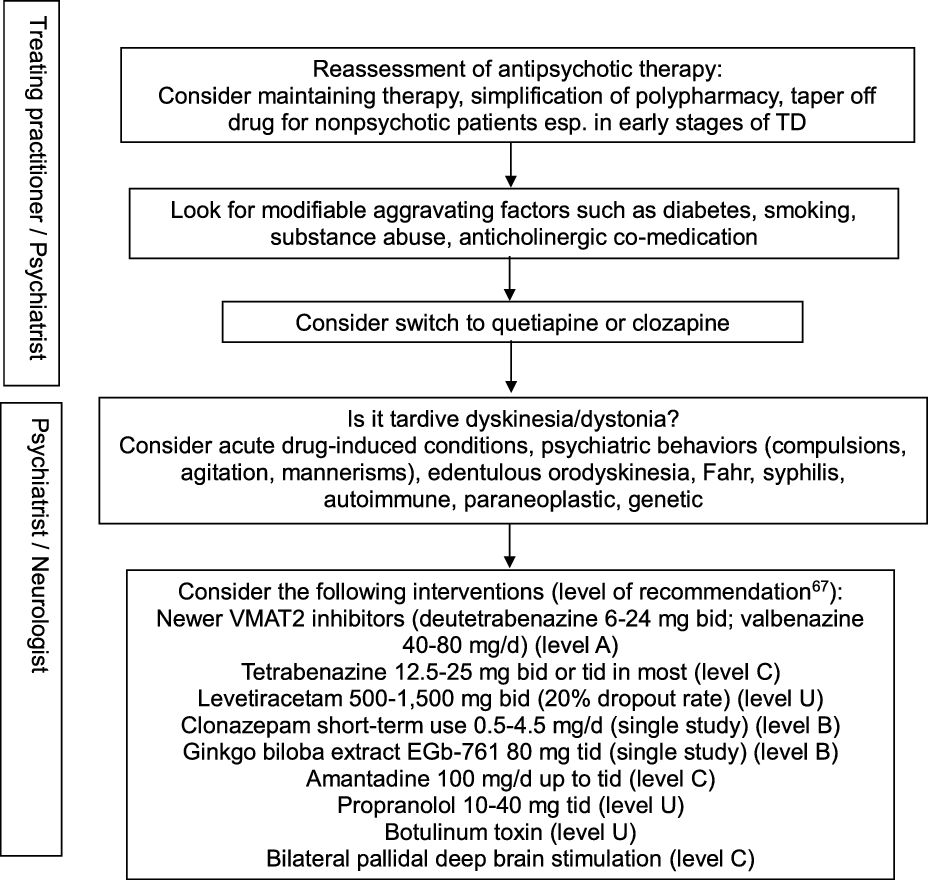
In view of the existing evidence, preventive measures clearly remain a key aspect of TD management, particularly in older age groups. Antipsychotic drugs should only be prescribed for approved indications and carefully monitored by routine screening for abnormal movement disorders. Modifiable risk factors should be identified in order to minimize the incidence and intensity of TD. These include diabetes, smoking, substance abuse including alcohol and cocaine, and anticholinergic co-treatment. Reference Solmi, Pigato, Kane and Correll63 This step should be part of any management algorithm for TD and given due consideration before initiating antidyskinetic medications. Neurologists are typically asked to assess those with troublesome motor symptomatology and those displaying unusual TD variants such as tardive dystonia, tardive akathisia, tardive chorea, or tardive tics, suggest evidence-based therapeutic interventions, and monitor progress. The phenomenology and diagnosis must be ascertained in order to guide therapy (Figure 2). Other hyperkinetic movement disorders need to be ruled out, including acute, reversible drug-induced conditions, edentulous or denture orodyskinesia, psychiatric behaviors such as compulsions, agitation, or mannerisms, autoimmune and paraneoplastic disorders, neurosyphilis, Fahr syndrome, and hereditary diseases (Huntington’s disease, neuroacanthocytosis, McLeod syndrome, spinocerebellar ataxia-17, and neurodegeneration with brain iron accumulation). In cases with predominant oromandibular dystonia, primary dystonia, neuroacanthocytosis, Lesch-Nyhan syndrome, neurodegeneration with brain iron accumulation, Wilson’s disease, spinocerebellar ataxia-8, cerebrotendinous xanthomatosis, and GM1 or GM2 gangliosidosis are among the rare conditions that may need some consideration.
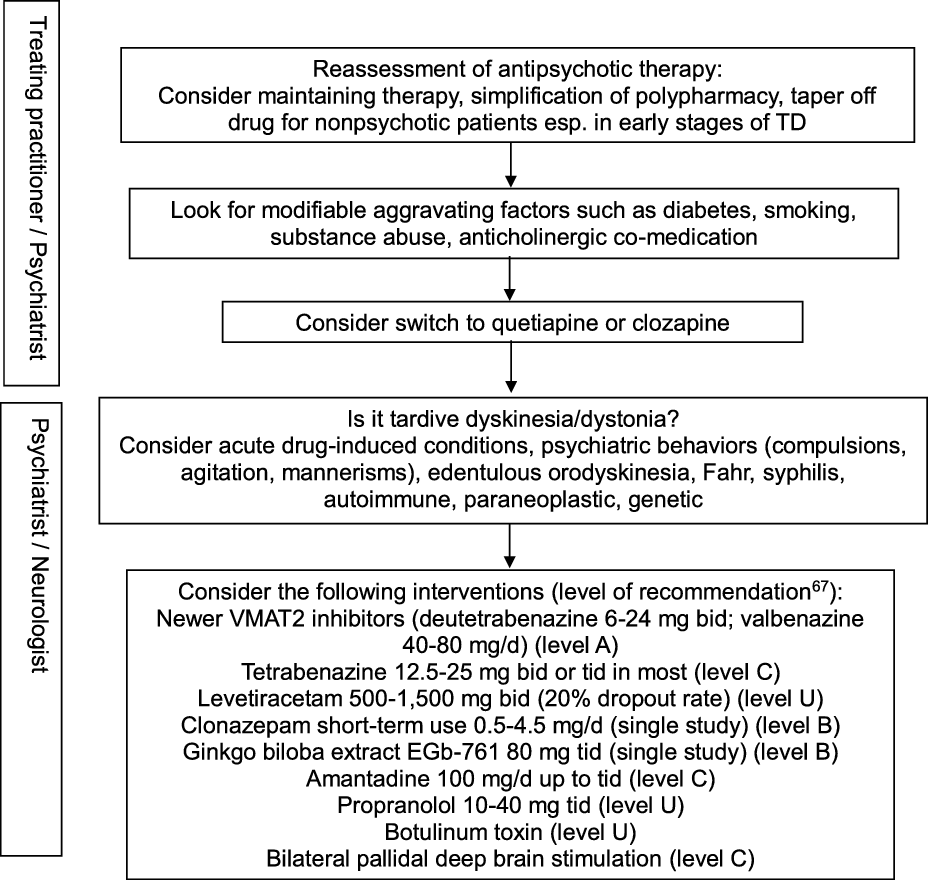
Figure 2: Practical management algorithm for troublesome tardive dyskinesia (inspired from Caroff et al.Reference Caroff, Campbell and Carroll64).
A Cochrane group conducted a systematic review of over 100 randomized trials on TD, concluding to the inability to construct an algorithm of treatment to manage TD. Reference Bergman, Walker, Nikolakopoulou, Soares-Weiser and Adams65 Typical management decisions were addressed, including a dose reduction or switch of the offending antipsychotic drug used, and use of adjunct therapies. According to the authors, the low quality of available evidence justified this outcome. Another authoritative review of all therapeutic interventions for TD published until 2011 was conducted by a subcommittee of the American Academy of Neurology. Reference Bhidayasiri, Fahn, Weiner, Gronseth, Sullivan and Zesiewicz66 The panel classified each selected study and graded evidence-based recommendations focused around five basic management issues: 1. Is withdrawal of dopamine receptor blocking agents an effective tardive syndromes treatment? 2. Does switching from typical to atypical dopamine receptor blocking agents reduce tardive syndromes symptoms? 3. What is the efficacy of pharmacologic agents in treating tardive syndromes? 4. Do patients with tardive syndromes benefit from chemodenervation with botulinum toxin? 5. Do patients with tardive syndromes benefit from surgical therapy? Given the paucity of well-designed, double-blind, randomized controlled trials, not a single intervention reached Level A recommendation. Insufficient evidence (Level U) or conflicting data made impossible to support or refute the use of many different putative antidyskinetic interventions, including botulinum toxin. The panel concluded that clonazepam and ginkgo biloba extracts (Level B) should be considered as treatment and that amantadine and tetrabenazine (Level C) might be considered as alternatives. Masking TD symptoms with risperidone or olanzapine found some support in the literature, but the safety of this strategy beyond one year has not been adequately addressed, and caution was advised when using these agents. An update of this systematic review identified a total of 687 publications relevant to the treatment of tardive syndromes between 2012 and 2017, of which 15 new articles were analyzed. Reference Bhidayasiri, Jitkritsadakul, Friedman and Fahn67 This time, the new VMAT2 inhibitors deutetrabenazine and valbenazine reached Level A recommendation as effective therapy, whereas the initial recommendations were maintained for the other pharmacological interventions and chemodenervation with botulinum toxin. The experts proposed an algorithm for management of troublesome TD involving initial readjustment of psychotropic medications, then consideration of suppressive agents first with newer VMAT2 inhibitors (Level A), then clonazepam and ginkgo biloba extracts (Level B), or eventually amantadine (Level C). Other GABAergic drugs tested in open-label trials or case series, such as kamishoyosan, Reference Lee, Shin and Lee68 zolpidem, Reference Waln and Jankovic69 and sodium oxybate, Reference D’Arrigo, Campiglio and Casellato70 await controlled studies before consideration. Among non-pharmacological interventions, bilateral pallidal deep brain stimulation reached Level C recommendation status, mainly based on a single class II study of 19 patients with severe TD or tardive dystonia. Reference Pouclet-Courtemanche, Rouaud and Thobois71 The participants were evaluated blindly with and without neurostimulation 6 months postoperatively, and 14 of them were rated along standard scales for periods of time extending between 6 and 11 years postoperatively. Sustained benefit in oral and extra-oral TD movements was observed with a reduction in TD scores >50% compared to preoperative values. Unfortunately, a recent multicentre, randomized, sham-controlled, but underpowered German study on tardive dystonia failed to reproduce those results, as the primary endpoint, which consisted of the percent change in blinded, video-based, Burke–Fahn–Marsden Dystonia Rating Scale (BFMDRS) severity score at 3 months following the start of neurostimulation compared to sham stimulation, was not met. Reference Gruber, Südmeyer and Deuschl72 Nonetheless, mean improvement in severity score at 3 months compared to preoperative values was significantly greater in the neurostimulation group (22.8%) than in the sham group (12.0%), and other secondary outcome measures favored neurostimulation. A single-center study involving a young set of 10 consecutive patients (all but one under age 40) with tardive dystonia who underwent bilateral subthalamic nucleus (STN) electrode placement reported remarkable benefit in nine participants, with a mean improvement of 87.3% in BFMDRS severity score at 6 months compared to preoperative values, sustained over an extended follow-up period. Reference Deng, Li and Zhang73 Of note, benefit was observed on postoperative day 1 in five patients, suggesting a microlesion effect. It remains unclear whether the STN is a better target for a subset of TD patients with predominant dystonia.
Other comprehensive reviews have been completed in order to document the best practice and feed an evidence-based algorithm. A Canadian group of experts recently conducted their own systematic review of 346 TD treatment articles available until June 2017, including 10 Cochrane reviews and publications targeting issues such as antipsychotic dose reduction, cessation, or switching to an atypical drug, or upward dose titration to mask troublesome TD manifestations. Reference Ricciardi, Pringsheim and Barnes74 In their Level B treatment recommendations, they considered antipsychotic drug cessation or upward titration not viable options in subjects with chronic schizophrenia, but careful drug withdrawal should be considered in other groups particularly in early stages of TD. They found insufficient evidence to recommend dose reduction, a strategy viewed as a legitimate way to prevent TD during maintenance antipsychotic drug therapy. Switching to a low-affinity dopamine D2 receptor blocker, such as clozapine and quetiapine, may be effective to reduce TD, but improvement may be delayed. The group also analyzed the evidence linking anticholinergic co-medications with TD and found very little support to support the withdrawal of these agents, which may in turn worsen parkinsonism. Keeping in mind the inconsistent outcome of this option and threat of worsened parkinsonism, they concluded that discontinuation of anticholinergic medications may be worth some consideration for the treatment of TD (Level C).
Other investigators collected the literature on drug treatment of TD published between 2007 and 2016 and identified 261 publications, of which 26 articles were reviewed including five meta-analyses. Reference Caroff, Campbell and Carroll64 The main findings were used to generate a stepwise management algorithm of troublesome TD implicating the treating psychiatrist and consultant neurologist, including initial assessment of the diagnosis and severity of TD, a review of the current antipsychotic drug treatment, including individual decision to discontinue, reduce, or switch antipsychotic, review of anticholinergic co-medication, and trials of antidyskinetic drugs with preference for the newly approved selective VMAT2 inhibitors. They then used the Delphi method, a systematic group communication process, to survey 29 North American experts (23 from the field of psychiatry, 6 from the field of neurology) in two rounds, who were asked to examine a selection of 18 recent publications reflecting the best evidence-based TD practice and research, and answer a maximum of 23 questions. Reference Caroff, Citrome and Meyer75 The degree of respondent agreement was assessed for targeted issues including TD screening methods, diagnosis, and treatment. Circumstances calling for a neurologist opinion were delineated. Unanimous consensus was reached on the need to periodically screen all patients under chronic antipsychotic drugs for TD, and on considering modifications to the antipsychotic drug regimen for patients with troublesome TD who cannot be safely withdrawn from antipsychotic therapy, with a high level of agreement for switching to an atypical second-generation drug or clozapine. A majority agreement was found for modifications of the anticholinergic co-medication, including options such as discontinuation, dose reduction, or switching to amantadine. Using different selective VMAT2 inhibitors was unanimously considered the first-line pharmacological option when an antidyskinetic strategy is necessary, and improvement of at least 30% relative to the baseline Abnormal Involuntary Movements Scale total score was considered clinically significant, representing in another study a two-point decrease in score for a minimal clinically important difference attributed to an intervention. Reference Stacy, Sajatovic and Kane76
Progress has been made in the last 5 years in the field of TD. The impact of the condition in everyday life and its current epidemiology are better understood. Experimental research has begun to unveil potentially important mechanisms suggestive of overactive striatal dopamine signaling along the direct striatonigral pathway in TD. New effective VMAT2 inhibitors have been approved following refined protocols that constitute stepping stones to generate sound data in the future. These drugs were approved by the US FDA and were added to a treatment algorithm emerging from the best experimental evidence and practice. These advances, together with cautious antipsychotic drug prescription, and development of a new generation of drugs Reference Dedic, Jones and Hopkins77 hopefully able to avoid or reverse some of the signaling alterations linked to TD, give hope to improved prevention and management of TD.
Acknowledgments
The preclinical TD program was supported by the Canadian Institutes of Health Research, grant MOP-81321, and grant MOP-130407 to Prof. Daniel Lévesque, University of Montreal. The author sincerely thanks Mrs. Marie-Luce Houde-Simard for her contribution to the graphic presentation, and Dr. Stanley N. Caroff, MD, Perelman School of Medicine, University of Pennsylvania, Philadelphia (PA, USA), who provided expert editorial comments.
Conflict of interest
The author was involved in the KINECT 3 studies sponsored by Neurocrine Biosciences, Inc., San Diego, CA, USA.