Malnutrition during fetal development and infancy – e.g. DHA, Fe and folic acid deficiencies(Reference Guesnet and Alessandri1–Reference Christian, Murray-Kolb and Khatry5) – can have severe negative implications for the brain and nervous system and may also affect the outcome of pregnancy, leading to premature births and the birth of weak or stillborn offspring(Reference Christian6, Reference McDonald, Han and Mulla7). We have hypothesised that vitamin C (vitC) is a pivotal factor in normal brain development(Reference Tveden-Nyborg and Lykkesfeldt8), and have recently reported that prenatal vitC deficiency leads to persistent postnatal hippocampal volume reduction and impaired neuronal migration(Reference Tveden-Nyborg, Vogt and Schjoldager9). Moreover, we have shown that postnatal vitC deficiency has detrimental effects on hippocampal neuron number and function(Reference Tveden-Nyborg, Johansen and Raida10), and that severe postnatal vitC depletion in weanling guinea pigs leads to oxidative stress in the brain(Reference Lykkesfeldt, Trueba and Poulsen11). In mice, a functional vitC transport (mediated by the Na-dependent vitC co-transporter 2; SVCT2) has been shown to be essential for perinatal survival(Reference Sotiriou, Gispert and Cheng12). However, knowledge of vitC deficiency's impact on in utero fetal development remains sparse.
Due to a mutation in the gene encoding for l-gulonolactone oxidase required for ascorbate synthesis, humans and guinea pigs are dependent on a dietary vitC source(Reference Frikke-Schmidt, Tveden-Nyborg, Lykkesfeldt, Hermann and Obeid13). Consequently, fetal supply of vitC relies on an adequate maternal dietary intake(Reference Duarte and Lunec14) and, expectedly, the maternal:fetal transport ratio has been shown to be dependent on maternal vitC status(Reference Teel, Burke and Draper15–Reference de Oliveira, Rondo and Barros17). In humans, vitC in plasma shows a declining maternal plasma concentration during pregnancy(Reference Teel, Burke and Draper15), while vitC levels in newborns are typically higher than those in their post-parturient mothers(Reference Dejmek, Ginter and Solansky16, Reference de Oliveira, Rondo and Barros17). This is most probably due to an active placental transport of ascorbate as indicated in both in vitro and in vivo studies(Reference Sotiriou, Gispert and Cheng12, Reference Prasad, Huang and Wang18, Reference Biondi, Pavan and Dalpiaz19). As with humans, the guinea pig has a haemochorial placenta and is a validated animal model for placental transfer(Reference Huppertz20–Reference Enders22). This makes the guinea pig a rational model for studying the putative effects of dietary-imposed chronic vitC deficiency on pregnancy outcome.
In humans, a daily intake of 200–400 mg vitC for adults results in plasma concentrations of about 70 μmol/l(Reference Frei, Birlouez-Aragon and Lykkesfeldt23, Reference Levine, Conry-Cantilena and Wang24), at which point tissue saturation is achieved. However, vitC deficiency (hypovitaminosis C) defined as a plasma concentration < 23 μmol/l and severe vitC deficiency (a plasma concentration < 11 μmol/l)(Reference Smith and Hodges25) remains surprisingly common. Even in the developed world, large cross-sectional studies have found that vitC deficiency affects between 7 and 40 % of the population, with high-risk subpopulations such as pregnant women, children and smokers being most vulnerable(Reference de Oliveira, Rondo and Barros17, Reference Schleicher, Carroll and Ford26–Reference Lykkesfeldt, Halliwell and Poulsen28). In spite of this high prevalence of vitC deficiency, its potential impact on pregnancy and newborns has only been the subject of little attention.
Using guinea pigs as an in vivo model, the present study investigated the impact of a prolonged subclinical maternal vitC deficiency during the second and third trimesters on maternal and early postnatal vitC status, birth weight and perinatal survival in guinea pig offspring.
Materials and methods
In vivo study
The study was approved by the Danish Animal Experimentation Inspectorate. Upon arrival, a total of eighty time-mated Dunkin Hartley guinea pigs (Charles River Laboratories), second-time breeders at gestational day 18, were weighed and tagged with a 12 mm microchip subcutaneously in the neck for identification (Pet-Id; Danworth Farm). The animals were randomly assigned to one of two weight-stratified groups that received special quality-controlled diets (Special Diets Services) either sufficient in vitC (923 mg/kg by analysis; product code: 829427-B#32 786; control (CTRL) group, n 30) or deficient in vitC (100 mg/kg by analysis; product code: 829415-B#32 785; deficient (DEF) group, n 50). The diets were produced to meet the nutritional requirements of guinea pigs and were identical in all aspects except vitC content. We have previously shown that a dose of 100 mg vitC/kg induces a non-scorbutic vitC deficiency in guinea pigs(Reference Lykkesfeldt and Moos29, Reference Tveden-Nyborg, Hasselholt and Miyashita30). Accordingly, none of the animals exhibited any clinical signs of scurvy during the study. The groups were allowed ad libitum feeding, dried hay (tested negative for vitC content) and drinking water. They were housed in large floor pens with a 12 h light cycle and tended several times daily by trained staff. All newborn pups (live: n 285, dead: n 58) were recorded within a maximum of 24 h from birth and viable pups were weighed, marked for identification and their sex determined.
Dams and pups were weighed once and twice weekly, respectively, and eight dams from each group (each time randomly chosen by a computer) were assigned for the collection of blood samples at arrival (ultimo first trimester, gestational day 18) and at the beginning of the third trimester (gestational day 40). Blood samples were collected from the saphenous vein on its superficial course on the tibia (300 μl). Only one pregnant female from the CTRL group died due to pregnancy toxaemia and five animals proved not to have conceived.
Euthanasia
At postnatal days 2–7 (pups) and postpartum days 7–10 (dams), four groups of newborn pups (n 10–11; females/males; CTRL/DEF) and two groups of dams (n 5–10; CTRL/DEF) were anaesthetised with inhalation of isoflurane (Isoba Vet 100 %; Intervet International), and blood (4 ml) was gently collected by cardiac puncture with an 18G hypodermic needle in a 5 ml syringe previously flushed with 15 % tripotassium-EDTA(Reference Lykkesfeldt31). Subsequently, while still anaesthetised, the animals were euthanised by exsanguination and decapitation. Plasma from dams and female/male pups and brains from female pups (either the right or left hemisphere, randomly assigned by a computer and divided by section through the cerebral longitudinal fissure) were immediately frozen at − 80°C until biochemical analyses were performed. The remaining brain tissues, pups and postpartum dams were allocated to other studies.
Biochemical analyses
Analysis of ascorbate and dehydroascorbic acid in a meta-phosphoric acid-stabilised plasma and brain homogenate was performed at HPLC with coulometric detection as described previously(Reference Lykkesfeldt32, Reference Lykkesfeldt33). As a measure of ascorbate oxidation, the ascorbate oxidation ratio in the brain was calculated as a percentage of dehydroascorbic acid of total vitC. Glutathione was measured spectrofluorometrically according to the method by Hissin & Hilf(Reference Hissin and Hilf34) modified for microtitre plates. Lipid oxidation was assessed by measuring malondialdehyde as described previously(Reference Lykkesfeldt35) and 8-F2-isoprostanes as described by the assay kit manufacturer (catalogue no. 516351; Cayman Chemicals).
Statistical analysis
Differences in body weight and plasma concentrations of ascorbic acid were evaluated by one-way ANOVA followed by Tukey's multiple comparison test to describe differences in means. In the case of a non-Gaussian distribution as established by the D'Agostino and Pearson omnibus normality test, evaluation was conducted by the Kruskal–Wallis test followed by Dunn's multiple comparisons. Data were analysed using Statistica (Statsoft). To assess the differences in perinatal mortality, logistic regression analysis corrected for overdispersion was carried out in SAS version 9.2 (SAS Institute, Inc.). The significance level for all statistical analyses was P< 0·05 and data are expressed as means and standard deviations unless otherwise stated.
Results
Maternal weight gain and birth weight
No signs of pregnancy-associated disease or dystocia were recorded for dams brought to term. Maternal weight gain during gestation was similar between the groups (Fig. 1). Due to group housing in large pens, individual litter sizes could not be recorded and because of a few incidences of cannibalism, the recorded number of perinatally dead pups could be slightly flawed. There was no significant difference in the birth weight of live-born pups between the groups (CTRL 80·0 (sd 18·1) g v. DEF 80·7 (sd 15·9) g) or sex (females 80·06 (sd 16·59) g v. males 80·97 (sd 16·91) g).
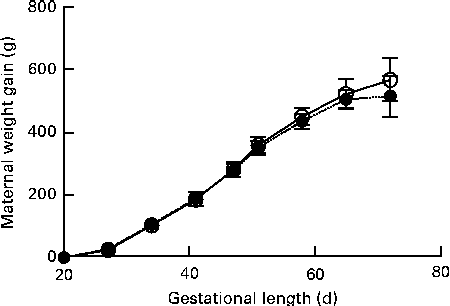
Fig. 1 Maternal weight gain during gestation. Body weights were recorded once weekly. Data are shown as weight gain normalised to body weight on arrival. Data are presented using body-weight gestational day 18 as baseline. Values are means, with their standard errors represented by vertical bars. There was no significant difference between the deficient (![]() , n 45) and control (
, n 45) and control (![]() , n 29) dams at any of the recorded time points.
, n 29) dams at any of the recorded time points.
Plasma levels of total vitamin C concentration
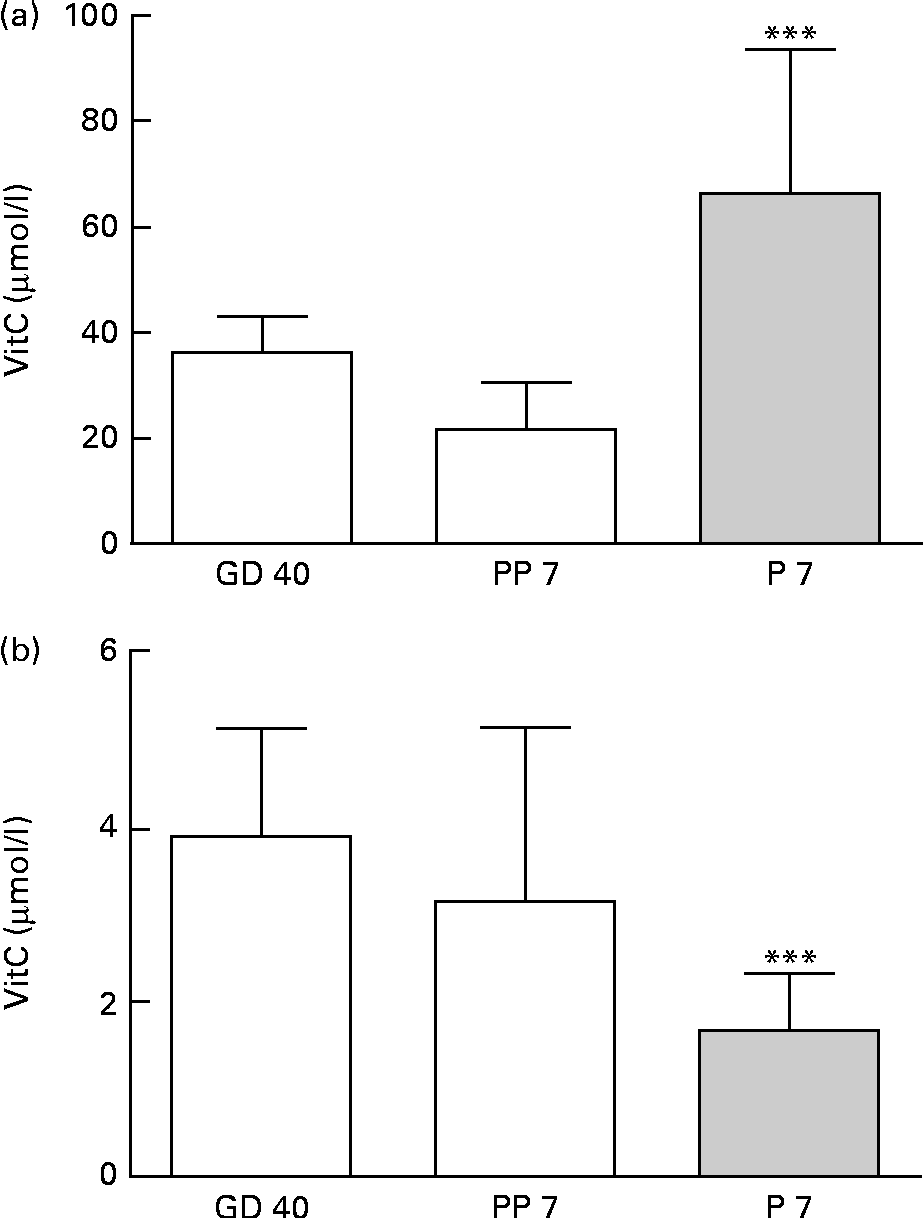
Upon arrival at gestational day 18, dams displayed plasma levels of total vitC of 39·8 (sd 11·8) μmol/l. As expected, the dietary regimen was reflected in the plasma levels of total vitC, CTRL dams had significantly higher plasma vitC levels than DEF dams at both gestational day 40 (CTRL: 36·3 (sd 6·8) v. DEF: 3·9 (sd 1·2) μmol/l) and at postpartum days 7–10 (CTRL: 21·8 (sd 9·0) v. DEF: 3·1 (sd 2·0) μmol/l), as displayed in Fig. 2. Towards parturition, vitC status in the CTRL dams declined significantly (P< 0·01; Fig. 2). In the DEF dams, plasma vitC levels declined significantly within 3 weeks after arrival (to 3·9 (sd 1·2) μmol/l) and remained at this low level during pregnancy and parturition (P< 0·001; Fig. 2). The dietary regimen was also reflected in plasma vitC concentrations of newborn pups. However, as no effect of sex on vitC concentration could be detected in either group of newborns, vitC data from both sexes were pooled within the groups. The newborn CTRL pups had significantly higher plasma vitC concentrations (CTRL pups: 66·3 (sd 27·4) μmol/l) compared with the CTRL dams at parturition (P< 0·001; Fig. 3(a)); however, in contrast, the newborn DEF pups had a significantly lower plasma level of vitC (1·6 (sd 0·7) μmol/l) compared with their mothers (P< 0·001; Fig. 3(b)).
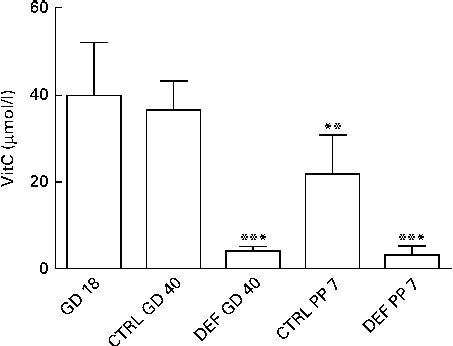
Fig. 2 Plasma vitamin C (vitC) levels in dams. Initial vitC levels were recorded upon arrival at gestational day (GD) 18. On both GD 40 and postpartum day (PP) 7, plasma vitC levels in the deficient (DEF) dams were significantly lower compared with the control (CTRL) dams (*** P< 0·001), reflecting the two applied dietary regimens. Also, plasma vitC levels in the CTRL dams declined significantly during gestation (** P< 0·01). Data are obtained from randomly chosen pregnant/postpartum dams (n 5–10; CTRL/DEF) at each time point. Values are means, with standard deviations represented by vertical bars.

Fig. 3 Plasma vitamin C (vitC) levels in dams and offspring. (a) Newborn control (CTRL) female (n 10) and male (n 10) pups at postnatal day (P) 7 had significantly higher vitC levels than the CTRL dams at postpartum day(PP) 7 (*** P< 0·001). P7 female and male data are pooled as no effect of sex was observed. (b) Newborn deficient (DEF) female (n 11) and male (n 10) pups at postnatal day (P) 7 had significantly lower plasma vitC than the DEF dams during gestation (*** P< 0·001). P7 female and male data are pooled as no effect of sex was observed. Values are means, with standard deviations represented by vertical bars.
Oxidative stress markers in the brain
In the newborn DEF female pups, vitC concentrations in the brain decreased by 60 % as a result of the maternal dietary regimen (P< 0·001; Table 1). No effects on the measured oxidative stress and damage markers, i.e. ascorbate oxidation ratio, glutathione, malondialdehyde or 8-F2-isoprostanes, were found in the brain (Table 1).
Table 1 Antioxidant status and lipid oxidation markers in the brains of newborn guinea pig pups (Mean values and standard deviations)

AOR, ascorbate oxidation ratio; vitC, vitamin C.
*** Mean value was significantly different compared with the control animals (P< 0·001; one-way ANOVA using Tukey's post hoc test of planned comparisons).
† AOR is calculated as dehydroascorbic acid divided by total vitC in percentage.
Perinatal survival
A total of 143 CTRL (n 29 dams) and 200 DEF (n 45 dams) pups were delivered. Of these, eighteen CTRL and forty DEF pups were recorded as dead perinatally (i.e. dead in utero or within a maximum of 24 h after birth). All dead pups appeared to be delivered at or very close to term. No macroscopic causes of death could be identified, except from one mummified fetus (DEF) and five moribund newborns (DEF) that were immediately euthanised by cervical dislocation. To evaluate the association between the maternal diet and the offspring's risk of perinatal death, data were fitted to a logistic regression model corrected for overdispersion between day and diet. When corrected for overdispersion, the effect of diet was not significant (P= 0·18) with an OR for perinatal death being (DEF v. CTRL) 1·7 (95 % CI 0·8, − 3·9).
Discussion
In both humans and guinea pigs, maternal vitC intake affects vitC concentrations in offspring(Reference Pate, Lukert and Kipp36–Reference Norkus, Bassi and Rosso39). During gestation, maternal plasma levels decline even when maternal vitC intake is high(Reference Teel, Burke and Draper15). In agreement, the present study also reports of a declining maternal vitC concentration in both investigated dietary groups during gestation. Interestingly, DEF pups were significantly lower in vitC than DEF dams at gestational day 40, i.e. during the third trimester.
It has been proposed that the placental–fetal consolidation is genetically programmed to alter maternal physiology to benefit the fetus and to ensure survival until birth, sometimes at the expense of the mother(Reference Haig40). Conditions such as gestational hypertension and pre-eclampsia have been suggested to develop when the mother is unable to meet fetal nutritional requirements(Reference Haig40) and in cases of insufficient Ca, fetal supply is ensured by maternal release from the bone, ultimately decreasing maternal skeletal strength(Reference Haig41). In humans, an insufficient maternal vitC intake, defined from maternal blood samples taken at parturition or dietary information provided by pregnant women, has been reported to increase fetal:maternal plasma vitC ratios (i.e. fetal levels increase and maternal levels decrease) even under conditions associated with maternal deficiency(Reference Teel, Burke and Draper15, Reference Lund and Kimble42). This has led to the proposal of a selective retention of vitC by the placenta, ensuring adequate fetal levels even during severe maternal deficiency. However, it should be noted that the cited reports on maternal vitC deficiency date back to the late 1930s and early 1940s, where analytical methodology did not appropriately control for post-sampling and storage-induced degradation of ascorbate. Considering the labile nature of vitC(Reference Lykkesfeldt33), there is reason to believe that the authors may have significantly underestimated the vitC content of the blood samples in the absence of adequate technology. Moreover, the majority of the individuals included were in fact not severely vitC deficient by current definitions(Reference Smith and Hodges25). Consequently, it remains to be investigated to what extent the present data can be translated to human pregnancies.
Recent data emphasise SVCT2 as the dominant contributor to the placental transfer of vitC(Reference Sotiriou, Gispert and Cheng12, Reference Prasad, Huang and Wang18, Reference Biondi, Pavan and Dalpiaz19, Reference Harrison, Dawes and Meredith43) and in vivo studies in SVCT2( − / − ) mice have strongly suggested that placental uptake of vitC requires functional SVCT2 transporters(Reference Harrison, Dawes and Meredith43). Thus, regulatory mechanisms associated with a preferential transport across the placenta are most likely to be SVCT2-mediated. The present findings suggest that maternal deficiency during gestation may override such preferential maternal transport, sustaining a basal maternal vitC concentration preventing maternal scurvy at the expense of the offspring.
Natural selection favouring short-term survival over long-term health and reproduction through the allocation of scarce micronutrients by triage is a known hypothesis(Reference Ames44) and has been reported, for example, for vitamin K and Se(Reference Mccann and Ames45, Reference Mccann and Ames46). Reports have stated that vitC concentrations in fetal blood decrease towards birth(Reference Zalani, Rajalakshmi and Parekh47, Reference Das and Powers48) and up to 50 % perinatally(Reference Braestrup49–Reference Norkus and Rosso51); thus, postnatally obtained plasma samples might differ from the prenatal level. However, mean vitC concentrations in newborn CTRL pups were 50-fold higher in plasma and 3-fold higher in brain tissue compared with newborn DEF pups, making comparable fetal vitC exposure in utero highly unlikely.
VitC is well accepted to have a pivotal role as an antioxidant and in the maintenance of redox homeostasis, and serves as a cofactor in the formation and reuptake of neurotransmitters in the brain(Reference Tveden-Nyborg and Lykkesfeldt8). The developing brain is highly susceptible to oxidative damage due to immaturity and decreased levels and synthesis of antioxidant enzymes, high oxygen consumption and readily oxidisable PUFA(Reference Ikonomidou and Kaindl52). Increased oxidative stress in the brain has been shown in SVCT2( − / − ) mice delivered by caesarian sections close to term(Reference Harrison, Dawes and Meredith43) and in the offspring of gulonolactone oxidase-knockout mice unable to synthesise vitC(Reference Harrison, Meredith and Dawes53); thus, it is conceivable that maternal vitC deficiency leads to prenatal oxidative stress. In humans, oxidative stress has been suggested to play a role in the pathogenesis of a number of diseases in the pregnant mother and the newborn(Reference Gitto, Reiter and Karbownik54), including intraventricular haemorrhage leading to neonatal brain damage(Reference Buonocore, Perrone and Longini55). Furthermore, oxidative stress has been suggested as a plausible link between adverse fetal development and pregnancy outcome and the risk of developing chronic diseases later in life(Reference Luo, Fraser and Julien56). However, in the present study, no differences in oxidative stress markers – apart from vitC itself – were observed between the groups, suggesting that vitC deficiency imposed on these animals did not induce oxidative stress in the narrow time window studied here (Table 1), although the brain levels of vitC in the DEF newborns were decreased by approximately 60 % compared with the CTRL group. This observation is in accordance with previously obtained results(Reference Tveden-Nyborg, Vogt and Schjoldager9, Reference Tveden-Nyborg, Johansen and Raida10) indicating that alterations in redox homeostasis may not be the primary course underlying the detrimental effects of vitC deficiency on brain development(Reference Tveden-Nyborg, Vogt and Schjoldager9, Reference Tveden-Nyborg, Johansen and Raida10).
Mice devoid of SVCT2 die immediately after birth, displaying intraparenchymal brain haemorrhage emphasising an essential role of vitC in sustaining perinatal survival and normal brain development(Reference Sotiriou, Gispert and Cheng12). This finding has been confirmed by Harrison et al. (Reference Harrison, Dawes and Meredith43); SVCT2( − / − ) newborns died shortly after birth; however, fewer SVCT2( − / − ) and SVCT2(+/ − ) offspring (generated from SVCT2(+/ − ) stock) than expected were recorded at delivery, indicating that a lack of vitC in utero contributes to increased fetal mortality. This is suggested to be, at least in part, due to an impairment of collagen IV synthesis leading to fragile capillaries and a subsequent increased risk of intra-cortical haemorrhages and subsequent death in utero or at parturition(Reference Harrison, Dawes and Meredith43).
In human subjects, a low maternal intake of vitC has been associated with an increased risk of pregnancy-associated disorders such as pre-eclampsia(Reference Zhang, Williams and King57) and a variety of adverse pregnancy outcomes such as preterm delivery(Reference Siega-Riz, Promislow and Savitz58) and decreased birth weight(Reference Lee, Hong and Lee59). To reduce the risk of such adverse outcomes, intake of periconceptional multivitamins on a daily basis has been suggested(Reference Catov, Bodnar and Olsen60, Reference Hasan, Olshan and Herring61); however, other authors have failed to establish a positive correlation between maternal vitC intake and pregnancy outcome(Reference Roberts, Myatt and Spong62–Reference Wang, Ren and Liao66). Hence, whether supplementation should be offered on a regular basis during pregnancy or rather in the case of potentially ‘risk’ pregnancies remains controversial. Moreover, to evaluate the true effect of vitC deficiency and supplementation, inclusion criteria and vitC status must be well defined between groups which, in human trials, have often been neglected, leading to inconclusive and divergent data(Reference Lykkesfeldt and Poulsen67). Thus, as discussed above, there are presently insufficient data to conclude whether maternal vitC deficiency also leads to deficiency in the offspring of humans.
In conclusion, chronic maternal vitC intake during the second and third trimesters does not significantly influence pregnancy outcome with respect to gestational length, maternal weight gain, birth weight and perinatal survival in guinea pigs. However, the present data combined with previously reported results(Reference Sotiriou, Gispert and Cheng12, Reference Harrison, Dawes and Meredith43) suggest that an inadequate maternal vitC intake during pregnancy may indeed pose a risk to the fetus. The findings of reduced plasma vitC in newborn DEF pups compared with their mothers indicate that preferential transport of vitC from the mother to the fetus is overridden during a prolonged maternal vitC deficiency, hereby maintaining a basal maternal vitC concentration at the expense of the offspring. The present results contradict the notion that a fetus is protected from vitC deficiency and suggest that a prolonged maternal vitC deficiency may have a detrimental effect on fetal brain development in utero. Well-designed clinical studies of the impact of maternal vitC deficiency on fetal development are therefore warranted.
Acknowledgements
The authors wish to thank Annie Bjergby Kristensen, Elisabeth Veyhe Andersen and Joan Frandsen for their excellent technical assistance. The present study was supported by grants from the Danish National Research Council (#FSS271-08-0763) and The LIFEPHARM Centre for in vivo pharmacology to J. L. The authors' contributions were as follows: J. G. S., P. T.-N. and J. L. planned the study; J. G. S. and P. T.-N. conducted the in vivo experiment; J. G. S. and J. L. performed the data analysis; J. G. S., P. T.-N. and J. L. wrote the paper. The authors declare no conflict of interest which could influence the presented work.