


Introduction
The intestinal lumen contains a multitude of exogenous substances that include dietary antigens and micro-organisms, both commensal and sometimes pathogenic. The intestinal epithelium acts as interface and regulator, a selective barrier that separates the luminal contents from the underlying connective tissue of the host. The immune system safeguards the host from the translocation of harmful foreign substances and helps maintain the homeostatic balance between the internal and external environments of the intestinal tract( Reference Davies and Abreu 1 ). Inflammatory bowel disease (IBD) is a chronic condition that involves the disturbance of this balance.
IBD is comprised of a spectrum of disorders which include Crohn’s disease (CD) and ulcerative colitis (UC), chronic remittent or progressive disorders characterised by non-specific inflammation and intestinal tissue damage. The pathogenesis of IBD involves complex dysregulated interactions between various factors, with genetic predisposition, the intestinal microbiota and innate and adaptive immune responses appearing to be key elements. While Khan et al. ( Reference Khan, Kale and Bere 2 ) suggest that the failure of at least one component of this triad is sufficient to trigger the inflammatory changes necessary for the induction of IBD, other researchers disagree, claiming that genetic dysfunction of the intestinal innate immune system is a key precursor of the disease( Reference Wehkamp, Schwind and Herrlinger 3 – Reference Gersemann, Wehkamp and Stange 6 ). Gruber et al.( Reference Gruber, Lichti and Rath 7 ) state that the pathogenesis of IBD, its onset and its recurrence, is most likely triggered by unknown environmental agents( Reference Gruber, Lichti and Rath 7 , Reference Xavier and Podolsky 8 ). Many researchers have implicated other factors, such as the dysfunction of intercellular transport mechanisms, for example, PepT1( Reference Ingersoll, Ayyadurai and Charania 9 – Reference Dalmasso, Nguyen and Charrier-Hisamuddin 11 ), together with factors involved in the exacerbation of IBD such as diet( Reference de Medina, Daddaoua and Requena 12 – Reference Rutherfurd-Markwick and Moughan 15 ), cigarette smoking( Reference Jappar, Hu and Smith 16 ), stress( Reference Nassl, Rubio-Aliaga and Sailer 17 ), food additives( Reference Gruber, Lichti and Rath 7 ) and microbial dysbioses( Reference Frank, Zhu and Sartor 18 ). Although the course of the disease is variable, it commonly affects the intestinal mucosa and leads to both structural and functional impairment. In genetically susceptible individuals, a disturbed host–bacterial relationship leads to immunopathological changes in the mucosa that continue in chronic remitting–relapsing cycles( Reference Khan, Kale and Bere 2 ).
CD is an inflammatory condition associated with increased intestinal permeability( Reference Olaison, Sjodahl and Tagesson 19 , Reference Hollander 20 ), indicating a disturbance of the epithelial barrier, and may affect one or multiple areas of the intestinal tract, from mouth to anus( Reference Soderholm, Peterson and Olaison 21 ). It is unclear whether increased intestinal permeability precedes and contributes to intestinal inflammation( Reference Hollander, Vadheim and Brettholz 22 ) or is a result of the inflammatory process( Reference Travis and Menzies 23 , Reference Bjarnason, Macpherson and Hollander 24 ). CD has been described as being associated with Western societies that are ‘pathogen poor’, whereas in developing countries, cases of idiopathic IBD such as CD are rare( Reference Wehkamp and Stange 25 ). UC is an aggravated inflammatory response with accompanying ulceration of the colon, thought to result from the absorption of chemotactic bacterial peptides( Reference Smith, Clemencon and Hediger 26 ).
The intestinal tract of the mammalian neonate is permeable to large peptides and other molecules, allowing the absorption of intact immunoactive molecules from milk to supplement the immature immune system( Reference Brandsch and Brandsch 27 – Reference Meisel and FitzGerald 31 ). The permeability of the intestinal epithelium to these larger molecules normally ceases in the post-weaned mammal. If such permeability continues it may be a possible trigger of IBD. The absorption of intact macromolecules from the healthy intestine remains a controversial issue, as there is little unequivocal in vivo evidence (other than antigen sampling) demonstrating this phenomenon( Reference Miner-Williams, Stevens and Moughan 32 ). However, macromolecules, and indeed microbes, can be absorbed by the mucosal tissues via transport systems that predominantly involve the adaptive and innate immune responses of the intestinal mucosa and which are key to the absorption of proinflammatory proteins/peptides in IBD.
The aim of the present review is to assess the scientific evidence to support the hypothesis that defective transepithelial transport mechanisms and the increased absorption of antigenic proinflammatory oliogopeptides are important contributing factors in the pathogenesis of IBD.
Disruption to the intestinal environment
Epithelial barrier dysfunction
The intestinal epithelium regulates the flow of nutrients, ions and water between the lumen and underlying tissues, limiting contact between the host and the intraluminal quantities of exogenous antigens and microbes. In a healthy subject, the transepithelial transport of small amounts of food- and microbial-antigens participates in the induction of a homeostatic immune response that allows immune tolerance to such antigens( Reference Izcue, Coombes and Powrie 33 , Reference Strobel and Mowat 34 ), preventing the internalisation of both pathogenic and commensal microbes( Reference Menard, Cerf-Bensussan and Heyman 28 ). However, epithelial barrier dysfunction can lead to the entry of excessive dietary or microbe-derived macromolecules, which are putative contributors to the pathogenesis of a spectrum of human diseases including food allergies, coeliac disease (CeD), IBD, autoimmune diseases and the metabolic syndrome( Reference Goll and Granlund 35 , Reference Cani, Bibiloni and Knauf 36 ). Improving intestinal barrier function, particularly the paracellular pathway, may form a therapeutic strategy for the treatment or prevention of diseases driven by luminal antigens. Understanding how antigens are transported across the epithelium in both healthy and diseased states may assist in the development of appropriate therapies( Reference Menard, Cerf-Bensussan and Heyman 28 ).
The transport of molecules across the intestinal mucosa occurs through two distinct mechanisms: paracellular diffusion through tight junctions (TJ) between adjacent intestinal epithelial cells, and transcellular transport involving the transcytosis of materials which may or may not be mediated by membrane receptors (illustrated in Fig. 1).
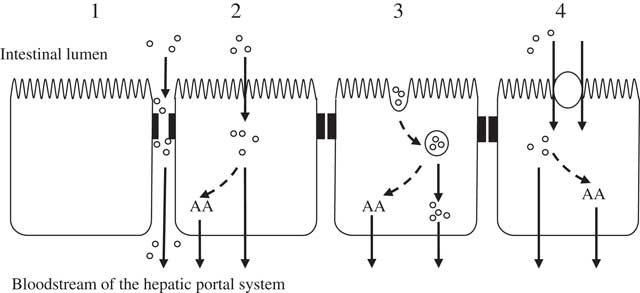
Fig. 1 Potential mechanisms of enterocytic uptake of peptides. (1) Paracellular: increased permeability of tight junctions may permit the passage of peptides. (2) Passive diffusion: cell-penetrating peptides are capable of transporting peptides as cargo. (3) Endocytosis: followed by endosomal release of the peptides. (4) Carrier-mediated transport: transport via the intestinal peptide transporter PEPT1 (H+/di- and tri-peptide symporter). Inside the enterocyte peptides can be hydrolysed to their constituent amino acids (AA) and transported across the basolateral membrane by specific AA transporters. It is thought that the transport of peptides across the basolateral membrane is mediated through other transporters such as those suggested by Terada et al. ( Reference Terada, Sawada and Saito 322 , Reference Terada and Inui 323 ), Shepherd et al. ( Reference Shepherd, Lister and Affleck 324 ) and Irie et al.( Reference Irie, Terada and Okuda 325 ).
Paracellular transport
The paracellular pathway involves structures joining adjacent intestinal epithelial cells and delineated by TJ, adherens junctions and desmosomes( Reference Menard, Cerf-Bensussan and Heyman 28 ). The rate-limiting factor in the paracellular diffusion of molecules are the TJ, a network of transmembrane proteins (claudins( Reference Furuse, Fujita and Hiiragi 37 ), occludin( Reference Furuse, Hirase and Itoh 38 ), junctional adhesion molecule A( Reference Mandell, McCall and Parkos 39 ) and tricellulin( Reference Ikenouchi, Furuse and Furuse 40 )) which control the TJ plasticity and permeability. TJ form pores that range in diameter between 0·4 and 0·9 nm to 5 and 6 nm in villi or crypts, respectively. Occludin interacts with the zonula occludens proteins (ZO-1, ZO-2) that regulate the actomyosin ring. TJ allow the diffusion of mostly cations and inert small molecules less than 600 Da such as water-soluble peptides( Reference Vermeirssen, Van Camp and Verstraete 41 ).
Increased paracellular permeability has been observed in IBD together with abnormal TJ structure and a down-regulation and redistribution of several TJ proteins or the subjacent adherens junction proteins( Reference Arrieta, Bistritz and Meddings 42 , Reference Madsen, Malfair and Gray 43 ). A variety of pathological conditions can increase paracellular permeability in which molecules of greater size can diffuse non-specifically across the intestinal epithelial layer( Reference Menard, Cerf-Bensussan and Heyman 28 ). The involvement of proinflammatory cytokines in the pathophysiology of IBD is well recognised( Reference Raddatz, Bockemuhl and Ramadori 44 , Reference Stallmach, Giese and Schmidt 45 ), and these cytokines are implicated in epithelial barrier dysfunction that leads to increased intestinal permeability along paracellular pathways( Reference Menard, Cerf-Bensussan and Heyman 28 ). Both interferon-γ (IFN-γ) and TNF-α are elevated in the mucosa of IBD patients and contribute to a proinflammatory cascade that includes barrier disruption( Reference Gassler, Rohr and Schneider 46 , Reference Kucharzik, Walsh and Chen 47 ). Bruewer et al. ( Reference Bruewer, Luegering and Kucharzik 48 ) have shown that the specificity of these mediators for the disruption of specific intercellular junctional proteins under inflammatory conditions is indicated, as the junctional proteins remained unaltered in non-inflamed areas of diseased tissue. Some of the mechanisms underlying the structural and functional modifications of TJ include the endocytosis of junctional proteins( Reference Utech, Ivanov and Samarin 49 , Reference Matsuda, Kubo and Furuse 50 ), epithelial apoptosis( Reference Bojarski, Gitter and Bendfeldt 51 – Reference Begue, Wajant and Bambou 53 ), reduced transcription of TJ proteins( Reference Mankertz, Tavalali and Schmitz 54 ) and the activation of myosin light-chain kinase phosphorylation to promote cytoskeletal contraction( Reference Zolotarevsky, Hecht and Koutsouris 55 ). Menard et al.( Reference Menard, Cerf-Bensussan and Heyman 28 ) suggest that myosin light-chain kinase is a key molecule that stimulates the opening of TJ by phosphorylating the myosin light chains. Increased claudin-2 expression increases the number of pores that allow the paracellular movement of small molecules. Myosin light-chain kinase activation and occludin down-regulation increase paracellular transport that is characteristic of both UC and CD( Reference Turner 56 ).
Probiotics are live bacteria which improve the health of the host beyond their inherent nutritional value( Reference Mennigen, Nolte and Rijcken 57 ). It has been hypothesised that probiotics have anti-inflammatory effects in human IBD and preserve intestinal epithelial integrity( Reference Gionchetti, Rizzello and Morselli 58 ). A number of in vitro studies using epithelial monolayers have demonstrated that probiotics have improved epithelial barrier function following Escherichia coli infection or incubation with proinflammatory cytokines( Reference Bai, Pak and Lee 59 – Reference Zyrek, Cichon and Helms 62 ). Several in vivo studies have demonstrated that probiotic therapy may change the expression of TJ proteins( Reference Mennigen, Nolte and Rijcken 57 , Reference Ukena, Singh and Dringenberg 63 ) and decrease paracellular permeability by increasing the phosphorylation of TJ proteins, such as ZO-1, claudin-1, or occludin in dextran-sodium sulfate-induced colitis (acute model) mice( Reference Mennigen, Nolte and Rijcken 57 , Reference Ukena, Singh and Dringenberg 63 , Reference Miyauchi, Morita and Tanabe 64 ) and IL-10–/– (chronic model) mice( Reference Chen, Yang and Zhang 65 , Reference Ewaschuk, Diaz and Meddings 66 ). However, the underlying molecular mechanisms by which probiotics diminish paracellular permeability remain unclear( Reference Chen, Yang and Zhang 65 ).
Transcellular transport
The transcellular transport of large particles, including microbes, has been ascribed to M cells located in the follicle-associated epithelium of Peyer’s patches( Reference Menard, Cerf-Bensussan and Heyman 28 ) and isolated lymphoid follicles in the distal part of the intestine( Reference Keita, Gullberg and Ericson 67 , Reference Keita, Salim and Jiang 68 ). Dendritic cells may sample bacteria in the intestinal lumen by extending dendrites between adjacent epithelial cells( Reference Rescigno, Rotta and Valzasina 69 ).
Lipid-soluble oliogopeptides may enter the enterocytes by passive diffusion where they are susceptible to hydrolytic degradation by cytosolic enzymes( Reference Vermeirssen, Van Camp and Verstraete 41 ). As large polar molecules (for example, peptide fragments >600 Da) cannot pass through the hydrophobic cell membrane of the enterocyte they may be captured by invagination of the apical membrane. Such vesicles normally fuse with lysosomes to form phagolysosomes, in which enzymic digestion of the macromolecules occurs. Only protein that escapes hydrolysis within these structures can be drawn through the enterocytes and cross the basolateral membrane.
The transcytosis of internalised vesicles may carry specifically bound ligands (receptor-mediated transcytosis), non-specifically adsorbed ligands (adsorptive transcytosis) or fluids (fluid-phase transcytosis) from the apical membrane across the cell to the basolateral membrane( Reference Shimizu, Tsunogai and Arai 70 , Reference Shen, Wan and Ekrami 71 ).
Epithelial cells of the intestinal mucosa also sample large molecules greater than 600 Da in size (such as food antigens) by endocytosis at the apical membrane and transcytosis toward the lamina propria. Within the epithelial cell, proteins/peptides are digested in acidic and lysosomal compartments before being released as amino acids or partly degraded products at the basolateral pole of the enterocytes( Reference Menard, Cerf-Bensussan and Heyman 28 ). Partially degraded food antigens in early endosomes bind to major histocompatibility complex (MHC) class II molecules in an intracellular endocytotic compartment (MIIC). Inward invagination of MIIC compartments leads to the formation of exosomes, small membrane vesicles (40–90 nm) bearing MHC class II/peptide complexes at their surface( Reference Menard, Cerf-Bensussan and Heyman 28 ). Antigen-loaded exosomes can then fuse with the basement membrane before being released into the extracellular medium to interact with local immune cells( Reference Van Niel, Mallegol and Bevilacqua 72 ). Exosome-bound peptides are much more potent than free peptides to interact with dendritic cells and stimulate peptide presentation to T cells( Reference Mallegol, Van Niel and Lebreton 73 ).
IgA is a dimeric protective mucosal immunoglobulin secreted into the intestinal lumen as secretory IgA (SIgA), and is the most representative immunoglobulin at the mucosal surface. The major role of SIgA in healthy subjects is to restrict potentially harmful food and microbial antigens from entering the intestinal epithelia( Reference Fernandez, Pedron and Tournebize 74 ). However, in some pathological conditions the abnormal apical-to-basal retrotransport of SIgA immune complexes can mediate the entry of noxious antigens into the intestinal epithelial cells( Reference Menard, Cerf-Bensussan and Heyman 28 ). In CeD, an enteropathy induced by the abnormal activation of T cells by gluten-derived gliadin peptides, SIgA allows the transcytosis of IgA–gliadin immune complexes through the intestinal barrier via the transferrin receptor CD71 at the apical surface( Reference Matysiak-Budnik, Moura and Arcos-Fajardo 75 ). In healthy individuals this receptor is confined to the basolateral membrane and gliadin peptides are taken up by non-specific endocytosis and almost exclusively degraded by the intestinal epithelia. In active CeD the retrotransport of IgA–gliadin immune complexes most likely triggers exacerbated adaptive and innate immune responses that result in mucosal lesions( Reference Menard, Cerf-Bensussan and Heyman 28 ).
Significant quantities of IgG are also secreted at the mucosal surface which suggests a protective role. The transcytosis of IgG is mediated via the neonatal Fc receptor at the surface of intestinal epithelial cells in an acidic environment. At the basolateral side of the enterocyte the neutral pH induces the dissociation of IgG immune complexes from the receptor. Although the role of the neonatal Fc receptor in humans has not been established, it has been reported to mediate passive immunity in suckling rats from the maternal milk( Reference Brambell 76 , Reference Jones and Waldmann 77 ). Bacteria as well as food antigens can be transported as IgG immune complexes via neonatal Fc receptors, a mechanism that is most likely involved in the defence against intestinal pathogens( Reference Menard, Cerf-Bensussan and Heyman 28 ), and a process that has been reported for commensal E. coli ( Reference Yoshida, Kobayashi and Kuo 78 ). However, the entry of bacteria or degraded bacterial components (for example, flagellin( Reference Kobayashi, Qiao and Yoshida 79 )) might precipitate an inappropriate immune response such as chronic inflammation( Reference Menard, Cerf-Bensussan and Heyman 28 ).
The overexpression of CD23, the low-affinity receptor for IgE, has been detected on both the apical and basolateral membranes of patients with gastrointestinal diseases such as IgE-dependent bovine milk allergy and enteropathy, autoimmune enteropathy, CD and UC( Reference Kaiserlian, Lachaux and Grosjean 80 ). Such an overexpression of CD23 at the apical surface of enterocytes can drive the transport of IgE–allergen immune complexes from the intestinal lumen (bypassing lysosomal degradation) to the lamina propria, which then triggers mast cell degranulation and the rapid onset of an allergic inflammatory response in subepithelial immunoreactive cells( Reference Menard, Cerf-Bensussan and Heyman 28 ).
Soderholm et al. ( Reference Soderholm, Peterson and Olaison 21 ) were the first to demonstrate that protein-sized macromolecules can permeate, at an increased rate, the non-inflamed ileal mucosa of patients with CD. Although they had previously demonstrated that molecular leakage can be induced by TJ dysfunction in patients with CD( Reference Soderholm, Olaison and Hedman 81 ), later studies indicated that the increased endosomal uptake of antigens was mediated by the proinflammatory cytokine TNF-α, which plays a pivotal role in CD pathogenesis. This demonstrates the importance of the transcellular route of antigen uptake in the barrier dysfunction of CD, and underlines the importance of immune–epithelial interaction in the development of mucosal inflammation( Reference Soderholm, Streutker and Yang 82 ), which suggests that anti-TNF-α therapy may produce positive effects in patients suffering from CD( Reference D’Haens 83 ).
Another cytokine abundant in CD is IFN-γ. This cytokine is also thought to enhance the transcytosis of macromolecules and was first demonstrated by Terpend et al. in 1998 using 3H-labelled horseradish peroxidase in an in vitro intestinal epithelial model. This suggests that IFN-γ may enhance both paracellular and transcellular leakage( Reference Terpend, Boisgerault and Blaton 84 ).
Antigen transcytosis into endocytic cell compartments and finally into the cytosol is strongly enhanced in rapid antigen uptake into the cytosol enterocyte (RACE) cells of patients with CD and UC. This suggests that in the inflamed intestine both paracellular and transcellular transport pathways are increased and contribute to overstimulation of the local immune system. Such overstimulation creates a vicious cycle in which luminal antigens reach the lamina propria, interact with subepithelial immunoreactive cells, and drive the secretion of permeability-enhancing factors that further increase epithelial barrier dysfunction( Reference Menard, Cerf-Bensussan and Heyman 28 ).
The microbiota
The host and intestinal microbiota are in a state of symbiotic mutualism, forming what Goodacre( Reference Goodacre 85 ) terms a human–microbe hybrid, where the human genome and the microbiome collectively define a ‘superorganism’. In the healthy human gut the composition of the microbiome is unique and consists of hundreds to thousands of bacterial species in largely four different phyla: Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria( Reference Wilson, Teft and Morse 86 ) although they are not distributed uniformly in number, species or metabolic activity. Diet and age are known to influence the composition of the gut microbiome( Reference Gill, Pop and DeBoy 87 , Reference Wu, Chen and Hoffmann 88 ). Although the intestinal tract is colonised by large numbers of commensal bacteria, intestinal infections and the translocation of bacterial proinflammatory antigens (for example, lipopolysaccharide and peptidoglycan) that can provoke NF-κB-dependent immune responses are uncommon in healthy individuals. However, the development of IBD appears to be dependent on the presence of a commensal microbiota, as mice raised in germ-free environments fail to develop IBD( Reference Sartor 89 ). While the classic interpretation of IBD pathogenesis is a loss of mucosal tolerance to proinflammatory bacteria, someone with IBD is more likely to contract an intestinal infection because of a weakened mucosal barrier, resulting from a defective immune defence system( Reference Wehkamp and Stange 25 , Reference Matsuoka and Kanai 90 ).
An excessive immune response to bacteria inhabiting the intestinal lumen and their degradation products is a common feature of IBD. Despite recent progress in defining factors that exacerbate or ameliorate these diseases, their precise causes remain poorly defined( Reference Jia, Whitehead and Griffiths 91 ). Nevertheless, there is broad agreement that luminal microbes are of particular relevance in the development of these pathologies( Reference Gersemann, Wehkamp and Stange 6 ). The composition of the microbiota is altered in IBD towards fewer anti-inflammatory and greater numbers of proinflammatory bacteria( Reference Bibiloni, Mangold and Madsen 92 – Reference Frank, Amand and Feldman 94 ). The treatment of IBD is often accompanied by substantial changes in the composition of intestinal microbiota and related immunoglobulins( Reference van der Waaij, Kroese and Visser 95 ). However, no single group of bacteria has been implicated to be uniquely causally related to these diseases( Reference Jia, Whitehead and Griffiths 91 ). It has been suggested that a toxin, such as H2O2 produced by bacteria, might play a significant role in provoking intestinal inflammation( Reference Jia, Whitehead and Griffiths 91 ). Although many studies have failed to link sulfate-reducing bacteria (SRB) and IBD( Reference Pitcher, Beatty and Cummings 96 – Reference Carmen Collado and Sanz 101 ), it has been proposed that SRB can exacerbate IBD by generating hydrogen sulfide, as well as depleting the production of beneficial butyrate( Reference Shatalin, Shatalina and Mironov 102 , Reference Marquet, Duncan and Chassard 103 ). Although the work of Jia et al.( Reference Jia, Whitehead and Griffiths 91 ) demonstrated some changes in the number of SRB in IBD patients pre- and post-treatment, their data were not unequivocal.
While there is little disagreement that patients suffering from IBD have antibodies against several microbes and microbial antigens( Reference Giaffer, Clark and Holdsworth 104 ), a number of researchers suggest that dysbiosis of the intestinal microbiome is a causal factor of inflammation in IBD patients, without reference to the genetic predisposition of IBD patients( Reference Gersemann, Wehkamp and Stange 6 , Reference Ingersoll, Ayyadurai and Charania 9 , Reference Charrier and Merlin 10 , Reference Sartor 89 , Reference Rescigno 105 , Reference Qin, Li and Raes 106 ). However, although commensal intraluminal microbiota are essential for the development and maintenance of IBD there is little evidence to support the hypothesis that intestinal microbiota are the single causal factor of the disease. Although the defence against these microbes is compromised in IBD it may be reasoned that this is due to a defective innate barrier where the production of α-defensins is reduced in ileal CD, that β-defensins are decreased in colonic CD, and the mucus layer is deficient in UC( Reference Gersemann, Wehkamp and Stange 6 ).
There is little evidence that the numbers of bacteria in the large intestine of human subjects with IBD are any different from those of normal subjects. However, a disequilibrium of inflammatory and non-inflammatory components of the intestinal microbiota (dysbioses) is associated with IBD( Reference Frank, Amand and Feldman 94 , Reference Qin, Li and Raes 106 – Reference Sokol, Lay and Seksik 111 ). Whether the changes in intestinal microbiota observed in IBD are simply a consequence of chronic inflammation and its treatment, or are necessary determinants of initiation and/or perpetuation of pathogenesis, is still open to question( Reference Frank, Zhu and Sartor 18 ). Microbiological investigations have so far failed to identify consistent alterations of microbiota composition in IBD patients relative to healthy controls( Reference Sokol, Lay and Seksik 111 ). While the commensal microbiota are largely tolerated by the mucosa and ignored by the systemic immune systems of normal hosts( Reference Feng and Elson 112 – Reference Slack, Hapfelmeier and Stecher 114 ), they are essential drivers of pathogenic mucosal and systemic inflammatory responses in genetically predisposed subjects( Reference Xavier and Podolsky 8 , Reference Sartor 89 , Reference Sartor 115 , Reference Abraham and Cho 116 ).
Such dysbiosis, widely reported in connection with CD( Reference Frank, Amand and Feldman 94 , Reference Qin, Li and Raes 106 – Reference Sokol, Lay and Seksik 111 ), is not solely the result of environmental effects, such as treatment history or diet. Complex interactions exist between the host genotype and the enteral microbial community( Reference Frank, Zhu and Sartor 18 ). Such interactions may arise as a consequence of direct genetic effects on microbial composition, perhaps through altered Paneth cell function( Reference Cadwell, Patel and Maloney 5 , Reference Cadwell, Liu and Brown 117 , Reference Wehkamp, Salzman and Porter 118 ), or as a direct result of the pathogenic process. However, the question of whether dysbiosis contributes to CD pathogenesis or is an innocuous by-product remains to be established. Furthermore, how dysfunction or inflammation of the mucosal barrier can lead to dysbiosis is also unclear. However, other than in exceptional cases, it is unlikely that disease-associated dysbioses will satisfy all of the criteria that have been proposed to prove causality( Reference Hill 119 – Reference Fredricks and Relman 121 ).
In a study of forty twin pairs focusing on disease incidence in genetically matched individuals that were concordant or discordant for either CD or UC, it was demonstrated that the microbiota of CD patients differed from those of healthy individuals, whereas the microbiota of patients with UC was similar to healthy controls( Reference Willing, Dicksved and Halfvarson 107 ). Also the microbial profiles of patients with CD, predominantly affecting the ileum, were different to those where the disease affected the colon. Although other studies have shown that the microbial profile of IBD patients differs in inflamed and non-inflamed states( Reference Sepehri, Kotlowski and Bernstein 122 – Reference Walker, Sanderson and Churcher 124 ), the faecal microbiota cannot be differentiated between patients with the active disease and those in remission( Reference Willing, Dicksved and Halfvarson 107 ). In terms of microbial profile the disease phenotype was a more significant factor than genotype. Although significant differences in the microbial profile between inflamed and non-inflamed mucosal biopsy sites have been demonstrated, such differences varied so greatly between individuals that no obvious bacterial signature could be positively associated with the inflamed intestine( Reference Walker, Sanderson and Churcher 124 ). Although a dysbiosis is observed in IBD patients, relative to healthy controls, it may result from a disturbed intestinal environment rather than be the direct cause of disease. The complex mutualistic interaction between the microbiota and the host suggests that the relationship is bi-directional( Reference Quigley 125 ) and that any observed inflammatory changes may be secondary to, rather than causative of, the disease process; this is a factor that must be considered in future studies( Reference Sartor 126 ).
It has previously been reported that a reduced abundance of Faecalibacterium prausnitzii, a bacterium thought to exert anti-inflammatory effects( Reference Sokol, Pigneur and Watterlot 127 ), is common in ileal CD patients( Reference Willing, Halfvarson and Dicksved 108 , Reference Swidsinski, Loening-Baucke and Vaneechoutte 128 ). However, F. prausnitzii only represents one of many core members of the microbiota that are less abundant in this disease phenotype. This has led to the suggestion that the oral replenishment of Roseburia, Alistipes, Collinsella and members of the Ruminococcaceae family may be beneficial( Reference Willing, Dicksved and Halfvarson 107 ).
In humans there are at least eighteen mucin-type glycoproteins; however, mucin 2 (MUC2) is the predominant component of the mucin layer in both the small and large intestines( Reference Maldonado-Contreras and McCormick 129 ). When comparing the thinning of the mucus layer in patients suffering from IBD with healthy controls there is evidence that Streptococcus is associated with CD (80% of all bacteria) and that Lactobacillus is associated with UC (90% of all bacteria)( Reference Fyderek, Strus and Kowalska-Duplaga 130 , Reference Corfield 131 ). In a study by Joossens et al. ( Reference Joossens, Huys and Cnockaert 132 ) a faecal microbiota dysbiosis was identified in patients with CD that was characterised by a decreased presence of butyrate-producing bacteria in conjunction with mucin degradation; although the patients’ relatives possessed similarly enhanced mucin degradation they did not share the depleted butyrate-producing bacteria.
As the mucosal barrier is the primary defence of the host against intestinal bacteria the shift from normobiosis observed in relatives of CD patients to the dysbiosis seen in CD patients might be an intermediate step towards CD and disease-associated dysbioses( Reference Joossens, Huys and Cnockaert 132 ). Although Joossens et al. ( Reference Joossens, Huys and Cnockaert 132 ) did not study the overall butyrate-producing or mucin degradation capacity of the microbiota in this cohort, there was a functional overlap between dysbiosis in patients with CD and their unaffected relatives at risk. Quigley emphasises the importance of this study suggesting that it indicates a role of the microbiota in CD that is independent of the genetic background and diet, features that the CD patients would have shared with their unaffected relatives( Reference Quigley 125 ). However, Quigley fails to mention the finding of Joossens et al.( Reference Joossens, Huys and Cnockaert 132 ) that the CD patients exhibited mucin degradation, a fact that weakens the argument that the microbiota in CD are independent of the genetic background and diet.
The immune system
Epithelial innate immunity includes pattern recognition receptors on the intestinal surfaces such as Toll-like receptors and nucleotide-binding oligomerisation domain (NOD)-containing molecules. Recognition of bacteria by the vertebrate innate immune systems relies upon the detection of invariant molecules by specialised receptors. The view is now emerging that activation of both Toll-like receptors and NOD by different bacterial agonists is important in an inflammatory response( Reference Kufer and Sansonetti 133 ). It appears that NOD1 and NOD2 receptors detect the peptidoglycan components of bacterial cell walls, and the nucleotide-binding domain, leucine-rich (NLR) proteins ipaf and Naip detect bacterial flagellin( Reference Wilmanski, Petnicki-Ocwieja and Kobayashi 134 , Reference Girardin, Travassos and Herve 135 ). NOD1 specifically senses diaminopimelic acid-containing muramyl peptides( Reference Girardin, Boneca and Carneiro 136 , Reference Chamaillard, Hashimoto and Horie 137 ) and NOD2 detects muramyl dipeptide (MDP)( Reference Girardin, Boneca and Viala 138 , Reference Inohara, Ogura and Fontalba 139 ), a motif found in almost all bacteria. Nod1 and Nod2 mutations have been associated with IBD in human subjects( Reference Le Bourhis, Benko and Girardin 140 ), with Nod2 being identified as the first susceptibility gene for CD( Reference Hugot, Chamaillard and Zouali 141 , Reference Ogura, Bonen and Inohara 142 ). It is hypothesised that any impairment to NOD2 function in innate immune responses to bacterial peptides, such as a dysfunctional PepT1 peptide transporter, may lead to defective sensing of bacterial proinflammatory peptides, abnormal bacterial survival and chronic inflammation of the intestinal mucosa( Reference Gruber, Lichti and Rath 7 ). The question of whether a defective PepT1 mechanism is a causative factor for the disease pathogenesis or severity of the IBD has yet to be determined( Reference Gruber, Lichti and Rath 7 ).
Constitutive and inducible antimicrobial peptides such as defensins and cathelicidins interact with secreted mucins and play an important role in intestinal defence( Reference Wehkamp, Schmid and Stange 143 , Reference Bevins, Martin-Porter and Ganz 144 ). Antimicrobial peptides, predominantly the defensins in mammals, possess a broad spectrum of antimicrobial activity. Antimicrobial peptides are hydrophobic peptides possessing positively charged domains that can interact with and disrupt cell membranes causing cell lysis, which leads to the efflux of ions and nutrients. In the small intestine, the most abundant constitutive defensins are the α-defensins HD5 and HD6 found in the Paneth cells. Following stimulation of the pattern recognition receptors by bacterial products (for example, lipopolysaccharide, which activates Toll-like receptor-4, and MDP, which activates NOD2)( Reference Ayabe, Satchell and Wilson 145 ), defensins are released into the lumen. In comparison with vertebrate cell membranes, bacterial cells possess high concentrations of negatively charged phospholipids, which defensins selectively and preferentially bind to( Reference Khan and Islam 146 ). Together with antimicrobial lysozymes, Paneth cells are key to intestinal defence.
Most dietary protein is completely digested and absorbed as amino acids, dipeptides or tripeptides. However, some proteins are resistant to both the acidic pH of the stomach and enzyme proteolysis( Reference Astwood, Leach and Fuchs 147 ), such that large immunogenic peptides or intact proteins may reach the small intestine( Reference Mahé, Messing and Thuillier 148 ). For example, β-lactoglobulin, a major bovine milk allergen, and gluten/gliadins, a major factor underlying CeD, are both partially resistant to digestive enzymes( Reference Menard, Cerf-Bensussan and Heyman 28 ). The ineffective digestion of gliadins which are high in proline content produce large irreducible immunogenic peptides that may activate the lamina propria CD4+ T cells in coeliac patients( Reference Shan, Molberg and Parrot 149 , Reference Shan, Qiao and Arentz-Hansen 150 ). The risk of developing food allergies from incomplete protein digestion has been reported in mice given antiulcer medication known to impair protein digestion where even low doses of ovalbumin resulted in the development of IgE-mediated food allergy( Reference Diesner, Knittelfelder and Krishnamurthy 151 ). Such observations indicate that immunogenic proteins and peptides present in the lumen may serve as potential candidates for intestinal absorption and immune stimulation( Reference Menard, Cerf-Bensussan and Heyman 28 ).
Intestinal alkaline phosphatase (IAP) is expressed throughout the gastrointestinal tract and has an essential role in intestinal homeostasis through interactions with the microbiota resident in the gut( Reference Estaki, DeCoffe and Gibson 152 ). IAP appears to have four major interactions in the gut: (a) the dephosphorylation of toxic pro-inflammatory microbial ligands such the lipopolysaccharides, components of the cell wall of Gram-negative bacteria, the presence of which in the blood can stimulate a strong inflammatory response; (b) the regulation of bicarbonate secretion and increasing the pH distal to the stomach; (c) modulation of long-chain fatty acid absorption; and (d) the regulation of the microbial ecosystem within the gut by forming a complex relationship between microbiota, diet and the intestinal mucosal surface, and the translocation of microbes across the gut wall( Reference Estaki, DeCoffe and Gibson 152 , Reference Lalles 153 ). IAP also dephosphorylates other pro-inflammatory ligands released from damaged cells such as extracellular nucleotides, for example, ATP. The detoxification of such ligands is essential in the prevention of inflammatory conditions such as IBD. IAP is reported as having a protective effect by ameliorating inflammation from increased permeability of the intestinal endothelia that results from vascular endothelial growth factor affecting the pericellular TJ. Such abnormal intestinal permeability can be partially reduced by IAP down-regulating vascular endothelial growth factor expression and regulating specific TJ proteins for example, claudin-2( Reference Wang, Chen and Zhu 154 ). Compared with normal subjects, epithelial IAP mRNA expression is reduced in patients with UC and CD( Reference Martínez-Augustin, López-Posadas and González 155 ) and is especially marked in severe cases of CeD( Reference Molnar, Vannay and Sziksz 156 ). Animal studies have shown that orally administered IAP may reduce inflammation by down-regulating the immune response, specifically reducing pro-inflammatory cytokines, for example, TNF-α( Reference Estaki, DeCoffe and Gibson 152 , Reference Tuin, Poelstra and de Jager-Krikken 157 ). In an uncontrolled trial by Lukas et al.( Reference Lukas, Drastich and Konecny 158 ), IAP was administered intraduodenally daily over a period of 7 d to patients with UC and was associated with short-term improvement in disease activity scores and clinical effects. The gastrointestinal administration of IAP appears to ameliorate both gut inflammation and favours intestinal tissue regeneration, whereas enteral and systemic administration of IAP attenuates systemic inflammation only( Reference Lalles 153 ).
Genetic disruption
IBD is a multigenic disease where an increased number of inherited risk alleles is associated with an increased risk of developing the disease, earlier onset and greater severity (for example, fibrostenotic or fistulising symptoms and/or the necessity for surgical intervention)( Reference Weersma, Stokkers and van Bodegraven 159 , Reference Ellinghaus, Bethune and Petersen 160 ). However, genetic susceptibility does not necessitate the development of the disease, as studies with monozygous twins discordant for IBD suggest that diverse environmental interactions (for example, diet and gastrointestinal tract microbiome) also play a role in the development of this disease( Reference Willing, Dicksved and Halfvarson 107 , Reference Willing, Halfvarson and Dicksved 108 , Reference Qin 161 ). Using ordinal regression analysis, Weersma et al. ( Reference Weersma, Stokkers and van Bodegraven 159 ) reported that individuals with six CD-associated risk alleles (OR 7·56) were unlikely to develop the condition whereas individuals with seven risk alleles (OR 25·6) were much more likely to develop CD. The genetic associations with IBD have been extensively reviewed by a number of researchers( Reference Ferguson 162 – Reference Boirivant and Cossu 169 ). Genome-wide association studies have resulted in the rapid discovery of susceptibility genes with over 163 IBD genes associated with these diseases( Reference Satokari 170 – Reference Thompson and Lees 172 ). Model-selection analysis has indicated that 110 of the 163 susceptibility loci are associated with both CD and UC, with thirty being specific to CD and twenty-three specific to UC( Reference Jostins, Ripke and Weersma 171 ). More recently thirty-six loci specific to CD( Reference Rivas, Beaudoin and Gardet 173 ) and 100 specific to UC have been reported( Reference Thompson and Lees 172 ). These loci encode genes which are involved in a number of homeostatic systems, the disruption of which provide a mechanistic description of IBD. Dysfunctional genes that are implicated in epithelial barrier function, bacterial recognition and adaptive immune response are given Table 1.
Table 1 Dysfunctional genes implicated in epithelial barrier function, bacterial recognition and adaptive immune responseFootnote *

IBD5, inflammatory bowel disease 5; DLG5, drosophila discs large homolog; ITLN1, intelectin-1; XBP1, X-box binding protein 1; MDR1, multidrug resistance protein 1; MALB1, Mal region B mutants; CDH1, cadherin 1; HNF4A, hepatocyte nuclear factor 4α; NOD1 and 2, nucleotide-binding oligomerisation domain; CARD9, caspase recruitment domain-containing protein 9; TLR4, Toll-like receptor 4; ATG16L1, autophagy-related protein 16-1; IRGM, immunity-related GTPase family M protein; LRRK2, leucine-rich repeat kinase 2; IL23R, IL23 receptor; JAK2, Janus kinase 2; STAT3, signal transducer and activator of transcription 3; HLA, human leucocyte antigen; MST1, macrophage stimulating 1; PTPN2, protein tyrosine phosphatase, non-receptor type 2.
* An additional sixteen genes and loci are implicated in inflammatory bowel disease after Graham & Xavier( Reference Graham and Xavier 164 ): RNF186 (ring finger protein 186); SP110 (nuclear body protein 110); SP140 (nuclear body protein 140); MST1 (macrophage stimulating 1 (hepatocyte growth factor-like)); FUT2 (fucosyltransferase); SLC22A4 (solute carrier family); GSDMB (gasdermin B); ORMDL3 (orosomucoid like 3); TNFAIP3 (TNFα-induced protein 3); SLC6A7 (solute carrier family); IL10RA (IL10 receptor α); IL18RAP (IL18 receptor accessory protein); MUC19 (mucin 19); CUL2 (cullin 2); PTPN22 (protein tyrosine phosphatase, non-receptor type 22); C1orf106 (chromosome 1 open reading frame 106).
Advanced genomic techniques have identified other loci and polymorphisms that are associated with IBD and highlight other cellular pathways that may contribute to the onset or progression of the disease( Reference Khor, Gardet and Xavier 174 ). A further sixteen genes and loci implicated in IBD suggest connections between cellular metabolism, inflammation and mucosal microbial communities (see notes for Table 1)( Reference Graham and Xavier 164 ). A comprehensive review of SNP with either susceptibility or protective effects in IBD has recently been published( Reference Neuman and Nanau 165 ). It appears there is significant overlap in genes associated with autoimmune and autoinflammatory diseases that indicate common immunological mechanisms and unique disease-specific pathways which lead to the complex pathophysiology of IBD( Reference Graham and Xavier 164 ). A number of the susceptibility genes identified with IBD (for example, ATG16L1) are common variants with high prevalence in the healthy population( Reference Weersma, Stokkers and van Bodegraven 159 ). Indeed many of the SNP implicated in IBD by genome-wide association studies are not independently causative of the disease phenotype; they exist as linked disequilibrium with as yet to be discovered variants that are functional( Reference Graham and Xavier 164 ). In the future, genetic screening for IBD-related SNP, combined with an assessment of the intestinal microbiome and other environmental factors (for example, diet), might allow clinicians to identify patients at risk of IBD and improve differential diagnosis and optimise treatment efficacy of the disease( Reference Neuman and Nanau 165 , Reference Viennois, Baker and Xiao 175 ).
Interestingly Hu et al.( Reference Hu, Smith and Ma 176 ) generated mice in which the PepT1 gene was disrupted by the insertion of a lacZ reporter gene under the control of the endogenous PepT1 promoter. Although the Pept1-null mice lacked expression of PepT1 protein in the intestine and kidney tissues in which this peptide transporter is normally expressed, the Pept1-deficient mice were found to be viable, fertile, grew to normal size and weight, and were without any obvious abnormalities( Reference Hu, Smith and Ma 176 ).
Other environmental factors
With the sudden emergence and dramatic increase in IBD during the last century( Reference Russel 177 ), a variety of environmental factors has been implicated with the onset of IBD, including: food storage in refrigerators, smoking, the use of non-steroidal anti-inflammatory drugs, and infections( Reference Steel, Nussberger and Romero 178 , Reference Boll, Markovich and Weber 179 ). Diet and food additives have long been suspected as major factors in IBD pathogenesis( Reference Gruber, Lichti and Rath 7 ) and in this context two new fields of study have emerged: nutrigenetics, which recognises the effect of genetic variation on nutrient requirements, and nutrigenomics, which describes the impact of nutrient regulation of gene expression( Reference Ferguson 162 ). Although Qin( Reference Qin 161 ) suggests a multitude of possible dietary factors affecting IBD, he singles out saccharin and/or sucralose to be a key causative factor in the disease. Proposing a unified hypothesis regarding the aetiology for IBD, Qin suggests that saccharin inhibits both the activity of β-glucuronidase itself as well as the growth of β-glucuronidase-positive bacteria in the gastrointestinal tract which are necessary for the deconjugation of biliary bilirubin( Reference Qin 180 ), that in turn leads to damage of the protective mucus layer and the underlying gut tissue by the poorly inactivated digestive proteases( Reference Qin 181 ) (the bacteria–protease–mucus–barrier hypothesis)( Reference Qin 161 ).
Peptide permeability
Peptide transporters
The principal transporter for the absorption of di- and tripeptides arising from the digestion of both exogenous and endogenous proteins in the intestinal lumen is the high-capacity, low-affinity PepT1 protein( Reference Smith, Clemencon and Hediger 26 ). Peptide transporters are integral membrane proteins that mediate the cellular uptake of di- and tripeptides. In vertebrates there are two peptide transporter proteins: PepT1 expressed predominantly in brush-border membranes of the small intestine and PepT2 in the kidney and lung. Although PepT1 is highly expressed in the small intestine( Reference Ogihara, Saito and Shin 182 ) there is little or no expression in the healthy colon( Reference Ismair, Vavricka and Kullak-Ublick 183 ). These transport proteins operate as electrogenic proton/peptide symporters with a broad substrate specificity, possibly transporting 400 dipeptides and 8000 tripeptides composed of l-α amino acids( Reference Weitz, Harder and Casagrande 184 ), but not free amino acids or peptides with more than three amino acid residues( Reference Daniel 185 , Reference Bailey, Boyd and Collier 186 ). The electrochemical gradient across the apical enterocyte membrane is dependent upon the Na-proton exchanger NHE3 and allows the absorption of di- and tripeptides against a concentration gradient, enabling higher intracellular than extracellular peptide concentrations( Reference Spanier 187 , Reference Watanabe, Kato and Ito 188 ).
Transport is enantio-selective and involves a variable proton-to-substrate stoichiometry for the uptake of neutral and mono- or polyvalently charged peptides. The peptide transporter proteins can also transport many therapeutic drugs (for example, β-lactam antibiotics, selected angiotensin-converting enzyme inhibitors, and peptidase inhibitors) and thereby determine their bioavailability and pharmacokinetics( Reference Weitz, Harder and Casagrande 184 ). In addition, PepT1 has an important role in the innate immune response to bacteria by mediating the transepithelial transport of bacterial antigens( Reference Chen, Yang and Zhang 65 ). Microbial peptides imported by PepT1, for example MDP, induce NOD2-dependent activation of the NF-κB pathway( Reference Gruber, Lichti and Rath 7 ) with submucosal macrophages that in turn release proinflammatory cytokines, for example, IL-8 and the monocyte chemoattractant protein-1 (MCP-1)( Reference Vavricka, Musch and Fujiya 189 , Reference Zucchelli, Torkvist and Bresso 190 ). PepT1 polymorphisms in the SLC15A1 gene have been associated with IBD( Reference Strober, Asano and Fuss 191 ) and NOD2 polymorphisms with CD( Reference Zucchelli, Torkvist and Bresso 190 ). The clinical relevance of intestinal uptake in disease has recently been reviewed by Freeman( Reference Freeman 192 ).
Despite the comprehensive analysis of the structure and functions of PepT1, with many hundreds of publications over the last 40 years, its overall importance in amino acid absorption from the gastrointestinal tract is still largely unknown( Reference Nassl, Rubio-Aliaga and Fenselau 193 ). Using mice lacking PepT1 (Pept1 –/–) the extent that PepT1 deletion is compensated for by changes in expression and function of the amino acid transporters in intestinal epithelial cells and the role of the transporter in amino acid absorption and metabolism have been characterised( Reference Nassl, Rubio-Aliaga and Fenselau 193 ). The intragastric administration of 15N-labelled proteins and the concomitant analysis of plasma and tissue amino acid levels have indicated that the role of PepT1 in the overall intestinal amino acid absorption is negligible when low amounts of protein are ingested. However, under conditions of a high protein load reaching the intestine, the maximum rate of hydrolysis in the lumen or at the brush-border membrane may be reached, leading to di- and tripeptides becoming available for PepT1 transport( Reference Nassl, Rubio-Aliaga and Fenselau 193 ). Nassl et al.( Reference Nassl, Rubio-Aliaga and Fenselau 193 ) suggested that when a high-protein diet is administered to Pept1 –/– mice it may induce changes in body amino acid homeostasis that resemble a state of amino acid imbalance, with amino acids that are related to the urea cycle being over-represented. This is suggestive of an altered hepatic detoxification capacity in animals deficient in PepT1.
Not all researchers share the same view; for example, the enteropathogenic E. coli (EPEC), a food-borne pathogen implicated in the pathophysiology of infantile diarrhoea( Reference Nataro and Kaper 194 ), may also induce PepT1 expression in colonocytes( Reference Nguyen, Dalmasso and Powell 195 ). Nguyen et al.( Reference Nguyen, Dalmasso and Powell 195 ) demonstrated that: EPEC transcriptionally induces functional PepT1 expression in the lipid rafts (LR) of colonocytes; that it induces PepT1 expression by intimately attaching to host cell membranes through LR; that the transcription factor Cdx2 is crucial for EPEC-induced PepT1 expression; and that PepT1 which are associated with LR have a role in bacterial–epithelial interaction and bacteria-induced intestinal inflammation. It is proposed that EPEC is a causal factor of human colonic PepT1 expression, activating signalling molecules within the LRs, resulting from changes in conformation and/or composition of LR, and consequently reducing the binding affinity of EPEC for LR. PepT1 appears to attenuate EPEC-triggered proinflammatory responses in intestinal epithelial cells, and therefore colonic PepT1 expression might be a host protective mechanism that modulates bacterial–epithelial interaction and inflammatory responses to pathogens( Reference Nguyen, Dalmasso and Powell 195 ), a finding that is in line with that of other researchers( Reference Smith, Clemencon and Hediger 26 , Reference Merlin, Steel and Gewirtz 196 – Reference Foster and Zheng 199 ).
Apically expressed colonic PepT1 may be a host defence mechanism via its ability to modulate bacterial–epithelial interactions and colonic inflammation( Reference Smith, Clemencon and Hediger 26 ). Colonic PepT1 expressed in IBD may absorb small proinflammatory peptides derived from bacterial peptidoglycans (for example, N-formylmethionylleucyl-phenylalanine (fMLP)( Reference Merlin, Steel and Gewirtz 196 ), MDP( Reference Vavricka, Musch and Chang 197 ) and l-Ala-γ-d-Glu-meso-diaminopimelic acid (Tri-DAP)( Reference Dalmasso, Hang and Ingersoll 198 )) that interact with NOD-like receptors and determine the activation level of inflammatory pathways such as the NF-κB and MAPK( Reference Dalmasso, Hang and Ingersoll 198 ). These pathways lead to proinflammatory cytokine/chemokine production and the subsequent migration of neutrophils into regions of inflammation and bacterial infection( Reference Smith, Clemencon and Hediger 26 , Reference Foster and Zheng 199 ).
Dalmasso et al. ( Reference Dalmasso, Nguyen and Charrier-Hisamuddin 11 ) used Tri-DAP to induce inflammation in human colonic HT29-Cl.19A cells. Similar to fMLP and MDP, Tri-DAP is a natural peptide released during peptidoglycan degradation of Gram-negative bacteria, a bacterial tripeptide that may pass through the PepT1 transporter. Although it is still unclear if the peptide induces PepT1 expression in colonocytes, this suggests that bacterial products might induce or regulate colonic PepT1 expression( Reference Dalmasso, Yan and Charrier 200 ), and that once PepT1 is expressed in the colon in IBD, then PepT1 could then be involved in the transport of bacterial peptides that aggravate inflammation. However, the studies of Dalmasso et al. ( Reference Dalmasso, Nguyen and Charrier-Hisamuddin 11 ) did indicate that colonic epithelia only respond to peptidoglycan motifs such as Tri-DAP when such products are present in the cytosol and most importantly that colonocytes fail to transport Tri-DAP or are inert to Tri-DAP under normal physiological conditions when PepT1 is not expressed in the colon.
Although the peptide/histidine transporters PhT1 and PhT2 have been found in the villous epithelia of the human small intestine( Reference Bhardwaj, Herrera-Ruiz and Eltoukhy 201 ), their relevance in the absorption of peptides and peptidomimetics has not been established( Reference Smith, Clemencon and Hediger 26 ). However, it appears that neither of these transporters is involved in the absorption of the proinflammatory peptide fMLP( Reference Wu and Smith 202 ).
The multidrug resistance 1 gene (MDR1), which encodes for the membrane-bound efflux transporter P-glycoprotein 170 (P-gp), has been associated with IBD and thought to protect the intestinal epithelia from the uptake of endogenous and exogenous toxins by transporting drugs and xenobiotics into the lumen( Reference Gruber, Lichti and Rath 7 , Reference Tsuji, Nakashima and Deguchi 203 ). Although several SNP of this gene have been reported, its relevance to the pathogenesis of IBD varies across ethnic groups( Reference Walker, Thwaites and Simmons 204 – Reference Potocnik, Ferkolj and Glavac 207 ).
Epithelial barrier dysfunction, peptides and Crohn’s disease
The studies of Cadwell et al. ( Reference Cadwell, Patel and Maloney 5 ) have demonstrated that a common enteric viral pathogen, norovirus, can induce a mutation in the CD susceptibility gene Atg16L1, producing intestinal pathologies in mice. These pathologies, activated by virus-plus-susceptibility gene interaction that mimic aspects of CD, were dependent on IFN-γ and TNF-α, and were preventable by treatment with broad-spectrum antibiotics( Reference Cadwell, Patel and Maloney 5 ). Sabbah et al.( Reference Sabbah, Chang and Harnack 208 ) have demonstrated that NOD2 can also function as a cytoplasmic viral pattern recognition receptor that can sense viral single-stranded RNA and activate IFN production. As a result it has been suggested that the CD-associated gene NOD2 may recognise viral RNA in addition to bacterial peptidoglycan and raises the possibility that a viral infection can interact with CD susceptibility genes( Reference Cadwell, Patel and Maloney 5 ). Both Garrett et al. ( Reference Garrett, Gallini and Yatsunenko 209 ) and Cadwell et al. ( Reference Cadwell, Patel and Maloney 5 ) give clear insight into the complex interaction between gene and pathogen, which individually may display only poor association with disease incidence and severity. In animal models with induced pathology, reproducing the full disease may require combinations of specific alleles of multiple genes with certain environmental agents. Not all patients with CD present with identical symptoms or pathologies; the disease varies with time and also with age, sex, ethnicity, temporal trends and geographical distributions( Reference Russel 177 , Reference Molodecky, Soon and Rabi 210 ). Some therapeutic interventions may alleviate symptoms of one patient but not in others( Reference Cadwell, Patel and Maloney 5 ). Such complex IBD diseases may represent a combinatorial confluence of pathological responses, each with overlapping but non-identical genetic and environmental causes and therefore require different therapeutic responses( Reference Cadwell, Patel and Maloney 5 ).
In CD, a transcription factor-4-mediated defect in Paneth cell differentiation has been linked to a specific absence of the α-defensins, especially in patients with Nod2 mutations( Reference Gersemann, Wehkamp and Stange 6 ). Consequently, the deficient mucosal barrier allows luminal microbes to invade the mucosa and trigger a secondary inflammatory response( Reference Wehkamp and Stange 25 ).
The up-regulated expression of PepT1 in patients with IBD( Reference Merlin, Si-Tahar and Sitaraman 211 ) and NOD2 mutations associated with CD( Reference Girardin, Boneca and Viala 138 ) may result from defective sensing of bacterial peptidoglycan-derived peptides such as MDP (a NOD2 agonist)( Reference Ismair, Vavricka and Kullak-Ublick 183 ). As a result of NOD2 mutations, which result in a NOD2 deficiency, a loss of microbial surveillance and the unmonitored import of microbial proinflammatory peptides may be caused that contribute to the onset of CD.
The expression of PepT1 in the colon of patients with UC or CD may be a response to the absorption of chemotactic bacterial di- and tripeptides that cause an aggravated inflammation/immune response( Reference Charrier and Merlin 10 , Reference Smith, Clemencon and Hediger 26 ). The expression of PepT1 in human colonocytes has also been linked to leptin, an adipocyte-secreted hormone. High concentrations of leptin were found in inflamed colonic mucosa which in turn triggered the colonic expression of PepT1 via the cAMP response element-binding (CREB) and Cdx2 transcription factors. Such increased expression of colonic PepT1 may thus enhance the uptake of the small bacterial di- and tripeptides that perpetuate intestinal inflammation. Such findings may provide important new insights into the mechanisms of intestinal inflammation and its treatment( Reference Nduati, Yan and Dalmasso 212 ).
Although the mechanism of colonic PepT1 expression in IBD remains unknown, it has been suggested that its expression is most probably induced at a transcriptional level, where specific transcriptional regulation by signalling pathway(s) may be activated( Reference Nguyen, Dalmasso and Powell 195 , Reference Merlin, Si-Tahar and Sitaraman 211 , Reference Ziegler, Fernández-Estívariz and Gu 213 , Reference Shimakura, Terada and Shimada 214 ). However, Vavricka et al.( Reference Vavricka, Musch and Fujiya 189 ) demonstrated both in vitro (in human colonic Caco2/bbe monolayers) and in vivo (in mouse intestine), that TNF-α and IFN-γ increased the activity and the total and apical membrane protein expression of PepT1 protein in a concentration- and time-dependent fashion. As no changes in PepT1 mRNA were observed, it may be concluded that the increased PepT1 activity and expression were post-transcriptionally regulated( Reference Vavricka, Musch and Fujiya 189 ). Current research into the expression of PepT1 transporters in the colon of patients with some form of IBD suggests that the presence of PepT1 in the colon is due to the bacterial load of the colon being higher than that of the ileum, and that such a high bacterial load creates a concomitantly high luminal concentration of bacterial peptides, which PepT1 transporters absorb, thus stimulating an exaggerated proinflammatory immune response.
It has also been suggested that PepT1 expression is normally restricted to the small intestine because the concentrations of bacterial di- and tripeptides are much lower in the small intestine than in the colon and that the human small intestine contains only low numbers of prokaryotes( Reference Ingersoll, Ayyadurai and Charania 9 ). However, this statement can be challenged in light of the evidence that the numbers of bacteria present in the distal ileum may be as high as 109/ml of digesta( Reference Metges 215 ). Although less than the densities reported by Whitman et al. for the large intestine (1011–1012/ml digesta( Reference Whitman, Coleman and Wiebe 216 )), they are in sufficient numbers that the presence of substantial quantities of bacterial di- and tripeptides in the ileum cannot be discounted, and are not evidential as to the lack of inflammation in the healthy human small intestine. Ingersol et al. ( Reference Ingersoll, Ayyadurai and Charania 9 ) suggest that the presence of colonic PepT1 transporters is solely due to the higher numbers of bacteria in the large bowel as the profile of PepT1 expression along the normal human digestive tract is such that bacterial peptides have little access to PepT1 and minimises the intracellular uptake of bacterial peptides. As PepT1 expression is altered in patients with IBD and commensal bacteria colonising the human colon produce significant amounts of proinflammatory di- and tripeptides, the transport of the peptides by PepT1 may lead to an increased intracellular accumulation of prokaryotic peptides that trigger downstream proinflammatory effects( Reference Ingersoll, Ayyadurai and Charania 9 ).
Epithelial barrier dysfunction, peptides and ulcerative colitis
In experiments with conventionally raised T-bet –/– ×Rag2 –/– knockout mice lacking an adaptive immune system, the loss of the transcription factor T-bet results in a spontaneous and highly penetrant colitis that shares histological features with UC in humans( Reference Garrett, Lord and Punit 217 ). T-bet –/– × Rag2 –/– UC (TRUC) is associated with altered colonic barrier function, elevated TNF-α levels and dysfunctional dendritic cells. Both the T-bet-deficient genetic background and the microbiota are required for disease initiation( Reference Garrett, Lord and Punit 217 ). Once the disease is established, the microbiota from the afflicted mice are vertically transmissible and cause intestinal inflammation in wild-type mice. TRUC is transmissible to wild-type hosts when they are cross-fostered or co-housed with TRUC mice. In a later paper, Garrett et al. ( Reference Garrett, Gallini and Yatsunenko 209 ) demonstrated that the presence of Proteus mirabilis and Klebsiella pneumoniae contributes to disease pathogenesis of colitis in TRUC mice and that TRUC-derived strains, in conjunction with an endogenous microbial community, incite colitis in wild-type mice( Reference Garrett, Gallini and Yatsunenko 209 ). Their results may provide mechanistic insights about how intestinal microbial communities, working in concert with specific colitogenic agents, contribute to the initiation and perpetuation of IBD in susceptible human hosts, and provide the foundation for proof-of-concept tests of preventative or therapeutic measures( Reference Garrett, Gallini and Yatsunenko 209 ).
Dietary protein is, to the host, foreign protein that may contain peptides that would trigger an immune response if exposed to the immune system in the enterocyte/lamina propria/bloodstream. The bacterial di- and tripeptides fMLP and MDP have been found to elicit an immune response; therefore it is probably safe to assume that there are other hydrolysis-resistant dietary peptides which could also elicit a similar response. Therefore any leakage of the apical/basolateral membranes would also elicit a similar response that results in inflammation. A high intake of dairy products or low dietary fibre intake has been reported to be associated with the relapse of patients with UC( Reference Davies and Rhodes 218 – Reference Samuelsson, Ekbom and Zack 221 ). However, more recently Jowett et al. ( Reference Jowett, Seal and Phillips 14 ) detected no association between the intake of milk or dairy products and relapse of UC, neither did they find any protective effect from increased dietary fibre. However, they did find that the consumption of meat (particularly red meat and processed meat), protein, and alcohol were linked to increased relapse in patients with UC( Reference Jowett, Seal and Phillips 14 ). Speculation that the high sulfur or sulfate compounds in many of these foods was the trigger associated with the relapse of UC has led to a number of studies( Reference Jowett, Seal and Phillips 14 , Reference Pithadia and Jain 222 ) not least of which were those of Marquet et al. ( Reference Marquet, Duncan and Chassard 103 ), Shatalin et al. ( Reference Shatalin, Shatalina and Mironov 102 ) and Jia et al. ( Reference Jia, Whitehead and Griffiths 91 ) that highlighted the toxicity of hydrogen sulfide, possibly mediated through the impaired utilisation of butyrate in colonocytes( Reference Roediger, Duncan and Kapaniris 223 ). Carbohydrates have also been reported to result in colonic inflammation( Reference Sakamoto, Kono and Wakai 224 ) and promote UC in some individuals( Reference Pithadia and Jain 222 ). Although enteral feeding to control dietary intake has been effective in the treatment of CD, it is ineffective in UC( Reference King, Woolner and Hunter 225 ).
Large numbers of peptides have been isolated from both milk( Reference Choi, Sabikhi and Hassan 226 – Reference Meisel 228 ) and meat( Reference Schmid 229 – Reference Minkiewicz, Dziuba and Michalska 232 ) and the possible existence of small proinflammatory peptides similar to the previously identified proinflammatory bacterial peptides cannot be excluded. Recently, Chatterton et al.( Reference Chatterton, Nguyen and Bering 233 ) reviewed the anti-inflammatory mechanisms of milk proteins that assist in the prevention of a severe form of intestinal inflammation known as necrotising enterocolitis which is associated with a high mortality in neonates. In this review, the authors commented that although raw human milk contains many anti-inflammatory proteins (for example, immunoglubulins that chelate bacterial and viral proinflammatory antigens), bovine milk may have much fewer anti-inflammatory components due to digestive proteolysis and pasteurisation( Reference Chatterton, Nguyen and Bering 233 ). Various growth factors present in both human and bovine milk have been reported as having anti-inflammatory properties( Reference Rutherfurd-Markwick and Moughan 15 , Reference Chatterton, Nguyen and Bering 233 ). Transforming growth factor-β1 (TGF-β1) was reported by Letterio et al.( Reference Letterio, Geiser and Kulkarni 234 ) to have anti-inflammatory properties, which were later attributed to the modulation of inflammatory responses( Reference Boll, Markovich and Weber 179 ). TGF-β regulates the differentiation of T helper 17 cells (Th17-cells) which maintain intestinal barrier integrity and produce the anti-inflammatory cytokine IL-10( Reference Fei, Kanai and Nussberger 235 ). Heparin-binding epidermal growth factor-like growth factor was found to attenuate bacterial binding to the intestinal mucosa( Reference Tanida, Kataoka and Mizoshita 236 ), repress the cytokine-induced activation of NF-κB and release of proinflammatory cytokines( Reference Mehta and Besner 237 , Reference Mehta and Besner 238 ).
The hypothesis that dietary proteins and their hydrolysates contain peptides that may affect mucin secretion has been studied by a number of researchers( Reference Montagne, Toullec and Formal 239 – Reference Han, Deglaire and Sengupta 242 ). The casomorphins, a family of bioactive peptides derived from milk β-casein, are opioid agonists known to affect the secretion of mucin, a protective response that also stimulates the production of epidermal growth factor, that in turn promotes epithelial cell proliferation( Reference Miner-Williams 243 ). The effect of various opioid-acting casomorphins on mucin secretion has been reported by a number of researchers( Reference Claustre, Toumi and Trompette 241 , Reference Trompette, Claustre and Caillon 244 ) who found that the intraluminal administration of β-casomorphin-7 provoked a 500% increase (over the controls) in the secretion of mucin. β-Casomorphin-7 seems unique in this respect as little or no increase in mucin secretion was observed from any of the other opioid peptides tested. Milk-borne opioid receptor ligands have been extensively reviewed by Clare & Swaisgood( Reference Clare and Swaisgood 245 ). Zoghbi et al.( Reference Zoghbi, Trompette and Claustre 246 ) reported that β-casomorphin-7 increased the expression of rMuc2 and may contribute significantly to mucin production through a direct effect on intestinal goblet cells and the activation of μ-opioid receptors. Because intestinal mucins are an integral part of epithelial barrier function, dietary supplements containing β-casomorphin-7 are worthy of investigation for their potential to improve intestinal protection in IBD.
Secretory mucin MUC2 is the predominant structural component of the mucus layer and is abundantly expressed by goblet cells in the colon( Reference van Klinken, Einerhand and van der Wal 247 , Reference Van Klinken, Van der Wal and Einerhand 248 ). MUC2 synthesis is decreased in both human and animal models of IBD( Reference Tytgat, Opdam and Einerhand 249 , Reference Tytgat, vanderWal and Einerhand 250 ), and the expression of MUC2 is considered as a phenotypic marker, which can be inversely correlated with the severity of inflammation( Reference Shaoul, Okada and Cutz 251 ). Quantitative changes in mucin secretion occur in IBD that include structural changes in the glycoprotein core together with the sulfation and sialylation of the oligosaccharide residues. Such changes are associated with a dysfunctional mucous barrier( Reference Boltin, Perets and Vilkin 252 ). In UC, defensin synthesis and activity are not disturbed, even in inflamed mucosa; however, deficiencies in the mucus layer of UC patients are indicative of defects in goblet cell differentiation. With a diminished mucus layer the secreted defensins in physiologically normal concentrations are not retained and this allows bacteria to pass through the epithelium and induce inflammation( Reference Gersemann, Wehkamp and Stange 6 ). There is evidence that matrix metalloproteinases (MMP) are the predominant extracellular proteinases within the mucosa disorders such as IBD and peptic ulcer disease( Reference Xie and Costello 253 – Reference Kirkegaard, Hansen and Bruun 255 ). Both serum and tissue levels of MMP-9, localised in the colonic mucosa( Reference Castaneda, Walia and Vijay-Kumar 256 ), are known to correlate with disease activity in UC( Reference Baugh, Perry and Hollander 254 , Reference Baugh, Evans and Hollander 257 , Reference Kossakowska, Medlicott and Edwards 258 ). Garg et al. ( Reference Garg, Ravi and Patel 259 ) demonstrated that MMP-9 modulates MUC2 expression by regulating goblet cell differentiation. Overexpression of MMP-9 inhibits goblet cell differentiation with a concomitant decrease in MUC2 mucin( Reference Shields, Christy and Yang 260 , Reference Ng, Waring and Ristevski 261 ). The aberrant expression of MMP-9, observed in inflammatory conditions, leads to the impaired differentiation and a consequent decrease in goblet cell function, known to be associated with increased susceptibility to bacterial infection/inflammation( Reference Garg, Ravi and Patel 259 ). A target for future IBD therapies may be to strengthen the mucous barrier through the up-regulation/down-regulation of MUC genes, the manipulation of post-transcriptional processing, or targeting the mucin molecule itself( Reference Boltin, Perets and Vilkin 252 ).
Epithelial barrier dysfunction, peptides and coeliac disease
CeD is a chronic immune-mediated disorder that primarily affects the mucosa of the small intestine. The condition is a food antigen-triggered autoimmune disorder that involves an immune response (both innate and adaptive) following exposure to dietary gluten-containing foods in genetically predisposed individuals. The pathogenesis of CeD involves a triad of predisposing genes, gluten and other environmental factors. The inception of CeD follows the deamination of gliadins, monomeric proteins contained in gluten, by tissue transglutaminase. Gliadin immunogenic fragments, resistant to endopeptidases, then bind to the chemokine CXCR3 present on the luminal surface of the enterocytes. Zonulin, one of the transmembrane proteins that regulate epithelial barrier permeability, may then be released from the TJ as a result of gliadin binding to CXCR3( Reference Lammers, Lu and Brownley 262 ). The MyD88 adapter protein-dependent release of zonulin, highly expressed in CeD, results in the disassembly of the TJ and a subsequent increase in intestinal permeability( Reference Fasano, Not and Wang 263 ) and activation of antigen-presenting cells (for example, macrophages, dendritic cells and B cells). These cells in turn display the gliadin peptides and interact with gluten-specific CD4+ T cells in the lamina propria. CD4+ T cells release inflammatory cytokines (for example, IFN-γ and IL-15) that facilitate the transformation of intraepithelial lymphocytes into cytotoxic CD8+ T cells that kill the intestinal epithelial cells. The cumulative effect of this inflammatory cascade is the manifestation of villous atrophy and crypt hyperplasia( Reference Green, Lebwohl and Greywoode 264 ). Inflammation of the intestinal epithelia consequent to the gliadin assault on the enterocytes may then exacerbate intestinal barrier dysfunction leading to the increased passage of antigens involved in the pathogenesis of CeD. In genetically predisposed individuals (those carrying the HLA-DQ2 or less commonly HLA-DQ8 haplotype) environmental factors, such as rotavirus infection, toxins or Fe-deficiency anaemia, are thought to initiate the development of CeD( Reference Menard, Cerf-Bensussan and Heyman 28 ). It is interesting to note that constitutive abnormalities in intestinal permeability are not the hallmarks of food allergy; increased epithelial permeability is more the consequence of immunological changes producing villous atrophy, rather than the cause of food sensitisation( Reference Heyman 265 ). For example, significant increases in the trans-epithelial transport of horseradish peroxidase (about 44 kDa) are observed in children with active cows’ milk allergy, which return to normal levels following a cows’ milk-free diet( Reference Menard, Cerf-Bensussan and Heyman 28 ). However, increases in epithelial permeability are the cause of a self-perpetuating cycle that maintains allergic inflammation( Reference Heyman, Abed and Lebreton 266 ).
A number of non-dietary therapies are currently under investigation that target specific aspects of CeD pathogenesis including intraluminal agents, immunomodulators and vaccination( Reference Perez, de Villasante and Ruiz 267 , Reference Crowe 268 ). Larazotide acetate (AT-1001), a peptide derived from cholera toxin, is thought to regulate intestinal paracellular permeability by inhibiting the disassembly of intestinal TJ. However, current clinical trials with larazotide have not demonstrated any decrease in intestinal permeability in CeD patients taking the drug, although decreased tissue transglutaminase IgA levels and improved clinical symptoms were observed( Reference Leffler, Kelly and Abdallah 269 , Reference Kelly, Green and Murray 270 ). Other proposed treatments include: reduced gluten exposure by genetic modification of the cereal grains containing gluten( Reference Spaenij-Dekking, Kooy-Winkelaar and Van Veelen 271 , Reference Carroccio, Di Prima and Noto 272 ); or using co-polymeric binders of gluten( Reference Pinier, Fuhrmann and Galipeau 273 ); pre-digestion of gluten before intestinal epithelial cell uptake using prolyl-endopeptidases( Reference Mitea, Havenaar and Drijfhout 274 , Reference Tack, Van de Water and Bruins 275 ); transglutaminase inhibitors or the blockade of HLA-DQ2/DQ8( Reference Schuppan, Junker and Barisani 276 – Reference Xia, Siegel and Bergseng 278 ); and immune tolerance induction( Reference Keech, Dromey and Chen 279 , Reference Brown, Daveson and Marjason 280 ).
Peptide permeability: is it the cause or consequence of intestinal disorders?
That intestinal barrier dysfunction is the primary cause of intestinal disorders, such as food allergy, CeD and IBD, involving the transformation of antigenic tolerance into antigenic sensitisation following the excessive absorption of antigens, is a longstanding hypothesis to explain the pathogenesis of these diseases( Reference Menard, Cerf-Bensussan and Heyman 28 ). However, in food allergy, constitutive abnormality in intestinal permeability is unrecorded, and permeability to horseradish peroxidase in infants with active bovine milk allergy returns to normal following treatment with a milk-free diet( Reference Heyman, Grasset and Ducroc 281 ). Infection might be the triggering factor in CeD where inflammation increases intestinal permeability to gliadin peptides and thus immune responses in susceptible individuals. Rotavirus infection is associated with a higher risk of CeD in early childhood( Reference Stene, Honeyman and Hoffenberg 282 ). Increased permeability to intact gliadin peptides observed in active CeD( Reference Matysiak-Budnik, Candalh and Dugave 283 ) ceases in most coeliac patients once treated with a gluten-free diet.
The hypothesis that a primary defect is the cause of intestinal barrier dysfunction in patients with IBD arises from observations of hyperpermeability in CD patients before phenotypic symptoms arise and an increased permeability in the healthy relatives of IBD patients( Reference Hollander, Vadheim and Brettholz 22 ). A genetic susceptibility to CD has been indicated by genome-wide association studies where a number of distinct genomic loci are involved in the maintenance of epithelial barrier integrity( Reference Xavier and Podolsky 8 , Reference Van Limbergen, Wilson and Satsangi 284 ). Variants of the myosin IXB (myo 9B) gene, that codes for the myosin IX motor protein, have been associated with IBD( Reference Latiano, Palmieri and Valvano 285 , Reference Van Bodegraven, Curley and Hunt 286 ) and CeD( Reference Monsuur, de Bakker and Alizadeh 287 ), and may suggest a primary defect in intestinal permeability in these diseases( Reference Menard, Cerf-Bensussan and Heyman 28 ). Foster & Zheng( Reference Foster and Zheng 199 ) demonstrated that the inflammatory cytokine IFN-γ increases intestinal PepT1 expression, which in turn mediates the absorption of the proinflammatory bacterial peptide fMLP( Reference Foster, Landowski and Welage 288 ). They demonstrated that cephalexin inhibits fMLP transport across cultured intestinal monolayers, which partially attenuates polymorphonuclear leucocyte-induced intestinal hyperpermeability. From their data it may be concluded that the use of pharmacological PepT1 substrates such as cephalexin may represent a novel means of preserving intestinal barrier integrity( Reference Foster and Zheng 199 ).
Current therapy
Steroids remain the conventional treatment in acute inflammation of CD( Reference Bryant, Brain and Travis 289 ). The two main immunosuppressant drugs are azathioprine and methotrexate( Reference Teml, Schaeffeler and Herrlinger 290 ). Unfortunately, current anti-inflammatory therapy remains unsatisfactory due to substantial side effects and uncontrolled relapses( Reference Wehkamp and Stange 25 ). Treatment with antibiotics are only effective in limited situations probably due to the modification of commensal microbiota( Reference Wehkamp and Stange 25 ). Probiotics can modify luminal microbiota to suppress pathogens by producing inhibitory substances including H2O2, organic acids and bacteriocins, substances which inhibit both the proliferation of pathogens, toxin production and bacterial metabolism( Reference Khan, Kale and Bere 2 ). Although the treatment of colitis in human patients with Lactobacillus plantarum has yet to be established, it has been reported to be effective in the treatment and prevention of colitis in Il10 –/– mice( Reference de Medina, Daddaoua and Requena 12 , Reference Daniel 185 , Reference Darfeuille-Michaud, Boudeau and Bulois 291 ) by modulating the apical junctional complex proteins and PepT1-mediated transepithelial transport( Reference Chassaing and Darfeuille-Michaud 93 ). Probiotic bacteria like E. coli Nissle 1917 can induce antimicrobial peptides( Reference Wehkamp, Harder and Wehkamp 292 , Reference Moendel, Schroeder and Zimmermann 293 ) mediated by a specific flagellin( Reference Schlee, Wehkamp and Altenhoefer 294 ). It has been shown to be as effective as mesalazine in maintaining remission in UC( Reference Kruis, Frič and Pokrotnieks 295 ) although its effectiveness is much lower in the treatment of CD.
Since the advent of anti-TNF-α agents in the mid-1990s the treatment of IBD has changed significantly. Potent anti-inflammatory medications for the treatment of IBD have emerged. Infliximab has proved effective for the induction and maintenance of remission in CD and UC that leads to mucosal healing, the reduction of hospitalisations and surgeries, the closure of intestinal fistula and the improvement of patients’ quality of life( Reference Fiorino, Danese and Peyrin-Biroulet 296 ). Infliximab is a chimeric anti-TNF-α antibody that binds to proinflammatory TNF-α and induces complement activation and the apoptosis of inflammatory cells( Reference D’Haens 83 ). However, the high cost of treatment and the uncertainty of long-term safety have led to limited clinical use. A number of toxicity issues related to anti-TNF-α therapies have been reported including cases of heart failure and mycobacterial infections with the possible development of autoimmune disease, lymphomas and neurological disorders( Reference Blonski and Lichtenstein 297 – Reference Siegel, Hur and Korzenik 300 ).
Many researchers report that although antibiotics and some probiotics may alleviate some of the symptoms of IBD they do not reduce inflammation completely( Reference Dalmasso, Hang and Ingersoll 198 , Reference Pithadia and Jain 222 , Reference Khan, Ullman and Ford 301 – Reference Triantafillidis, Merikas and Georgopoulos 304 ). Surely if the action of bacterial di- and tripeptides were the sole cause of inflammation, antibiotics would be expected to remove such inflammation completely. However, this is not the case and would indicate that bacterial di- and tripeptides are not the only cause of inflammation in IBD.
As PepT1 is up-regulated in IBD, and absorbs proinflammatory peptides such as fMLP (in UC) and is linked to NOD receptors, it is a worthy target for IBD therapeutics. Although a number of researchers have characterised novel high-affinity inhibitors for PepT1 and their use for the delivery of peptidomemitic drugs( Reference Nielsen, Brodin and Jorgensen 305 – Reference Knutter, Theis and Hartrodt 308 ), few have explored the application of PepT1 inhibitors in the treatment of IBD. However, a number of peptides have been identified as having PepT1-mediated anti-inflammatory activity in animal models and these include Lys-Pro-Val( Reference Kovacs-Nolan, Zhang and Ibuki 309 ) and Lys-Pro-Tyr( Reference Dalmasso, Charrier-Hisamuddin and Nguyen 310 ) peptides and the peptidomemitic drug cephalexin( Reference Foster and Zheng 199 ). In the future such enteral peptidomemitic drugs and nutritional oligopeptides may offer patients with IBD an alternative to conventional treatments.
IL-6, released by activated submucosal macrophages and CD4+ T cells, is a key factor in the uncontrolled inflammatory process of IBD and antibodies against this cytokine have shown promise in phase I and II clinical trials( Reference Ito, Takazoe and Fukuda 311 , Reference Choy, Isenberg and Garrood 312 ). In view of the severe side effects associated with TNF-α therapy there is a need to identify alternative IBD therapeutic strategies that do not entirely block cytokine responses. The application of the blocking protein sgp130 selectively inhibits IL-6 trans-signalling without affecting signals mediated via membrane-bound IL-6R and Mitsuyama et al.( Reference Mitsuyama, Sata and Rose-John 313 ) have suggested that the use of this strategy in inflammatory disease will lead to fewer side effects.
Recently exclusive enteral nutrition (EEN) has been shown to be more effective than corticosteroids in achieving mucosal healing for paediatric CD sufferers( Reference Navas-López, Blasco-Alonso and Maseri 314 ). Navas-López et al.( Reference Navas-López, Blasco-Alonso and Maseri 314 ) found that EEN administered for 6–8 weeks was effective in decreasing mucosal inflammation and inducing clinical remission. Although several studies have demonstrated the efficacy of EEN as a maintenance therapy, the long-term effectiveness of EEN has not been investigated fully. If EEN is an effective therapy for the maintenance of remission, it may reduce the use of steroids and immunosuppressive drugs together with reducing the serious adverse events associated with these medications( Reference Konno, Takahashi and Toita 315 ). In their study of the long-term administration of enteral aminosalicylates, Konno et al. ( Reference Konno, Takahashi and Toita 315 ) reduced the rate of intestinal surgery in paediatric CD. However, there are conflicting views on the efficacy of EEN and concomitant infliximab therapy. While Hirai et al. ( Reference Hirai, Ishihara and Yada 316 ) found that cumulative remission was considerably higher in patients receiving EEN than a non-EEN control, Yamamoto et al.( Reference Yamamoto, Nakahigashi and Umegae 317 ) found similar remission rates for both groups. In response, Chiba et al. ( Reference Chiba, Tsuji and Komatsu 318 ) pointed out a number of flaws in the previous studies and showed that the efficacy of EEN during infliximab maintenance therapy is dependent upon when infliximab is first administered. They demonstrated that although there is little benefit for patients with short disease duration, there were substantial benefits for patients of long disease duration( Reference Chiba, Tsuji and Komatsu 318 ). Research into the effectiveness of EEN therapy for maintaining remission in both child and adult CD sufferers requires long-term, multisite randomised controlled trials.
Conclusion
There can be little doubt that deleterious proinflammatory di- and tripeptides can cross the intestinal epithelia barrier in relatively small amounts (from a nutritional perspective) and produce an immune response that may result in inflammation. Furthermore, it can also be assumed that inflammation can induce further leakage of the intestinal epithelial barrier. Whereas the intestinal absorption of the bacterial peptides fMLP, MDP and Tri-DAP, via PepT1, cause an inflammatory response there is no reason to believe that these are the only small peptides that can be absorbed via the PepT1 transporter. Other food-borne di- and tripeptides may also resist acid hydrolysis and enzymic proteolysis, causing an immune response that leads to inflammation in IBD. Although a number of researchers have highlighted the potential of toxic food-derived peptides( Reference Picariello, Ferranti and Fierro 319 , Reference Schaafsma 320 ), the incidence of IBD even in the antigen-poor Western societies is small in comparison with the number of healthy individuals exposed to the same food antigens and must be considered evidential that in normal healthy subjects the transport of both small di- and tripeptides and/or macromolecules is (other than in antigen sampling) below the threshold necessary to cause an inflammatory disease. Other than in disease states, digestive processes both effectively and efficiently degrade dietary proteins so that only a few intact molecules come into contact with antibody molecules and any resultant complexes are unlikely to be absorbed( Reference Walker, Wu and Isselbacher 321 ).
The present review raises a number of questions that warrant further research. If the main cause of the IBD inflammation is faulty peptide transporters can these be inhibited or attenuated without any detrimental effects to the patients’ nutritional uptake from the intestinal tract? The work of Hu et al.( Reference Hu, Smith and Ma 176 ) with mice devoid of a functional PepT1 transporter indicates that this may be the case. As β-casomorphin-7 is known to increase the secretion of mucin in the small intestine( Reference Claustre, Toumi and Trompette 241 ), can this small peptide be utilised therapeutically to increase mucin protection in intestinal tissues denuded of mucins, such as in UC?
Acknowledgements
We would like to thank the Riddet Institute, a New Zealand Government-funded Centre of Research Excellence, for supporting this research.
There was no direct source of funding for this research.
The first draft of this manuscript was written by W. M. M.-W. and edited by P. J. M.
The authors report no conflicts of interest with this research.


